
Influence of Substitutions in the Binding Motif of Proline-Rich Antimicrobial Peptide ARV-1502 on 70S Ribosome Binding and Antimicrobial Activity


Abstract
:1. Introduction
2. Results
2.1. ARV-1502 Binds Well to E. coli 70S Ribosome
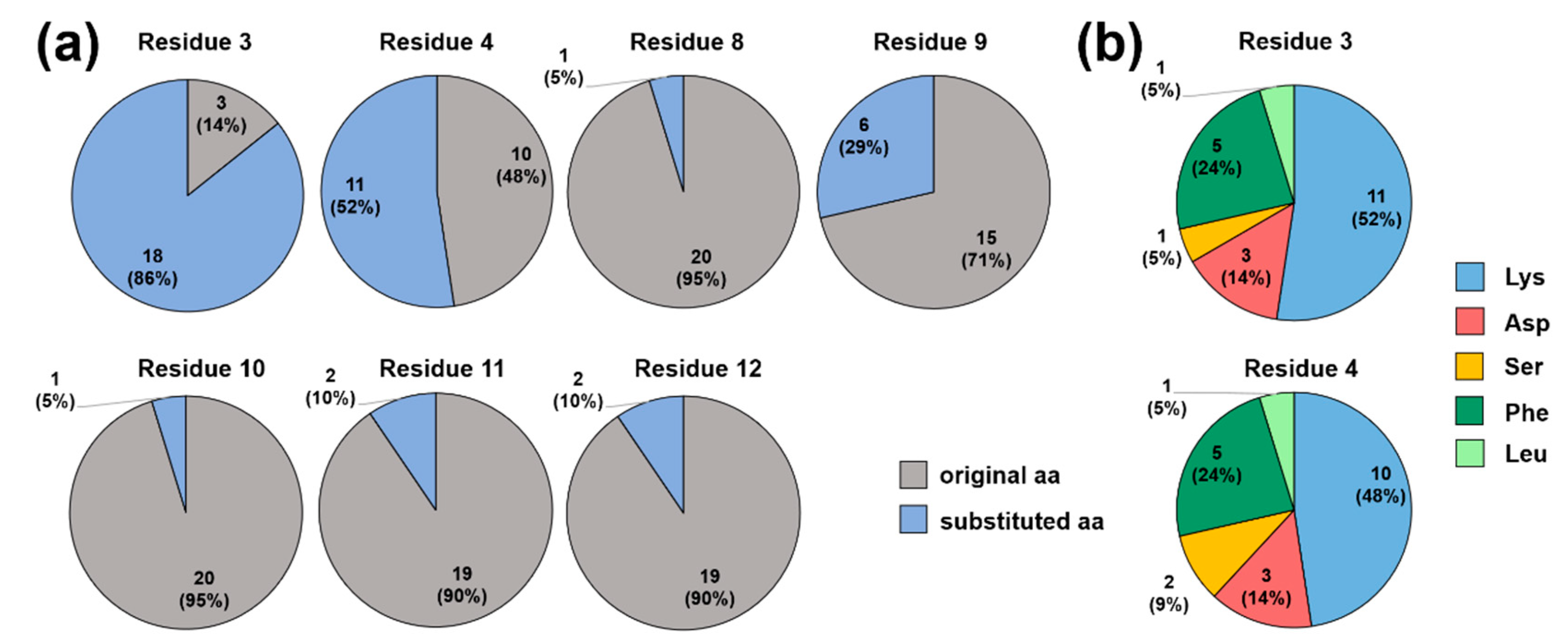
2.2. Screening for Competitive Peptide Binders
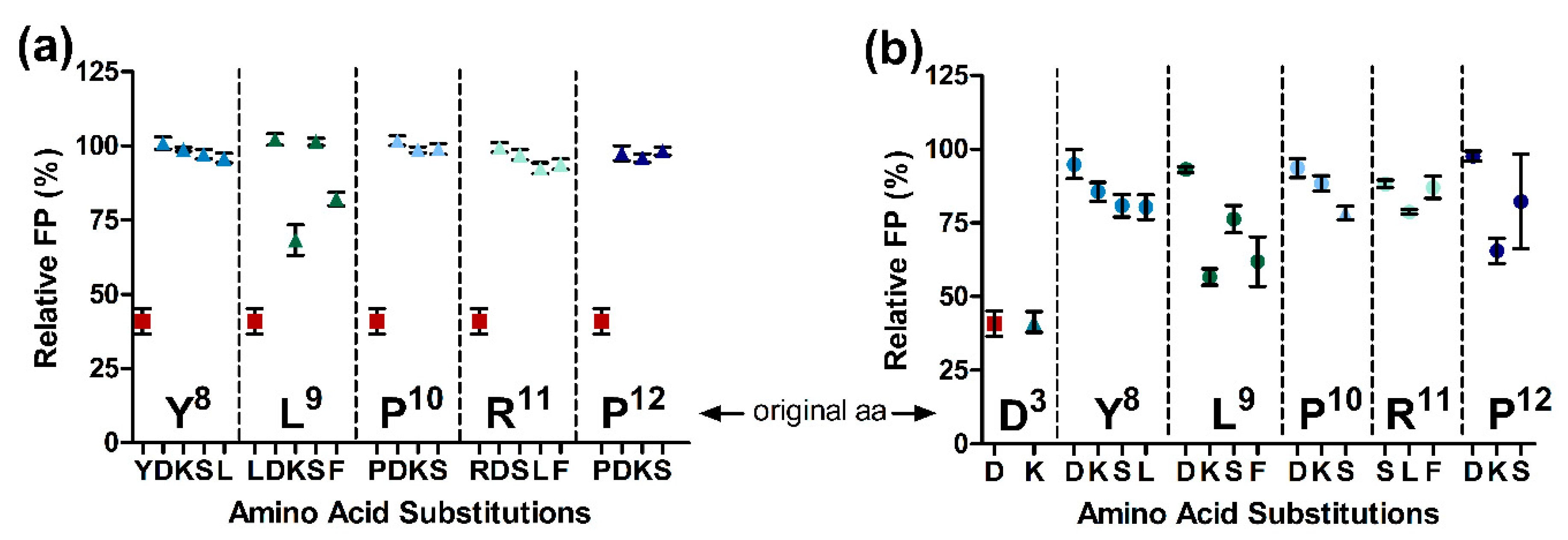
2.3. Substitutions in the Binding Motif YLPRP
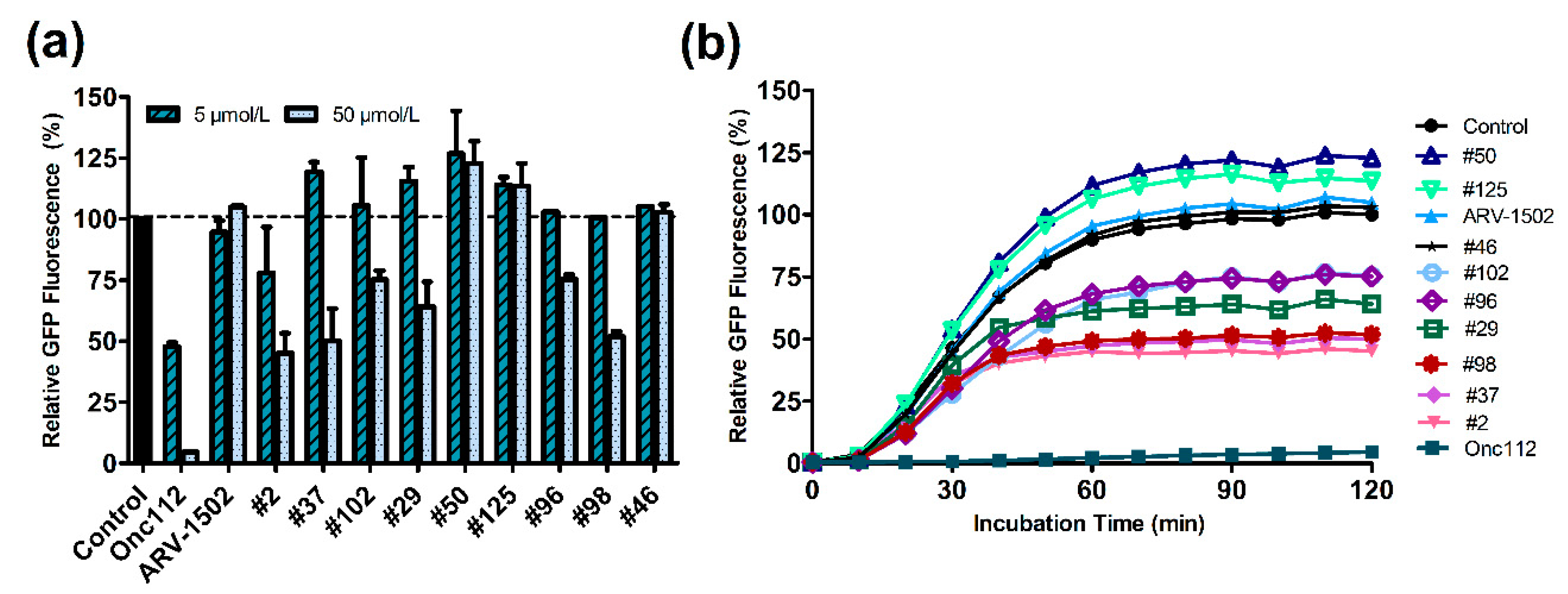
2.4. Inhibitory Effect on In Vitro Translation
2.5. Activity in ΔsbmA and ΔmdtM E. coli BW25113 Strains
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Preparation of E. coli 70S Ribosomes
4.3. Determination of Dissociation and Inhibitory Constants
4.4. Screening for Competitive Binder Peptides
4.5. Antimicrobial Activity
4.6. Cell-Free Protein Expression Assay
4.7. Checkerboard Assay
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Scocchi, M.; Tossi, A.; Gennaro, R. Proline-rich antimicrobial peptides: Converging to a non-lytic mechanism of action. Cell. Mol. Life Sci. 2011, 68, 2317–2330. [Google Scholar] [CrossRef]
- Li, W.; Tailhades, J.; O’Brien-Simpson, N.M.; Separovic, F.; Otvos, L.; Hossain, M.A.; Wade, J.D. Proline-rich antimicrobial peptides: Potential therapeutics against antibiotic-resistant bacteria. Amino Acids 2014, 46, 2287–2294. [Google Scholar] [CrossRef] [PubMed]
- Otvos, L., Jr. The short proline-rich antibacterial peptide family. Cell. Mol. Life Sci. 2002, 59, 1138–1150. [Google Scholar] [CrossRef]
- Krizsan, A.; Volke, D.; Weinert, S.; Sträter, N.; Knappe, D.; Hoffmann, R. Insect-derived proline-rich antimicrobial peptides kill bacteria by inhibiting bacterial protein translation at the 70S ribosome. Angew. Chem. 2014, 53, 12236–12239. [Google Scholar] [CrossRef] [PubMed]
