
Structural Plasticity of the Hippocampus in Neurodegenerative Diseases

 , ,
, ,
Abstract
:1. Introduction
2. Hippocampal Dysfunction in Neurodegenerative Diseases
2.1. Alzheimer’s Disease
2.2. Parkinson’s Disease
2.3. Huntington’s Disease
2.4. Multiple Sclerosis
2.5. Other Neurodegenerative Diseases
3. Structural Plasticity in the Hippocampus: General Overview
3.1. Dendritic Complexity
3.2. Dendritic Spine
4. Hippocampal Structural Plasticity in Neurodegenerative Diseases
4.1. Structural Plasticity in the Hippocampus with AD
4.1.1. Dendritic Complexity
4.1.2. Dendritic Spine Density and Morphology
4.2. Structural Plasticity in the Hippocampus with PD
4.2.1. Dendritic Complexity
4.2.2. Dendritic Spine Density and Morphology
4.3. Structural Plasticity in the Hippocampus with HD
4.3.1. Dendritic Complexity
4.3.2. Dendritic Spine Density and Morphology
4.4. Structural Plasticity in the Hippocampus with MS
4.4.1. Dendritic Complexity
4.4.2. Dendritic Spine Density and Morphology
4.5. Structural Plasticity in the Hippocampus with Other Neurodegenerative Diseases
4.5.1. Dendritic Complexity
4.5.2. Dendritic Spine Density and Morphology
5. Hippocampal Dysfunction and Structural Plasticity in the Aging Brain
6. Concluding Remarks
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Schaefers, A.T.; Teuchert-Noodt, G. Developmental neuroplasticity and the origin of neurodegenerative diseases. World J. Biol. Psychiatry 2016, 17, 587–599. [Google Scholar] [CrossRef] [PubMed]
- Keller, T.A.; Just, M.A. Structural and functional neuroplasticity in human learning of spatial routes. Neuroimage 2016, 125, 256–266. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sala, C.; Segal, M. Dendritic spines: The locus of structural and functional plasticity. Physiol. Rev. 2014, 94, 141–188. [Google Scholar] [CrossRef]
- Das, S.; Sadanandappa, M.K.; Dervan, A.; Larkin, A.; Lee, J.A.; Sudhakaran, I.P.; Priya, R.; Heidari, R.; Holohan, E.E.; Pimentel, A.; et al. Plasticity of local GABAergic interneurons drives olfactory habituation. Proc. Natl. Acad. Sci. USA 2011, 108, E646–E654. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuan, Q.; Xiang, Y.; Yan, Z.; Han, C.; Jan, L.Y.; Jan, Y.N. Light-induced structural and functional plasticity in Drosophila larval visual system. Science 2011, 333, 1458–1462. [Google Scholar] [CrossRef] [Green Version]
- Hotulainen, P.; Hoogenraad, C.C. Actin in dendritic spines: Connecting dynamics to function. J. Cell Biol. 2010, 189, 619–629. [Google Scholar] [CrossRef] [Green Version]
- Luo, L. Actin cytoskeleton regulation in neuronal morphogenesis and structural plasticity. Annu. Rev. Cell Dev. Biol. 2002, 18, 601–635. [Google Scholar] [CrossRef] [Green Version]
- Peche, V.; Shekar, S.; Leichter, M.; Korte, H.; Schröder, R.; Schleicher, M.; Holak, T.; Clemen, C.; Ramanath-y, B.; Pfitzer, G. CAP2, cyclase-associated protein 2, is a dual compartment protein. Cell. Mol. Life Sci. 2007, 64, 2702–2715. [Google Scholar] [CrossRef]
- Lai, K.O.; Ip, N.Y. Structural plasticity of dendritic spines: The underlying mechanisms and its dysregulation in brain disorders. Biochim. Biophys. Acta 2013, 1832, 2257–2263. [Google Scholar] [CrossRef] [Green Version]
- Dent, E.W.; Gertler, F.B. Cytoskeletal dynamics and transport in growth cone motility and axon guidance. Neuron 2003, 40, 209–227. [Google Scholar] [CrossRef] [Green Version]
- Van Aelst, L.; Cline, H.T. Rho GTPases and activity-dependent dendrite development. Curr. Opin. Neurobiol. 2004, 14, 297–304. [Google Scholar] [CrossRef] [PubMed]
- Verpelli, C.; Piccoli, G.; Zibetti, C.; Zanchi, A.; Gardoni, F.; Huang, K.; Brambilla, D.; Di Luca, M.; Battaglioli, E.; Sala, C. Synaptic activity controls dendritic spine morphology by modulating eEF2-dependent BDNF synthesis. J. Neurosci. 2010, 30, 5830–5842. [Google Scholar] [CrossRef] [PubMed]
- Rex, C.S.; Lin, C.Y.; Kramar, E.A.; Chen, L.Y.; Gall, C.M.; Lynch, G. Brain-derived neurotrophic factor promotes long-term potentiation-related cytoskeletal changes in adult hippocampus. J. Neurosci. 2007, 27, 3017–3029. [Google Scholar] [CrossRef] [PubMed]
- Vanderklish, P.W.; Edelman, G.M. Dendritic spines elongate after stimulation of group 1 metabotropic glutamate receptors in cultured hippocampal neurons. Proc. Natl. Acad. Sci. USA 2002, 99, 1639–1644. [Google Scholar] [CrossRef] [Green Version]
- Kutsarova, E.; Schohl, A.; Munz, M.; Wang, A.; Zhang, Y.Y.; Bilash, O.M.; Ruthazer, E.S. BDNF signaling in Hebbian and Stentian structural plasticity in the developing visual system. BioRxiv 2021. [Google Scholar] [CrossRef]
- Mattson, M.P. Glutamate and neurotrophic factors in neuronal plasticity and disease. Ann. N. Y. Acad. Sci. 2008, 1144, 97–112. [Google Scholar] [CrossRef] [Green Version]
- Xu, J.; Sriramula, S.; Lazartigues, E. Excessive Glutamate Stimulation Impairs ACE2 Activity Through ADAM17-Mediated Shedding in Cultured Cortical Neurons. Cell Mol. Neurobiol. 2018, 38, 1235–1243. [Google Scholar] [CrossRef]
- Gordon-Weeks, P.R.; Fournier, A.E. Neuronal cytoskeleton in synaptic plasticity and regeneration. J. Neurochem. 2014, 129, 206–212. [Google Scholar] [CrossRef]
- Hu, X.; Ballo, L.; Pietila, L.; Viesselmann, C.; Ballweg, J.; Lumbard, D.; Stevenson, M.; Merriam, E.; Dent, E.W. BDNF-induced increase of PSD-95 in dendritic spines requires dynamic microtubule invasions. J. Neurosci. 2011, 31, 15597–15603. [Google Scholar] [CrossRef] [Green Version]
- Ho, V.M.; Lee, J.A.; Martin, K.C. The cell biology of synaptic plasticity. Science 2011, 334, 623–628. [Google Scholar] [CrossRef] [Green Version]
- Mainardi, M.; Fusco, S.; Grassi, C. Modulation of hippocampal neural plasticity by glucose-related signaling. Neural. Plast. 2015, 2015, 657928. [Google Scholar] [CrossRef] [PubMed]
- Yamazaki, Y.; Fujiwara, H.; Kaneko, K.; Hozumi, Y.; Xu, M.; Ikenaka, K.; Fujii, S.; Tanaka, K.F. Short- and long-term functional plasticity of white matter induced by oligodendrocyte depolarization in the hippocampus. Glia 2014, 62, 1299–1312. [Google Scholar] [CrossRef] [PubMed]
- Magee, J.C.; Grienberger, C. Synaptic Plasticity Forms and Functions. Annu. Rev. Neurosci. 2020, 43, 95–117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Citri, A.; Malenka, R.C. Synaptic plasticity: Multiple forms, functions, and mechanisms. Neuropsychopharmacology 2008, 33, 18–41. [Google Scholar] [CrossRef] [Green Version]
- Ren, K.; Dubner, R. Pain facilitation and activity-dependent plasticity in pain modulatory circuitry: Role of BDNF-TrkB signaling and NMDA receptors. Mol. Neurobiol. 2007, 35, 224–235. [Google Scholar] [CrossRef] [PubMed]
- Ciccarelli, A.; Giustetto, M. Role of ERK signaling in activity-dependent modifications of histone proteins. Neuropharmacology 2014, 80, 34–44. [Google Scholar] [CrossRef] [PubMed]
- Alicea, D.; Perez, M.; Maldonado, C.; Dominicci-Cotto, C.; Marie, B. Cortactin Is a Regulator of Activity-Dependent Synaptic Plasticity Controlled by Wingless. J. Neurosci. 2017, 37, 2203–2215. [Google Scholar] [CrossRef] [Green Version]
- Reymann, K.G.; Frey, J.U. The late maintenance of hippocampal LTP: Requirements, phases, ‘synaptic tagging’, ‘late-associativity’ and implications. Neuropharmacology 2007, 52, 24–40. [Google Scholar] [CrossRef]
- Bliss, T.V.; Collingridge, G.L. A synaptic model of memory: Long-term potentiation in the hippocampus. Nature 1993, 361, 31–39. [Google Scholar] [CrossRef]
- Sajikumar, S.; Frey, J.U. Late-associativity, synaptic tagging, and the role of dopamine during LTP and LTD. Neurobiol. Learn. Mem. 2004, 82, 12–25. [Google Scholar] [CrossRef]
- Chidambaram, S.B.; Rathipriya, A.G.; Bolla, S.R.; Bhat, A.; Ray, B.; Mahalakshmi, A.M.; Manivasagam, T.; Thenmozhi, A.J.; Essa, M.M.; Guillemin, G.J.; et al. Dendritic spines: Revisiting the physiological role. Prog. Neuropsychopharmacol. Biol. Psychiatry 2019, 92, 161–193. [Google Scholar] [CrossRef] [PubMed]
- Hillman, C.H.; Erickson, K.I.; Kramer, A.F. Be smart, exercise your heart: Exercise effects on brain and cognition. Nat. Rev. Neurosci. 2008, 9, 58–65. [Google Scholar] [CrossRef] [PubMed]
- Mora, F. Successful brain aging: Plasticity, environmental enrichment, and lifestyle. Dialogues Clin. Neurosci. 2013, 15, 45–52. [Google Scholar]
- Leuner, B.; Falduto, J.; Shors, T.J. Associative memory formation increases the observation of dendritic spines in the hippocampus. J. Neurosci. 2003, 23, 659–665. [Google Scholar] [CrossRef]
- Beauquis, J.; Roig, P.; De Nicola, A.F.; Saravia, F. Short-term environmental enrichment enhances adult neurogenesis, vascular network and dendritic complexity in the hippocampus of type 1 diabetic mice. PLoS ONE 2010, 5, e13993. [Google Scholar] [CrossRef]
- Kang, S.; Son, Y.; Lee, S.; Kim, J.; Kim, J.C.; Kim, J.S.; Jung, U.; Kim, S.H.; Yang, M.; Moon, C. Changes in epigenetic markers, DNMT1 and HDAC1/2, in the adult mouse hippocampus after cranial irradiation. Neurosci. Lett. 2017, 657, 113–119. [Google Scholar] [CrossRef]
- Son, Y.; Kang, S.; Kim, J.; Lee, S.; Kim, J.C.; Kim, S.H.; Kim, J.S.; Jo, S.K.; Jung, U.; Youn, B.; et al. Possible involvement of hippocampal immediate-early genes in contextual fear memory deficit induced by cranial irradiation. Neurobiol. Learn Mem. 2016, 133, 19–29. [Google Scholar] [CrossRef]
- Sebastian, V.; Estil, J.B.; Chen, D.; Schrott, L.M.; Serrano, P.A. Acute physiological stress promotes clustering of synaptic markers and alters spine morphology in the hippocampus. PLoS ONE 2013, 8, e79077. [Google Scholar] [CrossRef] [PubMed]
- Cymerman, I.A.; Gozdz, A.; Urbanska, M.; Milek, J.; Dziembowska, M.; Jaworski, J. Structural Plasticity of Dendritic Spines Requires GSK3alpha and GSK3beta. PLoS ONE 2015, 10, e0134018. [Google Scholar] [CrossRef]
- Pyapali, G.K.; Turner, D.A. Increased dendritic extent in hippocampal CA1 neurons from aged F344 rats. Neurobiol. Aging 1996, 17, 601–611. [Google Scholar] [CrossRef]
- Graham, B.P.; van Ooyen, A. Mathematical modelling and numerical simulation of the morphological development of neurons. BMC Neurosci. 2006, 7 (Suppl. 1), S9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quackenbush, L.J.; Ngo, H.; Pentney, R.J. Evidence for nonrandom regression of dendrites of Purkinje neurons during aging. Neurobiol. Aging 1990, 11, 111–115. [Google Scholar] [CrossRef]
- Smith-Dijak, A.I.; Sepers, M.D.; Raymond, L.A. Alterations in synaptic function and plasticity in Huntington disease. J. Neurochem. 2019, 150, 346–365. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, M.; Wong, Y.C.; Ysselstein, D.; Severino, A.; Krainc, D. Synaptic, Mitochondrial, and Lysosomal Dysfunction in Parkinson’s Disease. Trends Neurosci. 2019, 42, 140–149. [Google Scholar] [CrossRef] [PubMed]
- Herms, J.; Dorostkar, M.M. Dendritic Spine Pathology in Neurodegenerative Diseases. Annu. Rev. Pathol. 2016, 11, 221–250. [Google Scholar] [CrossRef] [PubMed]
- Mikkonen, M.; Soininen, H.; Alafuzof, I.; Miettinen, R. Hippocampal plasticity in Alzheimer’s disease. Rev. Neurosci. 2001, 12, 311–325. [Google Scholar] [CrossRef] [PubMed]
- Verkhratsky, A.; Rodriguez, J.J.; Parpura, V. Astroglia in neurological diseases. Future Neurol. 2013, 8, 149–158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luebke, J.I.; Weaver, C.M.; Rocher, A.B.; Rodriguez, A.; Crimins, J.L.; Dickstein, D.L.; Wearne, S.L.; Hof, P.R. Dendritic vulnerability in neurodegenerative disease: Insights from analyses of cortical pyramidal neurons in transgenic mouse models. Brain Struct Funct. 2010, 214, 181–199. [Google Scholar] [CrossRef] [Green Version]
- Kweon, J.H.; Kim, S.; Lee, S.B. The cellular basis of dendrite pathology in neurodegenerative diseases. BMB Rep. 2017, 50, 5–11. [Google Scholar] [CrossRef] [Green Version]