- Runti, G.; Lopez Ruiz, M.D.C.; Stoilova, T.; Hussain, R.; Jennions, M.; Choudhury, H.G.; Benincasa, M.; Gennaro, R.; Beis, K.; Scocchi, M. Functional characterization of SbmA, a bacterial inner membrane transporter required for importing the antimicrobial peptide Bac7(1-35). J. Bacteriol. 2013, 195, 5343–5351. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kolano, L.; Knappe, D.; Volke, D.; Sträter, N.; Hoffmann, R. Ribosomal Target-Binding Sites of Antimicrobial Peptides Api137 and Onc112 Are Conserved among Pathogens Indicating New Lead Structures to Develop Novel Broad-Spectrum Antibiotics. Chembiochem 2020, 21, 2628–2634. [Google Scholar] [CrossRef]
- Otvos, L., Jr.; de Olivier Inacio, V.; Wade, J.D.; Cudic, P. Prior antibacterial peptide-mediated inhibition of protein folding in bacteria mutes resistance enzymes. Antimicrob. Agents Chemother. 2006, 50, 3146–3149. [Google Scholar] [CrossRef] [Green Version]
- Otvos, L.; Rogers, M.E.; Consolvo, P.J.; Condie, B.A.; Lovas, S.; Bulet, P.; Blaszczyk-Thurin, M. Interaction between Heat Shock Proteins and Antimicrobial Peptides. Biochemistry 2000, 39, 14150–14159. [Google Scholar] [CrossRef]
- Knappe, D.; Goldbach, T.; Hatfield, M.P.D.; Palermo, N.Y.; Weinert, S.; Sträter, N.; Hoffmann, R.; Lovas, S. Proline-rich Antimicrobial Peptides Optimized for Binding to Escherichia coli Chaperone DnaK. Protein Pept. Lett. 2016, 23, 1061–1071. [Google Scholar] [CrossRef]
- Zahn, M.; Berthold, N.; Kieslich, B.; Knappe, D.; Hoffmann, R.; Sträter, N. Structural studies on the forward and reverse binding modes of peptides to the chaperone DnaK. J. Mol. Biol. 2013, 425, 2463–2479. [Google Scholar] [CrossRef]
- Brakel, A.; Kolano, L.; Kraus, C.N.; Otvos, L.; Hoffmann, R. Functional Effects of ARV-1502 Analogs Against Bacterial Hsp70 and Implications for Antimicrobial Activity. Front. Chem. 2022, 10, 798006. [Google Scholar] [CrossRef] [PubMed]