- Penzes, P.; Cahill, M.E.; Jones, K.A.; VanLeeuwen, J.E.; Woolfrey, K.M. Dendritic spine pathology in neuropsychiatric disorders. Nat. Neurosci. 2011, 14, 285–293. [Google Scholar] [CrossRef] [Green Version]
- Gorrie, G.H.; Fecto, F.; Radzicki, D.; Weiss, C.; Shi, Y.; Dong, H.; Zhai, H.; Fu, R.; Liu, E.; Li, S.; et al. Dendritic spinopathy in transgenic mice expressing ALS/dementia-linked mutant UBQLN2. Proc. Natl. Acad. Sci. USA 2014, 111, 14524–14529. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, H.; Yi, J.H.; Choi, K.; Hong, S.; Shin, K.S.; Kang, S.J. Regional differences in acute corticosterone-induced dendritic remodeling in the rat brain and their behavioral consequences. BMC Neurosci. 2014, 15, 65. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ang, M.J.; Lee, S.; Wada, M.; Weerasinghe-Mudiyanselage, P.D.E.; Kim, S.H.; Shin, T.; Jeon, T.I.; Im, S.S.; Moon, C. SREBP-1c Deficiency Affects Hippocampal Micromorphometry and Hippocampus-Dependent Memory Ability in Mice. Int. J. Mol. Sci. 2021, 22, 6103. [Google Scholar] [CrossRef]
- Torres, M.D.; Garcia, O.; Tang, C.; Busciglio, J. Dendritic spine pathology and thrombospondin-1 deficits in Down syndrome. Free Radic Biol. Med. 2018, 114, 10–14. [Google Scholar] [CrossRef] [PubMed]
- Hou, Y.; Dan, X.; Babbar, M.; Wei, Y.; Hasselbalch, S.G.; Croteau, D.L.; Bohr, V.A. Ageing as a risk factor for neurodegenerative disease. Nat. Rev. Neurol. 2019, 15, 565–581. [Google Scholar] [CrossRef] [PubMed]
- Galea, L.A.; Leuner, B.; Slattery, D.A. Hippocampal plasticity during the peripartum period: Influence of sex steroids, stress and ageing. J. Neuroendocrinol. 2014, 26, 641–648. [Google Scholar] [CrossRef] [PubMed]
- Dickstein, D.L.; Weaver, C.M.; Luebke, J.I.; Hof, P.R. Dendritic spine changes associated with normal aging. Neuroscience 2013, 251, 21–32. [Google Scholar] [CrossRef] [Green Version]
- Boros, B.D.; Greathouse, K.M.; Gearing, M.; Herskowitz, J.H. Dendritic spine remodeling accompanies Alzheimer’s disease pathology and genetic susceptibility in cognitively normal aging. Neurobiol. Aging. 2019, 73, 92–103. [Google Scholar] [CrossRef] [PubMed]
- Moreno-Jimenez, E.P.; Flor-Garcia, M.; Terreros-Roncal, J.; Rabano, A.; Cafini, F.; Pallas-Bazarra, N.; Avila, J.; Llorens-Martin, M. Adult hippocampal neurogenesis is abundant in neurologically healthy subjects and drops sharply in patients with Alzheimer’s disease. Nat. Med. 2019, 25, 554–560. [Google Scholar] [CrossRef]
- Tada, T.; Sheng, M. Molecular mechanisms of dendritic spine morphogenesis. Curr. Opin. Neurobiol. 2006, 16, 95–101. [Google Scholar] [CrossRef]
- Leuner, B.; Gould, E. Structural plasticity and hippocampal function. Annu. Rev. Psychol. 2010, 61, 111–140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mateus-Pinheiro, A.; Pinto, L.; Bessa, J.M.; Morais, M.; Alves, N.D.; Monteiro, S.; Patricio, P.; Almeida, O.F.; Sousa, N. Sustained remission from depressive-like behavior depends on hippocampal neurogenesis. Transl. Psychiatry 2013, 3, e210. [Google Scholar] [CrossRef]
- Dioli, C.; Patricio, P.; Trindade, R.; Pinto, L.G.; Silva, J.M.; Morais, M.; Ferreiro, E.; Borges, S.; Mateus-Pinheiro, A.; Rodrigues, A.J.; et al. Tau-dependent suppression of adult neurogenesis in the stressed hippocampus. Mol. Psychiatry 2017, 22, 1110–1118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, B.; Halabisky, B.; Zhou, Y.; Palop, J.J.; Yu, G.; Mucke, L.; Gan, L. Imbalance between GABAergic and Glutamatergic Transmission Impairs Adult Neurogenesis in an Animal Model of Alzheimer’s Disease. Cell Stem Cell 2009, 5, 624–633. [Google Scholar] [CrossRef] [Green Version]
- Zhu, T.; Zhu, M.; Qiu, Y.; Wu, Z.; Huang, N.; Wan, G.; Xu, J.; Song, P.; Wang, S.; Yin, Y.; et al. Puerarin Alleviates Vascular Cognitive Impairment in Vascular Dementia Rats. Front Behav. Neurosci. 2021, 15, 717008. [Google Scholar] [CrossRef] [PubMed]
- Moodley, K.K.; Chan, D. The hippocampus in neurodegenerative disease. Front. Neurol. Neurosci. 2014, 34, 95–108. [Google Scholar] [CrossRef]
- Herzog, J.J.; Deshpande, M.; Shapiro, L.; Rodal, A.A.; Paradis, S. TDP-43 misexpression causes defects in dendritic growth. Sci. Rep. 2017, 7, 15656. [Google Scholar] [CrossRef] [PubMed]
- Duff, K.; Paulsen, J.; Mills, J.; Beglinger, L.J.; Moser, D.J.; Smith, M.M.; Langbehn, D.; Stout, J.; Queller, S.; Harrington, D.L.; et al. Mild cognitive impairment in prediagnosed Huntington disease. Neurology 2010, 75, 500–507. [Google Scholar] [CrossRef] [Green Version]
- Checkoway, H.; Lundin, J.I.; Kelada, S.N. Neurodegenerative diseases. IARC Sci. Publ. 2011, 163, 407–419. [Google Scholar]
- Kobelt, G.; Thompson, A.; Berg, J.; Gannedahl, M.; Eriksson, J.; Group, M.S.; Platform, E.M.S. New insights into the burden and costs of multiple sclerosis in Europe. Mult. Scler. J. 2017, 23, 1123–1136. [Google Scholar] [CrossRef]
- Hardiman, O.; Al-Chalabi, A.; Chio, A.; Corr, E.M.; Logroscino, G.; Robberecht, W.; Shaw, P.J.; Simmons, Z.; van den Berg, L.H. Amyotrophic lateral sclerosis. Nat. Rev. Dis. Primers 2017, 3, 17085. [Google Scholar] [CrossRef] [PubMed]
- Beeldman, E.; Raaphorst, J.; Klein Twennaar, M.; de Visser, M.; Schmand, B.A.; de Haan, R.J. The cognitive profile of ALS: A systematic review and meta-analysis update. J. Neurol. Neurosurg Psychiatry 2016, 87, 611–619. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chaudhuri, K.R.; Healy, D.G.; Schapira, A.H.; National Institute for Clinical, E. Non-motor symptoms of Parkinson’s disease: Diagnosis and management. Lancet Neurol. 2006, 5, 235–245. [Google Scholar] [CrossRef]
- Estrada-Bellmann, I.; Camara-Lemarroy, C.R.; Calderon-Hernandez, H.J.; Rocha-Anaya, J.J.; Villareal-Velazquez, H.J. Non-motor symptoms and quality of life in patients with Parkinson’s disease in Northeastern Mexico. Acta Neurol. Belg. 2016, 116, 157–161. [Google Scholar] [CrossRef]
- Liu, W.M.; Lin, R.J.; Yu, R.L.; Tai, C.H.; Lin, C.H.; Wu, R.M. The impact of nonmotor symptoms on quality of life in patients with Parkinson’s disease in Taiwan. Neuropsychiatr. Dis. Treat. 2015, 11, 2865–2873. [Google Scholar] [CrossRef] [Green Version]
- Bobholz, J.A.; Rao, S.M. Cognitive dysfunction in multiple sclerosis: A review of recent developments. Curr. Opin. Neurol. 2003, 16, 283–288. [Google Scholar] [CrossRef]
- Prakash, R.S.; Schirda, B.; Valentine, T.R.; Crotty, M.; Nicholas, J.A. Emotion dysregulation in multiple sclerosis: Impact on symptoms of depression and anxiety. Mult. Scler. Relat. Disord. 2019, 36, 101399. [Google Scholar] [CrossRef]
- Ransome, M.I.; Renoir, T.; Hannan, A.J. Hippocampal neurogenesis, cognitive deficits and affective disorder in Huntington’s disease. Neural. Plast. 2012, 2012, 874387. [Google Scholar] [CrossRef]
- Connolly, B.; Fox, S.H. Treatment of cognitive, psychiatric, and affective disorders associated with Parkinson’s disease. Neurotherapeutics 2014, 11, 78–91. [Google Scholar] [CrossRef] [Green Version]
- Sampath, D.; Sathyanesan, M.; Newton, S.S. Cognitive dysfunction in major depression and Alzheimer’s disease is associated with hippocampal-prefrontal cortex dysconnectivity. Neuropsychiatr. Dis. Treat. 2017, 13, 1509–1519. [Google Scholar] [CrossRef] [Green Version]
- McGregor, G.; Harvey, J. Regulation of Hippocampal Synaptic Function by the Metabolic Hormone, Leptin: Implications for Health and Neurodegenerative Disease. Front Cell Neurosci. 2018, 12, 340. [Google Scholar] [CrossRef] [PubMed]
- Mufson, E.J.; Mahady, L.; Waters, D.; Counts, S.E.; Perez, S.E.; DeKosky, S.T.; Ginsberg, S.D.; Ikonomovic, M.D.; Scheff, S.W.; Binder, L.I. Hippocampal plasticity during the progression of Alzheimer’s disease. Neuroscience 2015, 309, 51–67. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Irving, A.; Harvey, J. Regulation of hippocampal synaptic function by the metabolic hormone leptin: Implications for health and disease. Prog. Lipid. Res. 2021, 82, 101098. [Google Scholar] [CrossRef] [PubMed]
- Selkoe, D.J. Alzheimer’s disease: Genes, proteins, and therapy. Physiol. Rev. 2001, 81, 741–766. [Google Scholar] [CrossRef]
- Reitz, C.; Mayeux, R. Alzheimer disease: Epidemiology, diagnostic criteria, risk factors and biomarkers. Biochem. Pharmacol. 2014, 88, 640–651. [Google Scholar] [CrossRef] [Green Version]
- Goedert, M.; Spillantini, M.G. A century of Alzheimer’s disease. Science 2006, 314, 777–781. [Google Scholar] [CrossRef] [Green Version]
- Fotuhi, S.N.; Khalaj-Kondori, M.; Feizi, M.A.H.; Talebi, M. Memory-related process in physiological status and alzheimer’s disease. Mol. Biol. Rep. 2020, 47, 4651–4657. [Google Scholar] [CrossRef]
- Brun, A.; Englund, E. Regional pattern of degeneration in Alzheimer’s disease: Neuronal loss and histopathological grading. Histopathology 1981, 5, 549–564. [Google Scholar] [CrossRef]
- Wright, A.L.; Zinn, R.; Hohensinn, B.; Konen, L.M.; Beynon, S.B.; Tan, R.P.; Clark, I.A.; Abdipranoto, A.; Vissel, B. Neuroinflammation and neuronal loss precede Abeta plaque deposition in the hAPP-J20 mouse model of Alzheimer’s disease. PLoS ONE 2013, 8, e59586. [Google Scholar] [CrossRef] [Green Version]
- Scheff, S.W.; Price, D.A. Alzheimer’s disease-related alterations in synaptic density: Neocortex and hippocampus. J. Alzheimers Dis. 2006, 9, 101–115. [Google Scholar] [CrossRef]
- Jahn, H. Memory loss in Alzheimer’s disease. Dialogues Clin. Neurosci. 2013, 15, 445–454. [Google Scholar] [CrossRef] [PubMed]
- de Flores, R.; La Joie, R.; Chetelat, G. Structural imaging of hippocampal subfields in healthy aging and Alzheimer’s disease. Neuroscience 2015, 309, 29–50. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schönheit, B.; Zarski, R.; Ohm, T.G. Spatial and temporal relationships between plaques and tangles in Alzheimer-pathology. Neurobiol. Aging 2004, 25, 697–711. [Google Scholar] [CrossRef]
- Lace, G.; Savva, G.M.; Forster, G.; de Silva, R.; Brayne, C.; Matthews, F.E.; Barclay, J.J.; Dakin, L.; Ince, P.G.; Wharton, S.B.; et al. Hippocampal tau pathology is related to neuroanatomical connections: An ageing population-based study. Brain 2009, 132, 1324–1334. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kril, J.J.; Patel, S.; Harding, A.J.; Halliday, G.M. Patients with vascular dementia due to microvascular pathology have significant hippocampal neuronal loss. J. Neurol. Neurosurg Psychiatry 2002, 72, 747–751. [Google Scholar] [CrossRef] [Green Version]
- Padurariu, M.; Ciobica, A.; Mavroudis, I.; Fotiou, D.; Baloyannis, S. Hippocampal neuronal loss in the CA1 and CA3 areas of Alzheimer’s disease patients. Psychiatr. Danub. 2012, 24, 152–158. [Google Scholar]
- Price, J.L.; Ko, A.I.; Wade, M.J.; Tsou, S.K.; McKeel, D.W.; Morris, J.C. Neuron number in the entorhinal cortex and CA1 in preclinical Alzheimer disease. Arch Neurol. 2001, 58, 1395–1402. [Google Scholar] [CrossRef]
- Skaper, S.D.; Facci, L.; Zusso, M.; Giusti, P. Synaptic Plasticity, Dementia and Alzheimer Disease. CNS Neurol. Disord. Drug Targets 2017, 16, 220–233. [Google Scholar] [CrossRef]
- Scheff, S.W.; Price, D.A. Synaptic density in the inner molecular layer of the hippocampal dentate gyrus in Alzheimer disease. J. Neuropathol. Exp. Neurol. 1998, 57, 1146–1153. [Google Scholar] [CrossRef] [Green Version]
- Volpicelli-Daley, L.A.; Luk, K.C.; Patel, T.P.; Tanik, S.A.; Riddle, D.M.; Stieber, A.; Meaney, D.F.; Trojanowski, J.Q.; Lee, V.M. Exogenous alpha-synuclein fibrils induce Lewy body pathology leading to synaptic dysfunction and neuron death. Neuron 2011, 72, 57–71. [Google Scholar] [CrossRef] [Green Version]
- Talantova, M.; Sanz-Blasco, S.; Zhang, X.; Xia, P.; Akhtar, M.W.; Okamoto, S.; Dziewczapolski, G.; Nakamura, T.; Cao, G.; Pratt, A.E.; et al. Abeta induces astrocytic glutamate release, extrasynaptic NMDA receptor activation, and synaptic loss. Proc. Natl. Acad. Sci. USA 2013, 110, E2518–E2527. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yao, P.J.; Zhu, M.; Pyun, E.I.; Brooks, A.I.; Therianos, S.; Meyers, V.E.; Coleman, P.D. Defects in expression of genes related to synaptic vesicle traffickingin frontal cortex of Alzheimer’s disease. Neurobiol. Dis. 2003, 12, 97–109. [Google Scholar] [CrossRef]
- Counts, S.E.; Alldred, M.J.; Che, S.; Ginsberg, S.D.; Mufson, E.J. Synaptic gene dysregulation within hippocampal CA1 pyramidal neurons in mild cognitive impairment. Neuropharmacology 2014, 79, 172–179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cuestas Torres, D.M.; Cardenas, F.P. Synaptic plasticity in Alzheimer’s disease and healthy aging. Rev. Neurosci. 2020, 31, 245–268. [Google Scholar] [CrossRef]
- Soria Lopez, J.A.; Gonzalez, H.M.; Leger, G.C. Alzheimer’s disease. Handb. Clin. Neurol. 2019, 167, 231–255. [Google Scholar] [CrossRef]
- Scheff, S.W.; Price, D.A.; Schmitt, F.A.; Mufson, E.J. Hippocampal synaptic loss in early Alzheimer’s disease and mild cognitive impairment. Neurobiol. Aging 2006, 27, 1372–1384. [Google Scholar] [CrossRef]
- Scheff, S.W.; Price, D.A.; Schmitt, F.A.; DeKosky, S.T.; Mufson, E.J. Synaptic alterations in CA1 in mild Alzheimer disease and mild cognitive impairment. Neurology 2007, 68, 1501–1508. [Google Scholar] [CrossRef]
- Fol, R.; Braudeau, J.; Ludewig, S.; Abel, T.; Weyer, S.W.; Roederer, J.P.; Brod, F.; Audrain, M.; Bemelmans, A.P.; Buchholz, C.J.; et al. Viral gene transfer of APPsalpha rescues synaptic failure in an Alzheimer’s disease mouse model. Acta Neuropathol. 2016, 131, 247–266. [Google Scholar] [CrossRef]
- Zhang, W.; Gu, G.J.; Zhang, Q.; Liu, J.H.; Zhang, B.; Guo, Y.; Wang, M.Y.; Gong, Q.Y.; Xu, J.R. NSCs promote hippocampal neurogenesis, metabolic changes and synaptogenesis in APP/PS1 transgenic mice. Hippocampus 2017, 27, 1250–1263. [Google Scholar] [CrossRef]
- Peretti, D.; Bastide, A.; Radford, H.; Verity, N.; Molloy, C.; Martin, M.G.; Moreno, J.A.; Steinert, J.R.; Smith, T.; Dinsdale, D.; et al. RBM3 mediates structural plasticity and protective effects of cooling in neurodegeneration. Nature 2015, 518, 236–239. [Google Scholar] [CrossRef] [Green Version]
- Chen, R.; Zhang, J.; Wu, Y.; Wang, D.; Feng, G.; Tang, Y.P.; Teng, Z.; Chen, C. Monoacylglycerol lipase is a therapeutic target for Alzheimer’s disease. Cell Rep. 2012, 2, 1329–1339. [Google Scholar] [CrossRef] [Green Version]
- Steele, J.W.; Brautigam, H.; Short, J.A.; Sowa, A.; Shi, M.; Yadav, A.; Weaver, C.M.; Westaway, D.; Fraser, P.E.; St George-Hyslop, P.H.; et al. Early fear memory defects are associated with altered synaptic plasticity and molecular architecture in the TgCRND8 Alzheimer’s disease mouse model. J. Comp. Neurol. 2014, 522, 2319–2335. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsai, S.T.; Chen, S.Y.; Lin, S.Z.; Tseng, G.F. Rostral intralaminar thalamic deep brain stimulation ameliorates memory deficits and dendritic regression in beta-amyloid-infused rats. Brain Struct Funct. 2020, 225, 751–761. [Google Scholar] [CrossRef]
- Liu, Y.; Bian, H.; Xu, S.; Shu, S.; Jia, J.; Chen, J.; Cao, X.; Bao, X.; Gu, Y.; Xia, S.; et al. Muscone Ameliorates Synaptic Dysfunction and Cognitive Deficits in APP/PS1 Mice. J. Alzheimers Dis. 2020, 76, 491–504. [Google Scholar] [CrossRef] [PubMed]
- Hou, J.; Wang, C.; Zhang, M.; Ren, M.; Yang, G.; Qu, Z.; Hu, Y. Safflower Yellow Improves the Synaptic Structural Plasticity by Ameliorating the Disorder of Glutamate Circulation in Abeta1-42-induced AD Model Rats. Neurochem. Res. 2020, 45, 1870–1887. [Google Scholar] [CrossRef] [PubMed]
- Jacobsen, J.S.; Wu, C.C.; Redwine, J.M.; Comery, T.A.; Arias, R.; Bowlby, M.; Martone, R.; Morrison, J.H.; Pangalos, M.N.; Reinhart, P.H.; et al. Early-onset behavioral and synaptic deficits in a mouse model of Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2006, 103, 5161–5166. [Google Scholar] [CrossRef] [Green Version]
- Shankar, G.M.; Li, S.; Mehta, T.H.; Garcia-Munoz, A.; Shepardson, N.E.; Smith, I.; Brett, F.M.; Farrell, M.A.; Rowan, M.J.; Lemere, C.A.; et al. Amyloid-beta protein dimers isolated directly from Alzheimer’s brains impair synaptic plasticity and memory. Nat. Med. 2008, 14, 837–842. [Google Scholar] [CrossRef] [Green Version]
- Inestrosa, N.C.; Varela-Nallar, L. Wnt signaling in the nervous system and in Alzheimer’s disease. J. Mol. Cell Biol. 2014, 6, 64–74. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.N.; Qi, J.S.; Gao, R. Physical exercise reserved amyloid-beta induced brain dysfunctions by regulating hippocampal neurogenesis and inflammatory response via MAPK signaling. Brain Res. 2018, 1697, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Selkoe, D.J. Alzheimer’s disease is a synaptic failure. Science 2002, 298, 789–791. [Google Scholar] [CrossRef] [Green Version]
- Mucke, L.; Masliah, E.; Yu, G.-Q.; Mallory, M.; Rockenstein, E.M.; Tatsuno, G.; Hu, K.; Kholodenko, D.; Johnson-Wood, K.; McConlogue, L. High-level neuronal expression of Aβ1–42 in wild-type human amyloid protein precursor transgenic mice: Synaptotoxicity without plaque formation. J. Neurosci. 2000, 20, 4050–4058. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oddo, S.; Caccamo, A.; Shepherd, J.D.; Murphy, M.P.; Golde, T.E.; Kayed, R.; Metherate, R.; Mattson, M.P.; Akbari, Y.; LaFerla, F.M. Triple-transgenic model of Alzheimer’s disease with plaques and tangles: Intracellular Abeta and synaptic dysfunction. Neuron 2003, 39, 409–421. [Google Scholar] [CrossRef] [Green Version]
- Kamenetz, F.; Tomita, T.; Hsieh, H.; Seabrook, G.; Borchelt, D.; Iwatsubo, T.; Sisodia, S.; Malinow, R. APP processing and synaptic function. Neuron 2003, 37, 925–937. [Google Scholar] [CrossRef] [Green Version]
- Lang, A.E.; Lozano, A.M. Parkinson’s disease. Second of two parts. N. Engl. J. Med. 1998, 339, 1130–1143. [Google Scholar] [CrossRef] [PubMed]
- Dorsey, E.R.; Elbaz, A.; Nichols, E.; Abd-Allah, F.; Abdelalim, A.; Adsuar, J.C.; Ansha, M.G.; Brayne, C.; Choi, J.Y.J.; Collado-Mateo, D.; et al. Global, regional, and national burden of Parkinson’s disease, 1990-2016: A systematic analysis for the Global Burden of Disease Study 2016. Lancet Neurol. 2018, 17, 939–953. [Google Scholar] [CrossRef] [Green Version]
- Collaborators, G. GBD 2016 Dementia Collaborators Global, regional, and national burden of Alzheimer’s disease and other dementias, 1990–2016: A systematic analysis for the Global Burden of Disease Study 2016. Lancet Neurol. 2019, 18, 88–106. [Google Scholar]
- Leroi, I.; McDonald, K.; Pantula, H.; Harbishettar, V. Cognitive impairment in Parkinson disease: Impact on quality of life, disability, and caregiver burden. J. Geriatr. Psychiatry Neurol. 2012, 25, 208–214. [Google Scholar] [CrossRef]
- Barone, P.; Erro, R.; Picillo, M. Quality of Life and Nonmotor Symptoms in Parkinson’s Disease. Int. Rev. Neurobiol. 2017, 133, 499–516. [Google Scholar] [CrossRef]
- Calabresi, P.; Castrioto, A.; Di Filippo, M.; Picconi, B. New experimental and clinical links between the hippocampus and the dopaminergic system in Parkinson’s disease. Lancet Neurol. 2013, 12, 811–821. [Google Scholar] [CrossRef]
- Regensburger, M.; Prots, I.; Winner, B. Adult hippocampal neurogenesis in Parkinson’s disease: Impact on neuronal survival and plasticity. Neural. Plast. 2014, 2014, 454696. [Google Scholar] [CrossRef] [Green Version]
- Barone, P.; Antonini, A.; Colosimo, C.; Marconi, R.; Morgante, L.; Avarello, T.P.; Bottacchi, E.; Cannas, A.; Ceravolo, G.; Ceravolo, R.; et al. The PRIAMO study: A multicenter assessment of nonmotor symptoms and their impact on quality of life in Parkinson’s disease. Mov. Disord. 2009, 24, 1641–1649. [Google Scholar] [CrossRef] [PubMed]
- Leentjens, A.F.; Van den Akker, M.; Metsemakers, J.F.; Lousberg, R.; Verhey, F.R. Higher incidence of depression preceding the onset of Parkinson’s disease: A register study. Mov. Disord. 2003, 18, 414–418. [Google Scholar] [CrossRef] [PubMed]
- Ballanger, B.; Klinger, H.; Eche, J.; Lerond, J.; Vallet, A.E.; Le Bars, D.; Tremblay, L.; Sgambato-Faure, V.; Broussolle, E.; Thobois, S. Role of serotonergic 1A receptor dysfunction in depression associated with Parkinson’s disease. Mov. Disord. 2012, 27, 84–89. [Google Scholar] [CrossRef] [PubMed]
- Pavese, N.; Metta, V.; Bose, S.K.; Chaudhuri, K.R.; Brooks, D.J. Fatigue in Parkinson’s disease is linked to striatal and limbic serotonergic dysfunction. Brain 2010, 133, 3434–3443. [Google Scholar] [CrossRef]
- Goto, Y.; Grace, A.A. Dopaminergic modulation of limbic and cortical drive of nucleus accumbens in goal-directed behavior. Nat. Neurosci. 2005, 8, 805–812. [Google Scholar] [CrossRef]
- Costa, C.; Sgobio, C.; Siliquini, S.; Tozzi, A.; Tantucci, M.; Ghiglieri, V.; Di Filippo, M.; Pendolino, V.; de Iure, A.; Marti, M.; et al. Mechanisms underlying the impairment of hippocampal long-term potentiation and memory in experimental Parkinson’s disease. Brain 2012, 135, 1884–1899. [Google Scholar] [CrossRef] [Green Version]
- Esmaeili-Mahani, S.; Haghparast, E.; Nezhadi, A.; Abbasnejad, M.; Sheibani, V. Apelin-13 prevents hippocampal synaptic plasticity impairment in Parkinsonism rats. J. Chem. Neuroanat. 2021, 111, 101884. [Google Scholar] [CrossRef]
- Pendolino, V.; Bagetta, V.; Ghiglieri, V.; Sgobio, C.; Morelli, E.; Poggini, S.; Branchi, I.; Latagliata, E.C.; Pascucci, T.; Puglisi-Allegra, S.; et al. l-DOPA reverses the impairment of Dentate Gyrus LTD in experimental parkinsonism via beta-adrenergic receptors. Exp. Neurol. 2014, 261, 377–385. [Google Scholar] [CrossRef]
- Wang, Y.; Feng, L.; Liu, S.; Zhou, X.; Yin, T.; Liu, Z.; Yang, Z. Transcranial magneto-acoustic stimulation improves neuroplasticity in hippocampus of Parkinson’s disease model mice. Neurotherapeutics 2019, 16, 1210–1224. [Google Scholar] [CrossRef] [Green Version]
- Wu, Q.; Takano, H.; Riddle, D.M.; Trojanowski, J.Q.; Coulter, D.A.; Lee, V.M. alpha-Synuclein (alphaSyn) Preformed Fibrils Induce Endogenous alphaSyn Aggregation, Compromise Synaptic Activity and Enhance Synapse Loss in Cultured Excitatory Hippocampal Neurons. J. Neurosci. 2019, 39, 5080–5094. [Google Scholar] [CrossRef] [Green Version]
- Swanson-Park, J.; Coussens, C.; Mason-Parker, S.; Raymond, C.; Hargreaves, E.; Dragunow, M.; Cohen, A.; Abraham, W. A double dissociation within the hippocampus of dopamine D1/D5 receptor and β-adrenergic receptor contributions to the persistence of long-term potentiation. Neuroscience 1999, 92, 485–497. [Google Scholar] [CrossRef]
- Hansen, N.; Manahan-Vaughan, D. Dopamine D1/D5 receptors mediate informational saliency that promotes persistent hippocampal long-term plasticity. Cereb. Cortex. 2014, 24, 845–858. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kusuki, T.; Imahori, Y.; Ueda, S.; Inokuchi, K. Dopaminergic modulation of LTP induction in the dentate gyrus of intact brain. Neuroreport 1997, 8, 2037–2040. [Google Scholar] [CrossRef] [PubMed]
- O’Carroll, C.M.; Martin, S.J.; Sandin, J.; Frenguelli, B.; Morris, R.G. Dopaminergic modulation of the persistence of one-trial hippocampus-dependent memory. Learn Mem. 2006, 13, 760–769. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frey, U.; Morris, R.G. Synaptic tagging and long-term potentiation. Nature 1997, 385, 533–536. [Google Scholar] [CrossRef]
- Calabresi, P.; Ghiglieri, V.; Mazzocchetti, P.; Corbelli, I.; Picconi, B. Levodopa-induced plasticity: A double-edged sword in Parkinson’s disease? Philos. Trans. R. Soc. Lond. B Biol. Sci. 2015, 370, 20140184. [Google Scholar] [CrossRef] [Green Version]
- Borodovitsyna, O.; Flamini, M.; Chandler, D. Noradrenergic Modulation of Cognition in Health and Disease. Neural. Plast. 2017, 2017, 6031478. [Google Scholar] [CrossRef] [Green Version]