- Knappe, D.; Ruden, S.; Langanke, S.; Tikkoo, T.; Ritzer, J.; Mikut, R.; Martin, L.L.; Hoffmann, R.; Hilpert, K. Optimization of oncocin for antibacterial activity using a SPOT synthesis approach: Extending the pathogen spectrum to Staphylococcus aureus. Amino Acids 2016, 48, 269–280. [Google Scholar] [CrossRef] [PubMed]
- Czihal, P.; Knappe, D.; Fritsche, S.; Zahn, M.; Berthold, N.; Piantavigna, S.; Müller, U.; van Dorpe, S.; Herth, N.; Binas, A.; et al. Api88 is a novel antibacterial designer peptide to treat systemic infections with multidrug-resistant Gram-negative pathogens. ACS Chem. Biol. 2012, 7, 1281–1291. [Google Scholar] [CrossRef]
- Krizsan, A.; Prahl, C.; Goldbach, T.; Knappe, D.; Hoffmann, R. Short Proline-Rich Antimicrobial Peptides Inhibit Either the Bacterial 70S Ribosome or the Assembly of its Large 50S Subunit. Chembiochem 2015, 16, 2304–2308. [Google Scholar] [CrossRef] [PubMed]
- Seefeldt, A.C.; Graf, M.; Pérébaskine, N.; Nguyen, F.; Arenz, S.; Mardirossian, M.; Scocchi, M.; Wilson, D.N.; Innis, C.A. Structure of the mammalian antimicrobial peptide Bac7(1-16) bound within the exit tunnel of a bacterial ribosome. Nucleic Acids Res. 2016, 44, 2429–2438. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Florin, T.; Maracci, C.; Graf, M.; Karki, P.; Klepacki, D.; Berninghausen, O.; Beckmann, R.; Vázquez-Laslop, N.; Wilson, D.N.; Rodnina, M.V.; et al. An antimicrobial peptide that inhibits translation by trapping release factors on the ribosome. Nat. Struct. Mol. Biol. 2017, 24, 752–757. [Google Scholar] [CrossRef]
- Roy, R.N.; Lomakin, I.B.; Gagnon, M.G.; Steitz, T.A. The mechanism of inhibition of protein synthesis by the proline-rich peptide oncocin. Nat. Struct. Mol. Biol. 2015, 22, 466–469. [Google Scholar] [CrossRef] [Green Version]
- Gagnon, M.G.; Roy, R.N.; Lomakin, I.B.; Florin, T.; Mankin, A.S.; Steitz, T.A. Structures of proline-rich peptides bound to the ribosome reveal a common mechanism of protein synthesis inhibition. Nucleic Acids Res. 2016, 44, 2439–2450. [Google Scholar] [CrossRef] [Green Version]