- Kohl, Z.; Ben Abdallah, N.; Vogelgsang, J.; Tischer, L.; Deusser, J.; Amato, D.; Anderson, S.; Muller, C.P.; Riess, O.; Masliah, E.; et al. Severely impaired hippocampal neurogenesis associates with an early serotonergic deficit in a BAC alpha-synuclein transgenic rat model of Parkinson’s disease. Neurobiol. Dis. 2016, 85, 206–217. [Google Scholar] [CrossRef] [Green Version]
- Cubo, E.; Shannon, K.M.; Tracy, D.; Jaglin, J.A.; Bernard, B.A.; Wuu, J.; Leurgans, S.E. Effect of donepezil on motor and cognitive function in Huntington disease. Neurology 2006, 67, 1268–1271. [Google Scholar] [CrossRef]
- Paulsen, J.S. Cognitive impairment in Huntington disease: Diagnosis and treatment. Curr. Neurol. Neurosci. Rep. 2011, 11, 474–483. [Google Scholar] [CrossRef] [Green Version]
- Craufurd, D.; Thompson, J.C.; Snowden, J.S. Behavioral changes in Huntington Disease. Neuropsychiatry Neuropsychol. Behav. Neurol. 2001, 14, 219–226. [Google Scholar]
- Bates, G.; Tabrizi, S.; Jones, L. Huntington’s Disease; Oxford Monographs on Medical G; Oxford University Press: New York, NY, USA, 2014. [Google Scholar]
- The Huntington’s Disease Collaborative Research Group. A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington’s disease chromosomes. Cell 1993, 72, 971–983. [Google Scholar] [CrossRef]
- Waldvogel, H.J.; Kim, E.H.; Tippett, L.J.; Vonsattel, J.-P.G.; Faull, R.L. The Neuropathology of Huntington’s Disease. Curr. Top Behav. Neurosci. 2015, 22, 33–80. [Google Scholar] [CrossRef] [PubMed]
- Rubinstein, D. Molecular biology of Huntingtons Disease (HD) and HD-like disorders. In Genetics of Movement Disorders; Pulst, S., Ed.; Academic Press: Cambridge, MA, USA, 2003; pp. 365–377. [Google Scholar]
- Vonsattel, J.P.; DiFiglia, M. Huntington disease. J. Neuropathol. Exp. Neurol. 1998, 57, 369–384. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shehadeh, J.; Fernandes, H.B.; Zeron Mullins, M.M.; Graham, R.K.; Leavitt, B.R.; Hayden, M.R.; Raymond, L.A. Striatal neuronal apoptosis is preferentially enhanced by NMDA receptor activation in YAC transgenic mouse model of Huntington disease. Neurobiol. Dis. 2006, 21, 392–403. [Google Scholar] [CrossRef] [PubMed]
- Rikani, A.A.; Choudhry, Z.; Choudhry, A.M.; Rizvi, N.; Ikram, H.; Mobassarah, N.J.; Tulli, S. The mechanism of degeneration of striatal neuronal subtypes in Huntington disease. Ann. Neurosci. 2014, 21, 112–114. [Google Scholar] [CrossRef]
- Lebouc, M.; Richard, Q.; Garret, M.; Baufreton, J. Striatal circuit development and its alterations in Huntington’s disease. Neurobiol. Dis. 2020, 145, 105076. [Google Scholar] [CrossRef]
- Spargo, E.; Everall, I.P.; Lantos, P.L. Neuronal loss in the hippocampus in Huntington’s disease: A comparison with HIV infection. J. Neurol. Neurosurg. Psychiatry 1993, 56, 487–491. [Google Scholar] [CrossRef]
- Cheong, R.Y.; Gabery, S.; Petersen, A. The Role of Hypothalamic Pathology for Non-Motor Features of Huntington’s Disease. J. Huntingt. Dis. 2019, 8, 375–391. [Google Scholar] [CrossRef] [Green Version]
- Paulsen, J.S.; Langbehn, D.R.; Stout, J.C.; Aylward, E.; Ross, C.A.; Nance, M.; Guttman, M.; Johnson, S.; MacDonald, M.; Beglinger, L.J.; et al. Detection of Huntington’s disease decades before diagnosis: The Predict-HD study. J. Neurol. Neurosurg. Psychiatry. 2008, 79, 874–880. [Google Scholar] [CrossRef] [Green Version]
- Wilkie, C.M.; Barnes, J.R.; Benson, C.M.; Brymer, K.J.; Nafar, F.; Parsons, M.P. Hippocampal Synaptic Dysfunction in a Mouse Model of Huntington Disease Is Not Alleviated by Ceftriaxone Treatment. eNeuro 2020, 7, 19. [Google Scholar] [CrossRef] [PubMed]
- Usdin, M.T.; Shelbourne, P.F.; Myers, R.M.; Madison, D.V. Impaired synaptic plasticity in mice carrying the Huntington’s disease mutation. Hum. Mol. Genet. 1999, 8, 839–846. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murphy, K.P.; Carter, R.J.; Lione, L.A.; Mangiarini, L.; Mahal, A.; Bates, G.P.; Dunnett, S.B.; Morton, A.J. Abnormal synaptic plasticity and impaired spatial cognition in mice transgenic for exon 1 of the human Huntington’s disease mutation. J. Neurosci. 2000, 20, 5115–5123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cummings, D.M.; Milnerwood, A.J.; Dallerac, G.M.; Waights, V.; Brown, J.Y.; Vatsavayai, S.C.; Hirst, M.C.; Murphy, K.P. Aberrant cortical synaptic plasticity and dopaminergic dysfunction in a mouse model of Huntington’s disease. Hum. Mol. Genet. 2006, 15, 2856–2868. [Google Scholar] [CrossRef] [Green Version]
- Milnerwood, A.J.; Cummings, D.M.; Dallerac, G.M.; Brown, J.Y.; Vatsavayai, S.C.; Hirst, M.C.; Rezaie, P.; Murphy, K.P. Early development of aberrant synaptic plasticity in a mouse model of Huntington’s disease. Hum. Mol. Genet. 2006, 15, 1690–1703. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lynch, G.; Kramar, E.A.; Rex, C.S.; Jia, Y.; Chappas, D.; Gall, C.M.; Simmons, D.A. Brain-derived neurotrophic factor restores synaptic plasticity in a knock-in mouse model of Huntington’s disease. J. Neurosci. 2007, 27, 4424–4434. [Google Scholar] [CrossRef]
- Simmons, D.A.; Rex, C.S.; Palmer, L.; Pandyarajan, V.; Fedulov, V.; Gall, C.M.; Lynch, G. Up-regulating BDNF with an ampakine rescues synaptic plasticity and memory in Huntington’s disease knockin mice. Proc. Natl. Acad. Sci. USA 2009, 106, 4906–4911. [Google Scholar] [CrossRef] [Green Version]
- Brito, V.; Giralt, A.; Enriquez-Barreto, L.; Puigdellivol, M.; Suelves, N.; Zamora-Moratalla, A.; Ballesteros, J.J.; Martin, E.D.; Dominguez-Iturza, N.; Morales, M.; et al. Neurotrophin receptor p75(NTR) mediates Huntington’s disease-associated synaptic and memory dysfunction. J. Clin. Investig. 2014, 124, 4411–4428. [Google Scholar] [CrossRef] [Green Version]
- Kolodziejczyk, K.; Parsons, M.P.; Southwell, A.L.; Hayden, M.R.; Raymond, L.A. Striatal synaptic dysfunction and hippocampal plasticity deficits in the Hu97/18 mouse model of Huntington disease. PLoS ONE 2014, 9, e94562. [Google Scholar] [CrossRef]
- Giralt, A.; Brito, V.; Chevy, Q.; Simonnet, C.; Otsu, Y.; Cifuentes-Diaz, C.; de Pins, B.; Coura, R.; Alberch, J.; Gines, S.; et al. Pyk2 modulates hippocampal excitatory synapses and contributes to cognitive deficits in a Huntington’s disease model. Nat. Commun. 2017, 8, 15592. [Google Scholar] [CrossRef]
- Sepers, M.D.; Smith-Dijak, A.; LeDue, J.; Kolodziejczyk, K.; Mackie, K.; Raymond, L.A. Endocannabinoid-Specific Impairment in Synaptic Plasticity in Striatum of Huntington’s Disease Mouse Model. J. Neurosci. 2018, 38, 544–554. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Zhang, C.; Vincent, J.; Zala, D.; Benstaali, C.; Sainlos, M.; Grillo-Bosch, D.; Daburon, S.; Coussen, F.; Cho, Y.; et al. Modulation of AMPA receptor surface diffusion restores hippocampal plasticity and memory in Huntington’s disease models. Nat. Commun. 2018, 9, 4272. [Google Scholar] [CrossRef] [PubMed]
- Hodgson, J.G.; Agopyan, N.; Gutekunst, C.A.; Leavitt, B.R.; LePiane, F.; Singaraja, R.; Smith, D.J.; Bissada, N.; McCutcheon, K.; Nasir, J.; et al. A YAC mouse model for Huntington’s disease with full-length mutant huntingtin, cytoplasmic toxicity, and selective striatal neurodegeneration. Neuron 1999, 23, 181–192. [Google Scholar] [CrossRef] [Green Version]
- Quirion, J.G.; Parsons, M.P. The Onset and Progression of Hippocampal Synaptic Plasticity Deficits in the Q175FDN Mouse Model of Huntington Disease. Front. Cell Neurosci. 2019, 13, 326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anglada-Huguet, M.; Xifro, X.; Giralt, A.; Zamora-Moratalla, A.; Martin, E.D.; Alberch, J. Prostaglandin E2 EP1 receptor antagonist improves motor deficits and rescues memory decline in R6/1 mouse model of Huntington’s disease. Mol. Neurobiol. 2014, 49, 784–795. [Google Scholar] [CrossRef] [PubMed]
- Bruel-Jungerman, E.; Davis, S.; Laroche, S. Brain plasticity mechanisms and memory: A party of four. Neuroscientist 2007, 13, 492–505. [Google Scholar] [CrossRef] [PubMed]
- Bruel-Jungerman, E.; Rampon, C.; Laroche, S. Adult hippocampal neurogenesis, synaptic plasticity and memory: Facts and hypotheses. Rev. Neurosci. 2007, 18, 93–114. [Google Scholar] [CrossRef]
- Di Filippo, M.; Tozzi, A.; Picconi, B.; Ghiglieri, V.; Calabresi, P. Plastic abnormalities in experimental Huntington’s disease. Curr. Opin. Pharmacol. 2007, 7, 106–111. [Google Scholar] [CrossRef]
- Cattaneo, E.; Zuccato, C.; Tartari, M. Normal huntingtin function: An alternative approach to Huntington’s disease. Nat. Rev. Neurosci. 2005, 6, 919–930. [Google Scholar] [CrossRef]
- Cotsapas, C.; Mitrovic, M.; Hafler, D. Multiple sclerosis. Handb. Clin. Neurol. 2018, 148, 723–730. [Google Scholar] [CrossRef]
- Karussis, D. The diagnosis of multiple sclerosis and the various related demyelinating syndromes: A critical review. J. Autoimmun. 2014, 48–49, 134–142. [Google Scholar] [CrossRef]
- Trapp, B.D.; Peterson, J.; Ransohoff, R.M.; Rudick, R.; Mork, S.; Bo, L. Axonal transection in the lesions of multiple sclerosis. N. Engl. J. Med. 1998, 338, 278–285. [Google Scholar] [CrossRef] [PubMed]
- Lassmann, H. Pathology and disease mechanisms in different stages of multiple sclerosis. J. Neurol. Sci. 2013, 333, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Vercellino, M.; Plano, F.; Votta, B.; Mutani, R.; Giordana, M.T.; Cavalla, P. Grey matter pathology in multiple sclerosis. J. Neuropathol. Exp. Neurol. 2005, 64, 1101–1107. [Google Scholar] [CrossRef] [PubMed]
- Geurts, J.J.; Bo, L.; Roosendaal, S.D.; Hazes, T.; Daniels, R.; Barkhof, F.; Witter, M.P.; Huitinga, I.; van der Valk, P. Extensive hippocampal demyelination in multiple sclerosis. J. Neuropathol. Exp. Neurol. 2007, 66, 819–827. [Google Scholar] [CrossRef] [Green Version]
- Dutta, R.; Chang, A.; Doud, M.K.; Kidd, G.J.; Ribaudo, M.V.; Young, E.A.; Fox, R.J.; Staugaitis, S.M.; Trapp, B.D. Demyelination causes synaptic alterations in hippocampi from multiple sclerosis patients. Ann. Neurol. 2011, 69, 445–454. [Google Scholar] [CrossRef]
- Drake, M.A.; Carra, A.; Allegri, R.F.; Luetic, G. Differential patterns of memory performance in relapsing, remitting and secondary progressive multiple sclerosis. Neurol. India 2006, 54, 370–376. [Google Scholar] [CrossRef]
- Chiaravalloti, N.D.; DeLuca, J. Cognitive impairment in multiple sclerosis. Lancet Neurol. 2008, 7, 1139–1151. [Google Scholar] [CrossRef]
- Gaudino, E.A.; Chiaravalloti, N.D.; DeLuca, J.; Diamond, B.J. A comparison of memory performance in relapsing-remitting, primary progressive and secondary progressive, multiple sclerosis. Neuropsychiatry Neuropsychol. Behav. Neurol. 2001, 14, 32–44. [Google Scholar]
- Beatty, W.W.; Wilbanks, S.L.; Blanco, C.R.; Hames, K.A.; Tivis, R.; Paul, R.H. Memory disturbance in multiple sclerosis: Reconsideration of patterns of performance on the selective reminding test. J. Clin. Exp. Neuropsychol. 1996, 18, 56–62. [Google Scholar] [CrossRef]
- Litvan, I.; Grafman, J.; Vendrell, P.; Martinez, J.M. Slowed information processing in multiple sclerosis. Arch. Neurol. 1988, 45, 281–285. [Google Scholar] [CrossRef] [PubMed]
- Feinstein, A.; Magalhaes, S.; Richard, J.F.; Audet, B.; Moore, C. The link between multiple sclerosis and depression. Nat. Rev. Neurol. 2014, 10, 507–517. [Google Scholar] [CrossRef] [PubMed]
- Rocca, M.A.; Barkhof, F.; De Luca, J.; Frisen, J.; Geurts, J.J.G.; Hulst, H.E.; Sastre-Garriga, J.; Filippi, M.; Group, M.S. The hippocampus in multiple sclerosis. Lancet Neurol. 2018, 17, 918–926. [Google Scholar] [CrossRef]
- Pelletier, J.; Audoin, B.; Reuter, F.; Ranjeva, J. Plasticity in MS: From Functional Imaging to Rehabilitation. Int. MS J. 2009, 16, 26–31. [Google Scholar]
- Amato, M.P.; Zipoli, V.; Portaccio, E. Cognitive changes in multiple sclerosis. Expert Rev. Neurother. 2008, 8, 1585–1596. [Google Scholar] [CrossRef]