- Graf, M.; Mardirossian, M.; Nguyen, F.; Seefeldt, A.C.; Guichard, G.; Scocchi, M.; Innis, C.A.; Wilson, D.N. Proline-rich antimicrobial peptides targeting protein synthesis. Nat. Prod. Rep. 2017, 34, 702–711. [Google Scholar] [CrossRef]
- Holfeld, L.; Hoffmann, R.; Knappe, D. Correlating uptake and activity of proline-rich antimicrobial peptides in Escherichia coli. Anal. Bioanal. Chem. 2017, 409, 5581–5592. [Google Scholar] [CrossRef]
- Noto, P.B.; Abbadessa, G.; Cassone, M.; Mateo, G.D.; Agelan, A.; Wade, J.D.; Szabo, D.; Kocsis, B.; Nagy, K.; Rozgonyi, F.; et al. Alternative stabilities of a proline-rich antibacterial peptide in vitro and in vivo. Protein Sci. 2008, 17, 1249–1255. [Google Scholar] [CrossRef] [PubMed]
- Ostorhazi, E.; Holub, M.C.; Rozgonyi, F.; Harmos, F.; Cassone, M.; Wade, J.D.; Otvos, L. Broad-spectrum antimicrobial efficacy of peptide A3-APO in mouse models of multidrug-resistant wound and lung infections cannot be explained by in vitro activity against the pathogens involved. Int. J. Antimicrob. Agents 2011, 37, 480–484. [Google Scholar] [CrossRef] [PubMed]
- Kolano, L.; Knappe, D.; Berg, A.; Berg, T.; Hoffmann, R. Effect of Amino Acid Substitutions on 70S Ribosomal Binding, Cellular Uptake, and Antimicrobial Activity of Oncocin Onc112. Chembiochem 2021, 23, e202100609. [Google Scholar] [CrossRef]
- Graf, M.; Wilson, D.N. Intracellular Antimicrobial Peptides Targeting the Protein Synthesis Machinery. Adv. Exp. Med. Biol. 2019, 1117, 73–89. [Google Scholar] [CrossRef] [PubMed]
- Svetlov, M.S.; Plessa, E.; Chen, C.-W.; Bougas, A.; Krokidis, M.G.; Dinos, G.P.; Polikanov, Y.S. High-resolution crystal structures of ribosome-bound chloramphenicol and erythromycin provide the ultimate basis for their competition. RNA 2019, 25, 600–606. [Google Scholar] [CrossRef] [PubMed]