- Nistico, R.; Mori, F.; Feligioni, M.; Nicoletti, F.; Centonze, D. Synaptic plasticity in multiple sclerosis and in experimental autoimmune encephalomyelitis. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2014, 369, 20130162. [Google Scholar] [CrossRef] [Green Version]
- Michailidou, I.; Willems, J.G.; Kooi, E.J.; van Eden, C.; Gold, S.M.; Geurts, J.J.; Baas, F.; Huitinga, I.; Ramaglia, V. Complement C 1q-C 3–associated synaptic changes in multiple sclerosis hippocampus. Ann. Neurol. 2015, 77, 1007–1026. [Google Scholar] [CrossRef]
- Di Filippo, M.; Chiasserini, D.; Gardoni, F.; Viviani, B.; Tozzi, A.; Giampà, C.; Costa, C.; Tantucci, M.; Zianni, E.; Boraso, M.; et al. Effects of central and peripheral inflammation on hippocampal synaptic plasticity. Neurobiol. Dis. 2013, 52, 229–236. [Google Scholar] [CrossRef]
- Kim, D.Y.; Hao, J.; Liu, R.; Turner, G.; Shi, F.-D.; Rho, J.M. Inflammation-Mediated Memory Dysfunction and Effects of a Ketogenic Diet in a Murine Model of Multiple Sclerosis. PLoS ONE 2012, 7, e35476. [Google Scholar] [CrossRef] [Green Version]
- Di Filippo, M.; De Iure, A.; Giampà, C.; Chiasserini, D.; Tozzi, A.; Orvietani, P.L.; Ghiglieri, V.; Tantucci, M.; Durante, V.; Quiroga-Varela, A. Persistent activation of microglia and NADPH oxidase drive hippocampal dysfunction in experimental multiple sclerosis. Sci. Rep. 2016, 6, 1–16. [Google Scholar]
- Novkovic, T.; Shchyglo, O.; Gold, R.; Manahan-Vaughan, D. Hippocampal function is compromised in an animal model of multiple sclerosis. Neuroscience 2015, 309, 100–112. [Google Scholar] [CrossRef]
- Prochnow, N.; Gold, R.; Haghikia, A. An electrophysiologic approach to quantify impaired synaptic transmission and plasticity in experimental autoimmune encephalomyelitis. J. Neuroimmunol. 2013, 264, 48–53. [Google Scholar] [CrossRef] [PubMed]
- Nisticò, R.; Mango, D.; Mandolesi, G.; Piccinin, S.; Berretta, N.; Pignatelli, M.; Feligioni, M.; Musella, A.; Gentile, A.; Mori, F.; et al. Inflammation Subverts Hippocampal Synaptic Plasticity in Experimental Multiple Sclerosis. PLoS ONE 2013, 8, e54666. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Filippo, M.; de Iure, A.; Durante, V.; Gaetani, L.; Mancini, A.; Sarchielli, P.; Calabresi, P. Synaptic plasticity and experimental autoimmune encephalomyelitis: Implications for multiple sclerosis. Brain Res. 2015, 1621, 205–213. [Google Scholar] [CrossRef] [PubMed]
- Mori, F.; Nistico, R.; Mandolesi, G.; Piccinin, S.; Mango, D.; Kusayanagi, H.; Berretta, N.; Bergami, A.; Gentile, A.; Musella, A.; et al. Interleukin-1β Promotes Long-Term Potentiation in Patients with Multiple Sclerosis. NeuroMolecular Med. 2014, 16, 38–51. [Google Scholar] [CrossRef]
- Mosayebi, G.; Soleyman, M.R.; Khalili, M.; Mosleh, M.; Palizvan, M.R. Changes in Synaptic Transmission and Long-term Potentiation Induction as a Possible Mechanism for Learning Disability in an Animal Model of Multiple Sclerosis. Int. Neurourol. J. 2016, 20, 26–32. [Google Scholar] [CrossRef] [Green Version]
- Viviani, B.; Gardoni, F.; Bartesaghi, S.; Corsini, E.; Facchi, A.; Galli, C.L.; Di Luca, M.; Marinovich, M. Interleukin-1β Released by gp120 Drives Neural Death through Tyrosine Phosphorylation and Trafficking of NMDA Receptors. J. Biol. Chem. 2006, 281, 30212–30222. [Google Scholar] [CrossRef] [Green Version]
- Coogan, A.; O’Connor, J.J. Inhibition of NMDA receptor-mediated synaptic transmission in the rat dentate gyrus in vitro by IL-1β. Neuroreport 1997, 8, 2107–2110. [Google Scholar] [CrossRef]
- Lai, A.Y.; Swayze, R.D.; El-Husseini, A.; Song, C. Interleukin-1 beta modulates AMPA receptor expression and phosphorylation in hippocampal neurons. J. Neuroimmunol. 2006, 175, 97–106. [Google Scholar] [CrossRef]
- Mancini, A.; Gaetani, L.; Di Gregorio, M.; Tozzi, A.; Ghiglieri, V.; Calabresi, P.; Di Filippo, M. Hippocampal neuroplasticity and inflammation: Relevance for multiple sclerosis. Mult. Scler. Demyelinating Disord. 2017, 2, 2. [Google Scholar] [CrossRef] [Green Version]
- Luhder, F.; Gold, R.; Flugel, A.; Linker, R.A. Brain-derived neurotrophic factor in neuroimmunology: Lessons learned from multiple sclerosis patients and experimental autoimmune encephalomyelitis models. Arch. Immunol. Ther. Exp. 2013, 61, 95–105. [Google Scholar] [CrossRef] [PubMed]
- Frisoni, G.B.; Laakso, M.P.; Beltramello, A.; Geroldi, C.; Bianchetti, A.; Soininen, H.; Trabucchi, M. Hippocampal and entorhinal cortex atrophy in frontotemporal dementia and Alzheimer’s disease. Neurology 1999, 52, 91–100. [Google Scholar] [CrossRef] [PubMed]
- Laakso, M.P.; Frisoni, G.B.; Kononen, M.; Mikkonen, M.; Beltramello, A.; Geroldi, C.; Bianchetti, A.; Trabucchi, M.; Soininen, H.; Aronen, H.J. Hippocampus and entorhinal cortex in frontotemporal dementia and Alzheimer’s disease: A morphometric MRI study. Biol. Psychiatry. 2000, 47, 1056–1063. [Google Scholar] [CrossRef]
- Graff-Radford, J. Vascular Cognitive Impairment. Contin. Lifelong Learn. Neurol. 2019, 25, 147–164. [Google Scholar] [CrossRef] [PubMed]
- Appleton, J.P.; Scutt, P.; Sprigg, N.; Bath, P.M. Hypercholesterolaemia and vascular dementia. Clin. Sci. 2017, 131, 1561–1578. [Google Scholar] [CrossRef] [Green Version]
- Bocchetta, M.; Iglesias, J.E.; Scelsi, M.A.; Cash, D.M.; Cardoso, M.J.; Modat, M.; Altmann, A.; Ourselin, S.; Warren, J.D.; Rohrer, J.D. Hippocampal Subfield Volumetry: Differential Pattern of Atrophy in Different Forms of Genetic Frontotemporal Dementia. J. Alzheimers Dis. 2018, 64, 497–504. [Google Scholar] [CrossRef] [Green Version]
- Wilson, N.A.; Ramanan, S.; Roquet, D.; Goldberg, Z.L.; Hodges, J.R.; Piguet, O.; Irish, M. Scene construction impairments in frontotemporal dementia: Evidence for a primary hippocampal contribution. Neuropsychologia 2020, 137, 107327. [Google Scholar] [CrossRef]
- Christidi, F.; Karavasilis, E.; Velonakis, G.; Ferentinos, P.; Rentzos, M.; Kelekis, N.; Evdokimidis, I.; Bede, P. The Clinical and Radiological Spectrum of Hippocampal Pathology in Amyotrophic Lateral Sclerosis. Front. Neurol. 2018, 9, 523. [Google Scholar] [CrossRef] [Green Version]
- Liu, B.; Liu, J.; Zhang, J.; Mao, W.; Li, S. Effects of autophagy on synaptic-plasticity-related protein expression in the hippocampus CA1 of a rat model of vascular dementia. Neurosci. Lett. 2019, 707, 134312. [Google Scholar] [CrossRef]
- Huang, Y.; Liao, X.; Wang, H.; Luo, J.; Zhong, S.; Zhang, Z.; Zhang, F.; Chen, J.; Xie, F. Effects of imperatorin on apoptosis and synaptic plasticity in vascular dementia rats. Sci. Rep. 2021, 11, 8590. [Google Scholar] [CrossRef]
- Li, X.; Lu, F.; Li, W.; Qin, L.; Yao, Y.; Ge, X.; Yu, Q.; Liang, X.; Zhao, D.; Li, X.; et al. Edaravone injection reverses learning and memory deficits in a rat model of vascular dementia. Acta Biochim. Biophys. Sin. 2017, 49, 83–89. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, H.Y.; Liu, Y.; Xie, J.C.; Liu, N.N.; Tian, X. Effects of repetitive transcranial magnetic stimulation on synaptic plasticity and apoptosis in vascular dementia rats. Behav. Brain Res. 2015, 281, 149–155. [Google Scholar] [CrossRef] [PubMed]
- Meftahi, G.H.; Bayat, M.; Zarifkar, A.H.; Akbari, S.; Haghighi, A.B.; Naseh, M.; Nejad, A.Y.; Haghani, M. Treatment with edaravone improves the structure and functional changes in the hippocampus after chronic cerebral hypoperfusion in rat. Brain Res. Bull. 2021, 174, 122–130. [Google Scholar] [CrossRef]
- Levine, D.A.; Langa, K.M. Vascular cognitive impairment: Disease mechanisms and therapeutic implications. Neurotherapeutics 2011, 8, 361–373. [Google Scholar] [CrossRef]
- Radzicki, D.; Liu, E.; Deng, H.X.; Siddique, T.; Martina, M. Early Impairment of Synaptic and Intrinsic Excitability in Mice Expressing ALS/Dementia-Linked Mutant UBQLN2. Front Cell Neurosci. 2016, 10, 216. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ho, W.Y.; Navakkode, S.; Liu, F.; Soong, T.W.; Ling, S.C. Deregulated expression of a longevity gene, Klotho, in the C9orf72 deletion mice with impaired synaptic plasticity and adult hippocampal neurogenesis. Acta Neuropathol. Commun. 2020, 8, 155. [Google Scholar] [CrossRef] [PubMed]
- Christidi, F.; Karavasilis, E.; Rentzos, M.; Velonakis, G.; Zouvelou, V.; Xirou, S.; Argyropoulos, G.; Papatriantafyllou, I.; Pantolewn, V.; Ferentinos, P.; et al. Hippocampal pathology in amyotrophic lateral sclerosis: Selective vulnerability of subfields and their associated projections. Neurobiol. Aging 2019, 84, 178–188. [Google Scholar] [CrossRef]
- Brettschneider, J.; Del Tredici, K.; Toledo, J.B.; Robinson, J.L.; Irwin, D.J.; Grossman, M.; Suh, E.R.; Van Deerlin, V.M.; Wood, E.M.; Baek, Y.; et al. Stages of pTDP-43 pathology in amyotrophic lateral sclerosis. Ann. Neurol. 2013, 74, 20–38. [Google Scholar] [CrossRef]
- Bueno, A.P.A.; Pinaya, W.H.L.; Moura, L.M.; Bertoux, M.; Radakovic, R.; Kiernan, M.C.; Teixeira, A.L.; de Souza, L.C.; Hornberger, M.; Sato, J.R. Structural and functional papez circuit integrity in amyotrophic lateral sclerosis. Brain Imaging Behav. 2018, 12, 1622–1630. [Google Scholar] [CrossRef] [Green Version]
- Christidi, F.; Karavasilis, E.; Zalonis, I.; Ferentinos, P.; Giavri, Z.; Wilde, E.A.; Xirou, S.; Rentzos, M.; Zouvelou, V.; Velonakis, G.; et al. Memory-related white matter tract integrity in amyotrophic lateral sclerosis: An advanced neuroimaging and neuropsychological study. Neurobiol. Aging 2017, 49, 69–78. [Google Scholar] [CrossRef]
- Trojsi, F.; Di Nardo, F.; Caiazzo, G.; Siciliano, M.; D’Alvano, G.; Ferrantino, T.; Passaniti, C.; Ricciardi, D.; Esposito, S.; Lavorgna, L.; et al. Hippocampal connectivity in Amyotrophic Lateral Sclerosis (ALS): More than Papez circuit impairment. Brain Imaging Behav. 2021, 15, 2126–2138. [Google Scholar] [CrossRef] [PubMed]
- Rei, N.; Rombo, D.M.; Ferreira, M.F.; Baqi, Y.; Muller, C.E.; Ribeiro, J.A.; Sebastiao, A.M.; Vaz, S.H. Hippocampal synaptic dysfunction in the SOD1(G93A) mouse model of Amyotrophic Lateral Sclerosis: Reversal by adenosine A2AR blockade. Neuropharmacology 2020, 171, 108106. [Google Scholar] [CrossRef] [PubMed]
- Spalloni, A.; Geracitano, R.; Berretta, N.; Sgobio, C.; Bernardi, G.; Mercuri, N.B.; Longone, P.; Ammassari-Teule, M. Molecular and synaptic changes in the hippocampus underlying superior spatial abilities in pre-symptomatic G93A+/+ mice overexpressing the human Cu/Zn superoxide dismutase (Gly93→ ALA) mutation. Exp. Neurol. 2006, 197, 505–514. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Liu, J.; Yang, S.; Song, D.; Wang, C.; Chen, C.; Li, X.; Wang, Q.; Ge, S.; Yang, R.; et al. Ling-Yang-Gou-Teng-decoction prevents vascular dementia through inhibiting oxidative stress induced neurovascular coupling dysfunction. J. Ethnopharmacol. 2018, 222, 229–238. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Tang, J.; Li, S.; Zhang, J.; Yuan, M.; Wang, R. Autophagy activation aggravates neuronal injury in the hippocampus of vascular dementia rats. Neural Regen. Res. 2014, 9, 1288–1296. [Google Scholar] [CrossRef]
- He, Z.; Hu, M.; Zha, Y.H.; Li, Z.C.; Zhao, B.; Yu, L.L.; Yu, M.; Qian, Y. Piracetam ameliorated oxygen and glucose deprivation-induced injury in rat cortical neurons via inhibition of oxidative stress, excitatory amino acids release and P53/Bax. Cell Mol. Neurobiol. 2014, 34, 539–547. [Google Scholar] [CrossRef]
- Dong, J.; Zhao, J.; Lin, Y.; Liang, H.; He, X.; Zheng, X.; Sui, M.; Zhuang, Z.; Yan, T. Exercise improves recognition memory and synaptic plasticity in the prefrontal cortex for rats modelling vascular dementia. Neurol. Res. 2018, 40, 68–77. [Google Scholar] [CrossRef]
- Roman, G.C. Cholinergic dysfunction in vascular dementia. Curr. Psychiatry Rep. 2005, 7, 18–26. [Google Scholar] [CrossRef]
- Wang, J.; Zhang, H.Y.; Tang, X.C. Cholinergic deficiency involved in vascular dementia: Possible mechanism and strategy of treatment. Acta Pharmacol. Sin. 2009, 30, 879–888. [Google Scholar] [CrossRef]
- Mitsushima, D.; Sano, A.; Takahashi, T. A cholinergic trigger drives learning-induced plasticity at hippocampal synapses. Nat. Commun. 2013, 4, 2760. [Google Scholar] [CrossRef] [Green Version]
- Ribeiro, F.F.; Xapelli, S.; Miranda-Lourenco, C.; Tanqueiro, S.R.; Fonseca-Gomes, J.; Diogenes, M.J.; Ribeiro, J.A.; Sebastiao, A.M. Purine nucleosides in neuroregeneration and neuroprotection. Neuropharmacology 2016, 104, 226–242. [Google Scholar] [CrossRef] [PubMed]
- Mouro, F.M.; Rombo, D.M.; Dias, R.B.; Ribeiro, J.A.; Sebastiao, A.M. Adenosine A2A receptors facilitate synaptic NMDA currents in CA1 pyramidal neurons. Br. J. Pharmacol. 2018, 175, 4386–4397. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sreedharan, J.; Blair, I.P.; Tripathi, V.B.; Hu, X.; Vance, C.; Rogelj, B.; Ackerley, S.; Durnall, J.C.; Williams, K.L.; Buratti, E.; et al. TDP-43 Mutations in Familial and Sporadic Amyotrophic Lateral Sclerosis. Science 2008, 319, 1668–1672. [Google Scholar] [CrossRef]
- Alami, N.; Smith, R.B.; Carrasco, M.A.; Williams, L.A.; Winborn, C.S.; Han, S.S.; Kiskinis, E.; Winborn, B.; Freibaum, B.D.; Kanagaraj, A.; et al. Axonal Transport of TDP-43 mRNA Granules Is Impaired by ALS-Causing Mutations. Neuron 2014, 81, 536–543. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Polymenidou, M.; Lagier-Tourenne, C.; Hutt, K.R.; Huelga, S.C.; Moran, J.; Liang, T.Y.; Ling, S.-C.; Sun, E.; Wancewicz, E.; Mazur, C.; et al. Long pre-mRNA depletion and RNA missplicing contribute to neuronal vulnerability from loss of TDP-43. Nat. Neurosci. 2011, 14, 459–468. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Costessi, L.; Porro, F.; Iaconcig, A.; Muro, A.F. TDP-43 regulates beta-adducin (Add2) transcript stability. RNA Biol. 2014, 11, 1280–1290. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cembrowski, M.S.; Spruston, N. Heterogeneity within classical cell types is the rule: Lessons from hippocampal pyramidal neurons. Nat. Rev. Neurosci. 2019, 20, 193–204. [Google Scholar] [CrossRef]
- Dalton, M.A.; Zeidman, P.; Barry, D.N.; Williams, E.; Maguire, E.A. Segmenting subregions of the human hippocampus on structural magnetic resonance image scans: An illustrated tutorial. Brain Neurosci. Adv. 2017, 1, 2398212817701448. [Google Scholar] [CrossRef]
- Spruston, N. Pyramidal neurons: Dendritic structure and synaptic integration. Nat. Rev. Neurosci. 2008, 9, 206–221. [Google Scholar] [CrossRef]
- Claiborne, B.J.; Amaral, D.G.; Cowan, W.M. Quantitative, three-dimensional analysis of granule cell dendrites in the rat dentate gyrus. J. Comp. Neurol. 1990, 302, 206–219. [Google Scholar] [CrossRef]
- Kumar, V.; Zhang, M.X.; Swank, M.W.; Kunz, J.; Wu, G.Y. Regulation of dendritic morphogenesis by Ras-PI3K-Akt-mTOR and Ras-MAPK signaling pathways. J. Neurosci. 2005, 25, 11288–11299. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maletic-Savatic, M.; Malinow, R.; Svoboda, K. Rapid dendritic morphogenesis in CA1 hippocampal dendrites induced by synaptic activity. Science 1999, 283, 1923–1927. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cline, H.T. Dendritic arbor development and synaptogenesis. Curr. Opin. Neurobiol. 2001, 11, 118–126. [Google Scholar] [CrossRef]
- Dailey, M.E.; Smith, S.J. The dynamics of dendritic structure in developing hippocampal slices. J. Neurosci. 1996, 16, 2983–2994. [Google Scholar] [CrossRef] [PubMed]
- Parrish, J.Z.; Emoto, K.; Kim, M.D.; Jan, Y.N. Mechanisms that regulate establishment, maintenance, and remodeling of dendritic fields. Annu. Rev. Neurosci. 2007, 30, 399–423. [Google Scholar] [CrossRef]
- Sweet, E.S.; Tseng, C.-Y.; Firestein, B. To branch or not to branch: How PSD-95 regulates dendrites and spines. Bioarchitecture 2011, 1, 69–73. [Google Scholar] [CrossRef] [Green Version]
- Tata, D.A.; Anderson, B.J. The effects of chronic glucocorticoid exposure on dendritic length, synapse numbers and glial volume in animal models: Implications for hippocampal volume reductions in depression. Physiol. Behav. 2010, 99, 186–193. [Google Scholar] [CrossRef]
- Conrad, C.D.; McLaughlin, K.J.; Harman, J.S.; Foltz, C.; Wieczorek, L.; Lightner, E.; Wright, R.L. Chronic glucocorticoids increase hippocampal vulnerability to neurotoxicity under conditions that produce CA3 dendritic retraction but fail to impair spatial recognition memory. J. Neurosci. 2007, 27, 8278–8285. [Google Scholar] [CrossRef] [Green Version]
- Tolwani, R.J.; Buckmaster, P.S.; Varma, S.; Cosgaya, J.M.; Wu, Y.; Suri, C.; Shooter, E.M. BDNF overexpression increases dendrite complexity in hippocampal dentate gyrus. Neuroscience 2002, 114, 795–805. [Google Scholar] [CrossRef]
- Kulkarni, V.A.; Firestein, B.L. The dendritic tree and brain disorders. Mol. Cell Neurosci. 2012, 50, 10–20. [Google Scholar] [CrossRef]
- Kolomeets, N.S.; Orlovskaya, D.D.; Uranova, N.A. Decreased numerical density of CA3 hippocampal mossy fiber synapses in schizophrenia. Synapse 2007, 61, 615–621. [Google Scholar] [CrossRef] [PubMed]
- Anderton, B.H.; Callahan, L.; Coleman, P.; Davies, P.; Flood, D.; Jicha, G.A.; Ohm, T.; Weaver, C. Dendritic changes in Alzheimer’s disease and factors that may underlie these changes. Prog. Neurobiol. 1998, 55, 595–609. [Google Scholar] [CrossRef]
- Sousa, N.; Lukoyanov, N.; Madeira, M.; Almeida, O.; Paula-Barbosa, M. Reorganization of the morphology of hippocampal neurites and synapses after stress-induced damage correlates with behavioral improvement. Neuroscience 2000, 97, 253–266. [Google Scholar] [CrossRef]
- Sorra, K.E.; Harris, K.M. Overview on the structure, composition, function, development, and plasticity of hippocampal dendritic spines. Hippocampus 2000, 10, 501–511. [Google Scholar] [CrossRef]
- Ultanir, S.K.; Kim, J.E.; Hall, B.J.; Deerinck, T.; Ellisman, M.; Ghosh, A. Regulation of spine morphology and spine density by NMDA receptor signaling in vivo. Proc. Natl. Acad. Sci. USA 2007, 104, 19553–19558. [Google Scholar] [CrossRef] [Green Version]
- von Bohlen Und Halbach, O. Structure and function of dendritic spines within the hippocampus. Ann. Anat. 2009, 191, 518–531. [Google Scholar] [CrossRef]
- Kasai, H.; Matsuzaki, M.; Noguchi, J.; Yasumatsu, N.; Nakahara, H. Structure-stability-function relationships of dendritic spines. Trends Neurosci. 2003, 26, 360–368. [Google Scholar] [CrossRef]
- Bourne, J.N.; Harris, K.M. Balancing structure and function at hippocampal dendritic spines. Annu. Rev. Neurosci. 2008, 31, 47–67. [Google Scholar] [CrossRef] [Green Version]
- Bourne, J.; Harris, K.M. Do thin spines learn to be mushroom spines that remember? Curr. Opin. Neurobiol. 2007, 17, 381–386. [Google Scholar] [CrossRef]
- Trachtenberg, J.T.; Chen, B.E.; Knott, G.W.; Feng, G.; Sanes, J.R.; Welker, E.; Svoboda, K. Long-term in vivo imaging of experience-dependent synaptic plasticity in adult cortex. Nature 2002, 420, 788–794. [Google Scholar] [CrossRef]
- Skoff, R.P.; Hamburger, V. Fine structure of dendritic and axonal growth cones in embryonic chick spinal cord. J. Comp. Neurol. 1974, 153, 107–147. [Google Scholar] [CrossRef] [PubMed]
- Lohmann, C.; Bonhoeffer, T. A role for local calcium signaling in rapid synaptic partner selection by dendritic filopodia. Neuron 2008, 59, 253–260. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sorra, K.E.; Fiala, J.C.; Harris, K.M. Critical assessment of the involvement of perforations, spinules, and spine branching in hippocampal synapse formation. J. Comp. Neurol. 1998, 398, 225–240. [Google Scholar] [CrossRef]
- Leung, C.C.Y.; Wong, Y.H. Role of G Protein-Coupled Receptors in the Regulation of Structural Plasticity and Cognitive Function. Molecules 2017, 22, 1239. [Google Scholar] [CrossRef] [Green Version]
- Tsai, J.; Grutzendler, J.; Duff, K.; Gan, W.B. Fibrillar amyloid deposition leads to local synaptic abnormalities and breakage of neuronal branches. Nat. Neurosci. 2004, 7, 1181–1183. [Google Scholar] [CrossRef]
- Bellot, A.; Guivernau, B.; Tajes, M.; Bosch-Morato, M.; Valls-Comamala, V.; Munoz, F.J. The structure and function of actin cytoskeleton in mature glutamatergic dendritic spines. Brain Res. 2014, 1573, 1–16. [Google Scholar] [CrossRef] [Green Version]
- Einstein, G.; Buranosky, R.; Crain, B.J. Dendritic pathology of granule cells in Alzheimer’s disease is unrelated to neuritic plaques. J. Neurosci. 1994, 14, 5077–5088. [Google Scholar] [CrossRef]
- Moolman, D.L.; Vitolo, O.V.; Vonsattel, J.P.; Shelanski, M.L. Dendrite and dendritic spine alterations in Alzheimer models. J. Neurocytol. 2004, 33, 377–387. [Google Scholar] [CrossRef]
- Sun, G.Z.; He, Y.C.; Ma, X.K.; Li, S.T.; Chen, D.J.; Gao, M.; Qiu, S.F.; Yin, J.X.; Shi, J.; Wu, J. Hippocampal synaptic and neural network deficits in young mice carrying the human APOE4 gene. CNS Neurosci. Ther. 2017, 23, 748–758. [Google Scholar] [CrossRef] [Green Version]
- Kao, Y.C.; Wang, I.F.; Tsai, K.J. miRNA-34c Overexpression Causes Dendritic Loss and Memory Decline. Int. J. Mol. Sci. 2018, 19, 2323. [Google Scholar] [CrossRef] [Green Version]
- Pristerà, A.; Saraulli, D.; Vecchioli, S.F.; Strimpakos, G.; Costanzi, M.; di Certo, M.G.; Cannas, S.; Ciotti, M.T.; Tirone, F.; Mattei, E.; et al. Impact of N-tau on adult hippocampal neurogenesis, anxiety, and memory. Neurobiol. Aging 2013, 34, 2551–2563. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Androuin, A.; Potier, B.; Nägerl, U.V.; Cattaert, D.; Danglot, L.; Thierry, M.; Youssef, I.; Triller, A.; Duyckaerts, C.; El Hachimi, K.H.; et al. Evidence for altered dendritic spine compartmentalization in Alzheimer’s disease and functional effects in a mouse model. Acta Neuropathol. 2018, 135, 839–854. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Wu, L.; Pchitskaya, E.; Zakharova, O.; Saito, T.; Saido, T.; Bezprozvanny, I. Neuronal Store-Operated Calcium Entry and Mushroom Spine Loss in Amyloid Precursor Protein Knock-In Mouse Model of Alzheimer’s Disease. J. Neurosci. 2015, 35, 13275–13286. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dachsel, J.C.; Behrouz, B.; Yue, M.; Beevers, J.E.; Melrose, H.L.; Farrer, M.J. A comparative study of Lrrk2 function in primary neuronal cultures. Park. Relat. Disord. 2010, 16, 650–655. [Google Scholar] [CrossRef] [Green Version]
- Sepulveda, B.; Mesias, R.; Li, X.; Yue, Z.; Benson, D.L. Short- and long-term effects of LRRK2 on axon and dendrite growth. PLoS ONE 2013, 8, e61986. [Google Scholar] [CrossRef] [Green Version]
- Winner, B.; Melrose, H.; Zhao, C.; Hinkle, K.; Yue, M.; Kent, C.; Braithwaite, A.; Ogholikhan, S.; Aigner, R.; Winkler, J. Adult neurogenesis and neurite outgrowth are impaired in LRRK2 G2019S mice. Neurobiol. Dis. 2011, 41, 706–716. [Google Scholar] [CrossRef] [Green Version]
- Weerasinghe-Mudiyanselage, P.D.E.; Ang, M.J.; Wada, M.; Kim, S.H.; Shin, T.; Yang, M.; Moon, C. Acute MPTP Treatment Impairs Dendritic Spine Density in the Mouse Hippocampus. Brain Sci. 2021, 11, 833. [Google Scholar] [CrossRef]
- Froula, J.M.; Henderson, B.W.; Gonzalez, J.C.; Vaden, J.H.; McLean, J.W.; Wu, Y.; Banumurthy, G.; Overstreet-Wadiche, L.; Herskowitz, J.H.; Volpicelli-Daley, L.A. alpha-Synuclein fibril-induced paradoxical structural and functional defects in hippocampal neurons. Acta Neuropathol. Commun. 2018, 6, 35. [Google Scholar] [CrossRef] [Green Version]
- da Silva, M.O.; Santejo, M.; Babcock, I.; Magalhães, A.; Minamide, L.; Castillo, E.; Gerhardt, E.; Fahlbusch, C.; Swanson, R.; Outeiro, T. Cofilin pathology is a new player on α-synuclein-induced spine impairment in models of hippocampal synucleinopathy. Biorxiv 2021. [Google Scholar] [CrossRef]
- Anglada-Huguet, M.; Vidal-Sancho, L.; Giralt, A.; Barriga, G.G.-D.; Xifró, X.; Alberch, J. Prostaglandin E2 EP2 activation reduces memory decline in R6/1 mouse model of Huntington’s disease by the induction of BDNF-dependent synaptic plasticity. Neurobiol. Dis. 2016, 95, 22–34. [Google Scholar] [CrossRef]