- Seefeldt, A.C.; Nguyen, F.; Antunes, S.; Pérébaskine, N.; Graf, M.; Arenz, S.; Inampudi, K.K.; Douat, C.; Guichard, G.; Wilson, D.N.; et al. The proline-rich antimicrobial peptide Onc112 inhibits translation by blocking and destabilizing the initiation complex. Nat. Struct. Mol. Biol. 2015, 22, 470–475. [Google Scholar] [CrossRef] [PubMed]
- Ostorhazi, E.; Voros, E.; Nemes-Nikodem, E.; Pinter, D.; Sillo, P.; Mayer, B.; Wade, J.D.; Otvos, L. Rapid systemic and local treatments with the antibacterial peptide dimer A3-APO and its monomeric metabolite eliminate bacteria and reduce inflammation in intradermal lesions infected with Propionibacterium acnes and meticillin-resistant Staphylococcus aureus. Int. J. Antimicrob. Agents 2013, 42, 537–543. [Google Scholar] [CrossRef]
- Lu, J.; Kobertz, W.R.; Deutsch, C. Mapping the electrostatic potential within the ribosomal exit tunnel. J. Mol. Biol. 2007, 371, 1378–1391. [Google Scholar] [CrossRef]
- Charneski, C.A.; Hurst, L.D. Positively charged residues are the major determinants of ribosomal velocity. PLoS Biol. 2013, 11, e1001508. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Karki, C.; Xiao, Y.; Li, L. Electrostatics of Prokaryotic Ribosome and Its Biological Implication. Biophys. J. 2020, 118, 1205–1212. [Google Scholar] [CrossRef]
- Requião, R.D.; de Souza, H.J.A.; Domitrovic, T.; Palhano, F.L. Increased ribosome density associated to positively charged residues is evident in ribosome profiling experiments performed in the absence of translation inhibitors. RNA Biol. 2016, 13, 561–568. [Google Scholar] [CrossRef] [PubMed]
- Paulsen, V.S.; Mardirossian, M.; Blencke, H.-M.; Benincasa, M.; Runti, G.; Nepa, M.; Haug, T.; Stensvåg, K.; Scocchi, M. Inner membrane proteins YgdD and SbmA are required for the complete susceptibility of Escherichia coli to the proline-rich antimicrobial peptide arasin 1(1-25). Microbiology 2016, 162, 601–609. [Google Scholar] [CrossRef] [PubMed]
- Mattiuzzo, M.; Bandiera, A.; Gennaro, R.; Benincasa, M.; Pacor, S.; Antcheva, N.; Scocchi, M. Role of the Escherichia coli SbmA in the antimicrobial activity of proline-rich peptides. Mol. Microbiol. 2007, 66, 151–163. [Google Scholar] [CrossRef] [PubMed]
- Krizsan, A.; Knappe, D.; Hoffmann, R. Influence of the yjiL-mdtM Gene Cluster on the Antibacterial Activity of Proline-Rich Antimicrobial Peptides Overcoming Escherichia coli Resistance Induced by the Missing SbmA Transporter System. Antimicrob. Agents Chemother. 2015, 59, 5992–5998. [Google Scholar] [CrossRef] [Green Version]
- Holdsworth, S.R.; Law, C.J. The major facilitator superfamily transporter MdtM contributes to the intrinsic resistance of Escherichia coli to quaternary ammonium compounds. J. Antimicrob. Chemother. 2013, 68, 831–839. [Google Scholar] [CrossRef] [Green Version]
- Holdsworth, S.R.; Law, C.J. Functional and biochemical characterisation of the Escherichia coli major facilitator superfamily multidrug transporter MdtM. Biochimie 2012, 94, 1334–1346. [Google Scholar] [CrossRef]
- Tan, J.; Huang, J.; Huang, Y.; Chen, Y. Effects of single amino acid substitution on the biophysical properties and biological activities of an amphipathic α-helical antibacterial peptide against Gram-negative bacteria. Molecules 2014, 19, 10803–10817. [Google Scholar] [CrossRef] [Green Version]
- Li, W.; Sani, M.-A.; Jamasbi, E.; Otvos, L.; Hossain, M.A.; Wade, J.D.; Separovic, F. Membrane interactions of proline-rich antimicrobial peptide, Chex1-Arg20, multimers. Biochim. Biophys. Acta 2016, 1858, 1236–1243. [Google Scholar] [CrossRef]
- Mathias, U.; Jung, M. Determination of drug-serum protein interactions via fluorescence polarization measurements. Anal. Bioanal. Chem. 2007, 388, 1147–1156. [Google Scholar] [CrossRef]





 ) or leucine (
) or leucine (  ).
) or leucine ( ).
).
) or leucine ( ).