- Milnerwood, A.J.; Parsons, M.P.; Young, F.B.; Singaraja, R.R.; Franciosi, S.; Volta, M.; Bergeron, S.; Hayden, M.R.; Raymond, L.A. Memory and synaptic deficits in Hip14/DHHC17 knockout mice. Proc. Natl. Acad. Sci. USA 2013, 110, 20296–20301. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miguez, A.; Garcia-Diaz Barriga, G.; Brito, V.; Straccia, M.; Giralt, A.; Gines, S.; Canals, J.M.; Alberch, J. Fingolimod (FTY720) enhances hippocampal synaptic plasticity and memory in Huntington’s disease by preventing p75NTR up-regulation and astrocyte-mediated inflammation. Hum. Mol. Genet. 2015, 24, 4958–4970. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nithianantharajah, J.; Barkus, C.; Murphy, M.; Hannan, A.J. Gene-environment interactions modulating cognitive function and molecular correlates of synaptic plasticity in Huntington’s disease transgenic mice. Neurobiol. Dis. 2008, 29, 490–504. [Google Scholar] [CrossRef]
- Crombe, A.; Planche, V.; Raffard, G.; Bourel, J.; Dubourdieu, N.; Panatier, A.; Fukutomi, H.; Dousset, V.; Oliet, S.; Hiba, B.; et al. Deciphering the microstructure of hippocampal subfields with in vivo DTI and NODDI: Applications to experimental multiple sclerosis. NeuroImage 2018, 172, 357–368. [Google Scholar] [CrossRef] [PubMed]
- Planche, V.; Panatier, A.; Hiba, B.; Ducourneau, E.-G.; Raffard, G.; Dubourdieu, N.; Maitre, M.; Lesté-Lasserre, T.; Brochet, B.; Dousset, V.; et al. Selective dentate gyrus disruption causes memory impairment at the early stage of experimental multiple sclerosis. Brain Behav. Immun. 2017, 60, 240–254. [Google Scholar] [CrossRef] [PubMed]
- Baltan, S.; Jawaid, S.S.; Chomyk, A.M.; Kidd, G.J.; Chen, J.; Battapady, H.D.; Chan, R.; Dutta, R.; Trapp, B.D. Neuronal hibernation following hippocampal demyelination. Acta Neuropathol. Commun. 2021, 9, 34. [Google Scholar] [CrossRef]
- Chen, X.; Jiang, X.-M.; Zhao, L.-J.; Sun, L.-L.; Yan, M.-L.; Tian, Y.; Zhang, S.; Duan, M.-J.; Zhao, H.-M.; Li, W.-R.; et al. MicroRNA-195 prevents dendritic degeneration and neuron death in rats following chronic brain hypoperfusion. Cell Death Dis. 2017, 8, e2850. [Google Scholar] [CrossRef] [Green Version]
- Guo, S.; Xu, J.-J.; Wei, N.; Han, J.-Y.; Xue, R.; Xu, P.-S.; Gao, C.-Y. Honokiol Attenuates the Memory Impairments, Oxidative Stress, Neuroinflammation, and GSK-3 β Activation in Vascular Dementia Rats. J. Alzheimer’s Dis. 2019, 71, 97–108. [Google Scholar] [CrossRef]
- Zhu, Y.; Zhang, Q.; Zhang, W.; Li, N.; Dai, Y.; Tu, J.; Yang, F.; Brann, D.W.; Wang, R. Protective effect of 17β-estradiol upon hippocampal spine density and cognitive function in an animal model of vascular dementia. Sci. Rep. 2017, 7, 42660. [Google Scholar] [CrossRef] [Green Version]
- Jian, W.X.; Zhang, Z.; Zhan, J.H.; Chu, S.F.; Peng, Y.; Zhao, M.; Wang, Q.; Chen, N.H. Donepezil attenuates vascular dementia in rats through increasing BDNF induced by reducing HDAC6 nuclear translocation. Acta Pharmacol. Sin. 2020, 41, 588–598. [Google Scholar] [CrossRef]
- Fogarty, M.J.; Mu, E.W.H.; Lavidis, N.A.; Noakes, P.G.; Bellingham, M.C. Motor Areas Show Altered Dendritic Structure in an Amyotrophic Lateral Sclerosis Mouse Model. Front Neurosci. 2017, 11, 609. [Google Scholar] [CrossRef] [PubMed]
- Jack, C.R., Jr.; Knopman, D.S.; Jagust, W.J.; Shaw, L.M.; Aisen, P.S.; Weiner, M.W.; Petersen, R.C.; Trojanowski, J.Q. Hypothetical model of dynamic biomarkers of the Alzheimer’s pathological cascade. Lancet Neurol. 2010, 9, 119–128. [Google Scholar] [CrossRef] [Green Version]
- Blennow, K.; Hampel, H.; Weiner, M.; Zetterberg, H. Cerebrospinal fluid and plasma biomarkers in Alzheimer disease. Nat. Rev. Neurol. 2010, 6, 131–144. [Google Scholar] [CrossRef] [PubMed]
- Nisbet, R.M.; Polanco, J.C.; Ittner, L.M.; Gotz, J. Tau aggregation and its interplay with amyloid-beta. Acta Neuropathol. 2015, 129, 207–220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Canevari, L.; Abramov, A.Y.; Duchen, M.R. Toxicity of amyloid beta peptide: Tales of calcium, mitochondria, and oxidative stress. Neurochem. Res. 2004, 29, 637–650. [Google Scholar] [CrossRef] [PubMed]
- Mattson, M.P.; Chan, S.L. Neuronal and glial calcium signaling in Alzheimer’s disease. Cell Calcium. 2003, 34, 385–397. [Google Scholar] [CrossRef]
- Lee, G.; Neve, R.L.; Kosik, K.S. The microtubule binding domain of tau protein. Neuron 1989, 2, 1615–1624. [Google Scholar] [CrossRef]
- Kent, S.A.; Spires-Jones, T.L.; Durrant, C.S. The physiological roles of tau and Abeta: Implications for Alzheimer’s disease pathology and therapeutics. Acta Neuropathol. 2020, 140, 417–447. [Google Scholar] [CrossRef]
- Spires-Jones, T.; Knafo, S. Spines, plasticity, and cognition in Alzheimer’s model mice. Neural. Plast. 2012, 2012, 319836. [Google Scholar] [CrossRef]
- Toni, N.; Sultan, S. Synapse formation on adult-born hippocampal neurons. Eur. J. Neurosci. 2011, 33, 1062–1068. [Google Scholar] [CrossRef]
- Gong, X.; Lu, X.; Zhan, L.; Sui, H.; Qi, X.; Ji, Z.; Niu, X.; Liu, L. Role of the SNK-SPAR pathway in the development of Alzheimer’s disease. IUBMB Life 2010, 62, 214–221. [Google Scholar] [CrossRef] [PubMed]
- Hoover, B.R.; Reed, M.N.; Su, J.; Penrod, R.D.; Kotilinek, L.A.; Grant, M.K.; Pitstick, R.; Carlson, G.A.; Lanier, L.M.; Yuan, L.-L.; et al. Tau Mislocalization to Dendritic Spines Mediates Synaptic Dysfunction Independently of Neurodegeneration. Neuron 2010, 68, 1067–1081. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pluta, R.; Ułamek-Kozioł, M. Tau Protein-Targeted Therapies in Alzheimer’s Disease: Current State and Future Perspectives; Exon Publications: Brisbane, Australia, 2020; pp. 69–82. [Google Scholar]
- Xin, S.H.; Tan, L.; Cao, X.; Yu, J.T.; Tan, L. Clearance of Amyloid Beta and Tau in Alzheimer’s Disease: From Mechanisms to Therapy. Neurotox Res. 2018, 34, 733–748. [Google Scholar] [CrossRef] [PubMed]
- Polanco, J.C.; Li, C.; Bodea, L.-G.; Martinez-Marmol, R.; Meunier, F.A.; Götz, J. Amyloid-β and tau complexity—towards improved biomarkers and targeted therapies. Nat. Rev. Neurol. 2018, 14, 22–39. [Google Scholar] [CrossRef]
- McNeill, T.H.; Brown, S.A.; Rafols, J.A.; Shoulson, I. Atrophy of medium spiny I striatal dendrites in advanced Parkinson’s disease. Brain Res. 1988, 455, 148–152. [Google Scholar] [CrossRef]
- Day, M.; Wang, Z.; Ding, J.; An, X.; Ingham, C.A.; Shering, A.F.; Wokosin, D.; Ilijic, E.; Sun, Z.; Sampson, A.R.; et al. Selective elimination of glutamatergic synapses on striatopallidal neurons in Parkinson disease models. Nat. Neurosci. 2006, 9, 251–259. [Google Scholar] [CrossRef]
- Zaja-Milatovic, S.; Milatovic, D.; Schantz, A.M.; Zhang, J.; Montine, K.S.; Samii, A.; Deutch, A.Y.; Montine, T.J. Dendritic degeneration in neostriatal medium spiny neurons in Parkinson disease. Neurology 2005, 64, 545–547. [Google Scholar] [CrossRef]
- Stephens, B.; Mueller, A.; Shering, A.; Hood, S.; Taggart, P.; Arbuthnott, G.; Bell, J.; Kilford, L.; Kingsbury, A.; Daniel, S.; et al. Evidence of a breakdown of corticostriatal connections in Parkinson’s disease. Neuroscience 2005, 132, 741–754. [Google Scholar] [CrossRef]
- Ingham, C.A.; Hood, S.H.; Arbuthnott, G.W. Spine density on neostriatal neurones changes with 6-hydroxydopamine lesions and with age. Brain Res. 1989, 503, 334–338. [Google Scholar] [CrossRef]
- Borgkvist, A.; Malmlof, T.; Feltmann, K.; Lindskog, M.; Schilstrom, B. Dopamine in the hippocampus is cleared by the norepinephrine transporter. Int. J. Neuropsychopharmacol. 2012, 15, 531–540. [Google Scholar] [CrossRef] [Green Version]
- Park, J.H.; Enikolopov, G. Transient elevation of adult hippocampal neurogenesis after dopamine depletion. Exp. Neurol. 2010, 222, 267–276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gangarossa, G.; Longueville, S.; De Bundel, D.; Perroy, J.; Herve, D.; Girault, J.A.; Valjent, E. Characterization of dopamine D1 and D2 receptor-expressing neurons in the mouse hippocampus. Hippocampus 2012, 22, 2199–2207. [Google Scholar] [CrossRef] [PubMed]
- Shinohara, R.; Taniguchi, M.; Ehrlich, A.T.; Yokogawa, K.; Deguchi, Y.; Cherasse, Y.; Lazarus, M.; Urade, Y.; Ogawa, A.; Kitaoka, S.; et al. Dopamine D1 receptor subtype mediates acute stress-induced dendritic growth in excitatory neurons of the medial prefrontal cortex and contributes to suppression of stress susceptibility in mice. Mol. Psychiatry 2018, 23, 1717–1730. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neuner, J.; Ovsepian, S.V.; Dorostkar, M.; Filser, S.; Gupta, A.; Michalakis, S.; Biel, M.; Herms, J. Pathological alpha-synuclein impairs adult-born granule cell development and functional integration in the olfactory bulb. Nat. Commun. 2014, 5, 3915. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koch, J.C.; Bitow, F.; Haack, J.; D’Hedouville, Z.; Zhang, J.-N.; Tönges, L.; Michel, U.; Oliveira, L.M.A.; Jovin, T.M.; Liman, J.; et al. Alpha-Synuclein affects neurite morphology, autophagy, vesicle transport and axonal degeneration in CNS neurons. Cell Death Dis. 2015, 6, e1811. [Google Scholar] [CrossRef] [Green Version]
- Galvin, J.E.; Uryu, K.; Lee, V.M.; Trojanowski, J.Q. Axon pathology in Parkinson’s disease and Lewy body dementia hippocampus contains alpha-, beta-, and gamma-synuclein. Proc. Natl. Acad. Sci. USA 1999, 96, 13450–13455. [Google Scholar] [CrossRef] [Green Version]
- Scatton, B.; Javoy-Agid, F.; Rouquier, L.; Dubois, B.; Agid, Y. Reduction of cortical dopamine, noradrenaline, serotonin and their metabolites in Parkinson’s disease. Brain Res. 1983, 275, 321–328. [Google Scholar] [CrossRef]
- Deusser, J.; Schmidt, S.; Ettle, B.; Plotz, S.; Huber, S.; Muller, C.P.; Masliah, E.; Winkler, J.; Kohl, Z. Serotonergic dysfunction in the A53T alpha-synuclein mouse model of Parkinson’s disease. J. Neurochem. 2015, 135, 589–597. [Google Scholar] [CrossRef] [Green Version]
- Trakhtenberg, E.F.; Goldberg, J.L. The role of serotonin in axon and dendrite growth. Int. Rev. Neurobiol. 2012, 106, 105–126. [Google Scholar] [CrossRef]
- Lindgren, H.S.; Dunnett, S.B. Cognitive dysfunction and depression in Parkinson’s disease: What can be learned from rodent models? Eur. J. Neurosci. 2012, 35, 1894–1907. [Google Scholar] [CrossRef]
- Ji, Y.; Pang, P.T.; Feng, L.; Lu, B. Cyclic AMP controls BDNF-induced TrkB phosphorylation and dendritic spine formation in mature hippocampal neurons. Nat. Neurosci. 2005, 8, 164–172. [Google Scholar] [CrossRef] [PubMed]
- Norrholm, S.D.; Ouimet, C.C. Chronic fluoxetine administration to juvenile rats prevents age-associated dendritic spine proliferation in hippocampus. Brain Res. 2000, 883, 205–215. [Google Scholar] [CrossRef]
- Bender, H.; Fietz, S.A.; Richter, F.; Stanojlovic, M. Alpha-Synuclein Pathology Coincides With Increased Number of Early Stage Neural Progenitors in the Adult Hippocampus. Front. Cell Dev. Biol. 2021, 9, 691560. [Google Scholar] [CrossRef]
- Alim, M.A.; Ma, Q.-L.; Takeda, K.; Aizawa, T.; Matsubara, M.; Nakamura, M.; Asada, A.; Saito, T.; Xkaji, M.; Yoshii, M.; et al. Demonstration of a role for α-synuclein as a functional microtubule-associated protein. J. Alzheimer’s Dis. 2004, 6, 435–442. [Google Scholar] [CrossRef] [PubMed]
- Bonini, N.M.; Giasson, B.I. Snaring the function of alpha-synuclein. Cell 2005, 123, 359–361. [Google Scholar] [CrossRef] [PubMed]
- Stefanis, L. alpha-Synuclein in Parkinson’s disease. Cold Spring Harb. Perspect Med. 2012, 2, a009399. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wersinger, C.; Rusnak, M.; Sidhu, A. Modulation of the trafficking of the human serotonin transporter by human alpha-synuclein. Eur. J. Neurosci. 2006, 24, 55–64. [Google Scholar] [CrossRef] [PubMed]