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Peptide. | Sequence | Ki (nmol/L) |
|---|---|---|
| ARV-1502 | ChexRPDKPRPYLPRPRPPRPVR-NH2 | 135 ± 10 |
| Pyrrhocoricin | VDK GSYLPRPTPPRPIYNRN | 112 ± 7 |
| Drosocin | GKPRPYSPRPTSHPRPIRV | 3472 ± 158 |
| Onc112 | VDKPP YLPRPRPPRrIYNr-NH2 | 124 ± 2 |
| Api137 | gu-ONNRPVYIPRPRPPHPRL-OH | 267 ± 31 |
| Antibiotic | Ribosome Binding Site | Ki (nmol/L) |
| Kanamycin | 30S subunit | no fit |
| Streptomycin | 30S subunit | no fit |
| Chloramphenicol | 50S subunit | 1698 ± 481 |
| Erythromycin | 50S subunit | 23 ± 1.6 |
| Peptide | Sequence Motif | Minimal Inhibitory Concentration (µg/mL) | |||||
|---|---|---|---|---|---|---|---|
| 25% MHB2 | 33% TSB | ||||||
| wt | ∆sbmA | ∆sbmA ∆mdtM | wt | ∆sbmA | ∆sbmA ∆mdtM | ||
| ARV-1502 | DK YLPRP | 8 | 16–32 | 64 | 16 | 64 | 128 |
| #2 | KK YLPRP | 4 | 8 | 16 | 32–64 | 64 | 64 |
| #29 | KS YLPRP | 16 | 32 | 32 | 32 | 128 | 128 |
| #37 | KK YKPRP | 16 | 16 ** | 32 ** | 64 | ≥128 | >128 |
| #46 | KK YLPFP | 16 | 32 ** | 32 ** | 64 * | ≥128 * | >128 |
| #50 | SD YLPRP | 128 | >128 | >128 | 64 | >128 | >128 |
| #96 | FF YFPRP | 16 | 16–32 | 32 ** | 32 * | ≥128 | ≥128 |
| #98 | FF YLKRP | 8–16 | 32 | 32 | 32 | 128 | 128 |
| #99 | FF YLSRP | 16–32 | 32 | 32 | 64 | 128 | 128 |
| #102 | FF YLPLP | 32–64 | 64 | 64 | 64 | 64 | 64–128 |
| #123 | DK KFPRP | 32 | 64 | 128 | 128 | >128 | >128 |
| #125 | DK SKPRP | 128 | >128 | >128 | >128 | >128 | >128 |
| Onc112 | DK YLPRP | 8 | 16–32 | 64 | 16 | 64 | 128 |
| Peptide. | Sequence Motif | Ribosome Binding (%) | MIC (µg/mL) | In Vitro Translation (%) | Charge State |
|---|---|---|---|---|---|
| ARV-1502 | DK YLPRP | 40.9 ± 3.9 | 8 | 105.8 ± 0.9 | +6 |
| #2 | KK YLPRP | 40.3 ± 2.2 | 4 | 53.2 ± 8.2 | +8 |
| #29 | KS YLPRP | 78.5 ± 3.3 | 16 | 74.3 ± 10.1 | +7 |
| #37 | KK YKPRP | 56.7 ± 2.6 | 16 | 63.4 ± 13.3 | +9 |
| #46 | KK YLPFP | 87.1 ± 3.4 | 16–32 | 96.6 ± 3.1 | +7 |
| #50 | SD YLPRP | 67.9 ± 4.8 | 128 | 113.7 ± 9.1 | +5 |
| #96 | FF YFPRP | 62.9 ± 4.8 | 16 | 77.2 ± 1.9 | +6 |
| #98 | FF YLKRP | 98.3 ± 1.5 | 8–16 | 56.2 ± 2.1 | +7 |
| #102 | FF YLPLP | 76.5 ± 5.6 | 32–64 | 78.9 ± 3.5 | +5 |
| #125 | DK SKPRP | 86.3 ± 2.5 | 128 | 104.3 ± 9.3 | +7 |
| Onc112 | DK YLPRP | 32.1 ± 1.0 | 8 | 4.9 ± 0.3 | +5 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Brakel, A.; Krizsan, A.; Itzenga, R.; Kraus, C.N.; Otvos, L., Jr.; Hoffmann, R. Influence of Substitutions in the Binding Motif of Proline-Rich Antimicrobial Peptide ARV-1502 on 70S Ribosome Binding and Antimicrobial Activity. Int. J. Mol. Sci. 2022, 23, 3150. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23063150
Brakel A, Krizsan A, Itzenga R, Kraus CN, Otvos L Jr., Hoffmann R. Influence of Substitutions in the Binding Motif of Proline-Rich Antimicrobial Peptide ARV-1502 on 70S Ribosome Binding and Antimicrobial Activity. International Journal of Molecular Sciences. 2022; 23(6):3150. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23063150
Chicago/Turabian StyleBrakel, Alexandra, Andor Krizsan, Renke Itzenga, Carl N. Kraus, Laszlo Otvos, Jr., and Ralf Hoffmann. 2022. "Influence of Substitutions in the Binding Motif of Proline-Rich Antimicrobial Peptide ARV-1502 on 70S Ribosome Binding and Antimicrobial Activity" International Journal of Molecular Sciences 23, no. 6: 3150. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23063150