- Chaji, D.; Venkatesh, V.S.; Shirao, T.; Day, D.J.; Ellenbroek, B.A. Genetic Knockout of the Serotonin Reuptake Transporter Results in the Reduction of Dendritic Spines in In vitro Rat Cortical Neuronal Culture. J. Mol. Neurosci. 2021, 71, 2210–2218. [Google Scholar] [CrossRef]
- Nagano-Saito, A.; Habak, C.; Mejía-Constaín, B.; Degroot, C.; Monetta, L.; Jubault, T.; Bedetti, C.; Lafontaine, A.-L.; Chouinard, S.; Soland, V.; et al. Effect of mild cognitive impairment on the patterns of neural activity in early Parkinson’s disease. Neurobiol. Aging 2014, 35, 223–231. [Google Scholar] [CrossRef]
- Milnerwood, A.J.; Raymond, L.A. Early synaptic pathophysiology in neurodegeneration: Insights from Huntington’s disease. Trends Neurosci. 2010, 33, 513–523. [Google Scholar] [CrossRef]
- Raymond, L.A.; Andre, V.M.; Cepeda, C.; Gladding, C.M.; Milnerwood, A.J.; Levine, M.S. Pathophysiology of Huntington’s disease: Time-dependent alterations in synaptic and receptor function. Neuroscience 2011, 198, 252–273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmidt, M.E.; Buren, C.; Mackay, J.P.; Cheung, D.; Dal Cengio, L.; Raymond, L.A.; Hayden, M.R. Altering cortical input unmasks synaptic phenotypes in the YAC128 cortico-striatal co-culture model of Huntington disease. BMC Biol. 2018, 16, 58. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carmo, C.; Naia, L.; Lopes, C.; Rego, A.C. Mitochondrial Dysfunction in Huntington’s Disease. Adv. Exp. Med. Biol. 2018, 1049, 59–83. [Google Scholar] [CrossRef] [PubMed]
- Saavedra, A.; Garcia-Diaz Barriga, G.; Perez-Navarro, E.; Alberch, J. Huntington’s disease: Novel therapeutic perspectives hanging in the balance. Expert Opin. Ther. Targets 2018, 22, 385–399. [Google Scholar] [CrossRef]
- Li, X.J.; Orr, A.L.; Li, S. Impaired mitochondrial trafficking in Huntington’s disease. Biochim. Biophys. Acta 2010, 1802, 62–65. [Google Scholar] [CrossRef] [Green Version]
- Rangaraju, V.; Lewis, T.L., Jr.; Hirabayashi, Y.; Bergami, M.; Motori, E.; Cartoni, R.; Kwon, S.K.; Courchet, J. Pleiotropic Mitochondria: The Influence of Mitochondria on Neuronal Development and Disease. J. Neurosci. 2019, 39, 8200–8208. [Google Scholar] [CrossRef] [Green Version]
- Steib, K.; Schaffner, I.; Jagasia, R.; Ebert, B.; Lie, D.C. Mitochondria modify exercise-induced development of stem cell-derived neurons in the adult brain. J. Neurosci. 2014, 34, 6624–6633. [Google Scholar] [CrossRef]
- Magistretti, P.J.; Allaman, I. A cellular perspective on brain energy metabolism and functional imaging. Neuron 2015, 86, 883–901. [Google Scholar] [CrossRef] [Green Version]
- Gauthier, L.R.; Charrin, B.C.; Borrell, M.; Dompierre, J.P.; Rangone, H.; Cordelières, F.; De Mey, J.; MacDonald, M.E.; Leßmann, V.; Humbert, S.; et al. Huntingtin Controls Neurotrophic Support and Survival of Neurons by Enhancing BDNF Vesicular Transport along Microtubules. Cell 2004, 118, 127–138. [Google Scholar] [CrossRef] [Green Version]
- McAllister, A.K. Cellular and molecular mechanisms of dendrite growth. Cereb. Cortex. 2000, 10, 963–973. [Google Scholar] [CrossRef] [Green Version]
- Labbadia, J.; Morimoto, R.I. Huntington’s disease: Underlying molecular mechanisms and emerging concepts. Trends Biochem. Sci. 2013, 38, 378–385. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zuccato, C.; Valenza, M.; Cattaneo, E. Molecular mechanisms and potential therapeutical targets in Huntington’s disease. Physiol. Rev. 2010, 90, 905–981. [Google Scholar] [CrossRef]
- Flippo, K.H.; Strack, S. Mitochondrial dynamics in neuronal injury, development and plasticity. J. Cell Sci. 2017, 130, 671–681. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Z.; Okamoto, K.; Hayashi, Y.; Sheng, M. The importance of dendritic mitochondria in the morphogenesis and plasticity of spines and synapses. Cell 2004, 119, 873–887. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shirendeb, U.P.; Calkins, M.J.; Manczak, M.; Anekonda, V.; Dufour, B.; McBride, J.L.; Mao, P.; Reddy, P.H. Mutant huntingtin’s interaction with mitochondrial protein Drp1 impairs mitochondrial biogenesis and causes defective axonal transport and synaptic degeneration in Huntington’s disease. Hum. Mol. Genet. 2012, 21, 406–420. [Google Scholar] [CrossRef] [PubMed]
- Ettcheto, M.; Busquets, O.; Cano, A.; Sánchez-Lopez, E.; Manzine, P.R.; Espinosa-Jimenez, T.; Verdaguer, E.; Sureda, F.X.; Olloquequi, J.; Castro-Torres, R.D.; et al. Pharmacological Strategies to Improve Dendritic Spines in Alzheimer’s Disease. J. Alzheimer’s Dis. 2021, 82, S91–S107. [Google Scholar] [CrossRef]
- Grussu, F.; Schneider, T.; Tur, C.; Yates, R.L.; Tachrount, M.; Ianus, A.; Yiannakas, M.C.; Newcombe, J.; Zhang, H.; Alexander, D.C.; et al. Neurite dispersion: A new marker of multiple sclerosis spinal cord pathology? Ann. Clin. Transl. Neurol. 2017, 4, 663–679. [Google Scholar] [CrossRef]
- Gilmore, C.P.; DeLuca, G.C.; Bo, L.; Owens, T.; Lowe, J.; Esiri, M.M.; Evangelou, N. Spinal cord atrophy in multiple sclerosis caused by white matter volume loss. Arch Neurol. 2005, 62, 1859–1862. [Google Scholar] [CrossRef] [Green Version]
- D’Souza, S.; Alinauskas, K.; McCrea, E.; Goodyer, C.; Antel, J.P. Differential susceptibility of human CNS-derived cell populations to TNF-dependent and independent immune-mediated injury. J. Neurosci. 1995, 15, 7293–7300. [Google Scholar] [CrossRef]
- Akassoglou, K.; Bauer, J.; Kassiotis, G.; Pasparakis, M.; Lassmann, H.; Kollias, G.; Probert, L. Oligodendrocyte apoptosis and primary demyelination induced by local TNF/p55TNF receptor signaling in the central nervous system of transgenic mice: Models for multiple sclerosis with primary oligodendrogliopathy. Am. J. Pathol. 1998, 153, 801–813. [Google Scholar] [CrossRef]
- Andrews, T.; Zhang, P.; Bhat, N.R. TNFalpha potentiates IFNgamma-induced cell death in oligodendrocyte progenitors. J. Neurosci. Res. 1998, 54, 574–583. [Google Scholar] [CrossRef]
- Ben-Hur, T.; Ben-Menachem, O.; Furer, V.; Einstein, O.; Mizrachi-Kol, R.; Grigoriadis, N. Effects of proinflammatory cytokines on the growth, fate, and motility of multipotential neural precursor cells. Mol. Cell Neurosci. 2003, 24, 623–631. [Google Scholar] [CrossRef]
- Yoo, S.W.; Motari, M.G.; Susuki, K.; Prendergast, J.; Mountney, A.; Hurtado, A.; Schnaar, R.L. Sialylation regulates brain structure and function. FASEB J. 2015, 29, 3040–3053. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Q.; Yu, S.; Simonyi, A.; Sun, G.Y.; Sun, A.Y. Kainic acid-mediated excitotoxicity as a model for neurodegeneration. Molecular. Neurobiol. 2005, 31, 3–16. [Google Scholar] [CrossRef]
- Vallejo-Illarramendi, A.; Domercq, M.; Perez-Cerda, F.; Ravid, R.; Matute, C. Increased expression and function of glutamate transporters in multiple sclerosis. Neurobiol. Dis. 2006, 21, 154–164. [Google Scholar] [CrossRef]
- Crochemore, C.; Lu, J.; Wu, Y.; Liposits, Z.; Sousa, N.; Holsboer, F.; Almeida, O.F. Direct targeting of hippocampal neurons for apoptosis by glucocorticoids is reversible by mineralocorticoid receptor activation. Mol. Psychiatry 2005, 10, 790–798. [Google Scholar] [CrossRef] [Green Version]
- Lu, J.; Goula, D.; Sousa, N.; Almeida, O.F. Ionotropic and metabotropic glutamate receptor mediation of glucocorticoid-induced apoptosis in hippocampal cells and the neuroprotective role of synaptic N-methyl-D-aspartate receptors. Neuroscience 2003, 121, 123–131. [Google Scholar] [CrossRef] [Green Version]
- Uttner, I.; Muller, S.; Zinser, C.; Maier, M.; Süssmuth, S.; Claus, A.; Ostermann, B.; Elitok, E.; Ecker, D.; Brettschneider, J.; et al. Reversible impaired memory induced by pulsed methylprednisolone in patients with MS. Neurology 2005, 64, 1971–1973. [Google Scholar] [CrossRef]
- Roozendaal, B.; de Quervain, D.J. Glucocorticoid therapy and memory function: Lessons learned from basic research. Neurology 2005, 64, 184–185. [Google Scholar] [CrossRef]
- Brunner, R.; Schaefer, D.; Hess, K.; Parzer, P.; Resch, F.; Schwab, S. Effect of corticosteroids on short-term and long-term memory. Neurology 2005, 64, 335–337. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhang, H.; Wang, L.; Jiang, W.; Xu, H.; Xiao, L.; Bi, X.; Wang, J.; Zhu, S.; Zhang, R.; et al. Quetiapine enhances oligodendrocyte regeneration and myelin repair after cuprizone-induced demyelination. Schizophr. Res. 2012, 138, 8–17. [Google Scholar] [CrossRef] [PubMed]
- Abe, H.; Tanaka, T.; Kimura, M.; Mizukami, S.; Saito, F.; Imatanaka, N.; Akahori, Y.; Yoshida, T.; Shibutani, M. Cuprizone decreases intermediate and late-stage progenitor cells in hippocampal neurogenesis of rats in a framework of 28-day oral dose toxicity study. Toxicol. Appl. Pharmacol. 2015, 287, 210–221. [Google Scholar] [CrossRef] [PubMed]
- Bitzer-Quintero, O.K.; Gonzalez-Burgos, I. Immune system in the brain: A modulatory role on dendritic spine morphophysiology? Neural. Plast. 2012, 2012, 348642. [Google Scholar] [CrossRef] [PubMed]
- Donato, F.; Jacobsen, R.I.; Moser, M.B.; Moser, E.I. Stellate cells drive maturation of the entorhinal-hippocampal circuit. Science 2017, 355, eaai8178. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, J.; Olsen, R.H.; Sun, J.; Ming, G.L.; Song, H. Neuronal Circuitry Mechanisms Regulating Adult Mammalian Neurogenesis. Cold Spring Harb. Perspect. Biol. 2016, 8, a018937. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peterson, J.W.; Bo, L.; Mork, S.; Chang, A.; Trapp, B.D. Transected neurites, apoptotic neurons, and reduced inflammation in cortical multiple sclerosis lesions. Ann. Neurol. 2001, 50, 389–400. [Google Scholar] [CrossRef]
- Peterson, L.K.; Fujinami, R.S. Inflammation, demyelination, neurodegeneration and neuroprotection in the pathogenesis of multiple sclerosis. J. Neuroimmunol. 2007, 184, 37–44. [Google Scholar] [CrossRef] [Green Version]
- Spalloni, A.; Origlia, N.; Sgobio, C.; Trabalza, A.; Nutini, M.; Berretta, N.; Bernardi, G.; Domenici, L.; Ammassari-Teule, M.; Longone, P. Postsynaptic alteration of NR2A subunit and defective autophosphorylation of alphaCaMKII at threonine-286 contribute to abnormal plasticity and morphology of upper motor neurons in presymptomatic SOD1G93A mice, a murine model for amyotrophic lateral sclerosis. Cereb. Cortex. 2011, 21, 796–805. [Google Scholar]
- Sgobio, C.; Trabalza, A.; Spalloni, A.; Zona, C.; Carunchio, I.; Longone, P.; Ammassari-Teule, M. Abnormal medial prefrontal cortex connectivity and defective fear extinction in the presymptomatic G93A SOD1 mouse model of ALS. Genes Brain Behav. 2008, 7, 427–434. [Google Scholar] [CrossRef]
- Van Zundert, B.; Peuscher, M.H.; Hynynen, M.; Chen, A.; Neve, R.L.; Brown, R.H.; Constantine-Paton, M.; Bellingham, M.C. Neonatal neuronal circuitry shows hyperexcitable disturbance in a mouse model of the adult-onset neurodegenerative disease amyotrophic lateral sclerosis. J. Neurosci. 2008, 28, 10864–10874. [Google Scholar] [CrossRef]
- Martin, E.; Cazenave, W.; Cattaert, D.; Branchereau, P. Embryonic alteration of motoneuronal morphology induces hyperexcitability in the mouse model of amyotrophic lateral sclerosis. Neurobiol. Dis. 2013, 54, 116–126. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.H.; Ban, T.; Liu, C.D.; Chen, Q.X.; Wang, X.; Yan, M.L.; Hu, X.L.; Su, X.L.; Bao, Y.N.; Sun, L.L. Activation of Cdk5/p25 and tau phosphorylation following chronic brain hypoperfusion in rats involves micro RNA-195 down-regulation. J. Neurochem. 2015, 134, 1139–1151. [Google Scholar] [CrossRef] [PubMed]
- Ai, J.; Sun, L.-H.; Che, H.; Zhang, R.; Zhang, T.-Z.; Wu, W.-C.; Su, X.-L.; Chen, X.; Yang, G.; Li, K.; et al. MicroRNA-195 Protects Against Dementia Induced by Chronic Brain Hypoperfusion via Its Anti-Amyloidogenic Effect in Rats. J. Neurosci. 2013, 33, 3989–4001. [Google Scholar] [CrossRef] [PubMed]
- Tsai, T.H.; Sun, C.K.; Su, C.H.; Sung, P.H.; Chua, S.; Zhen, Y.Y.; Leu, S.; Chang, H.W.; Yang, J.L.; Yip, H.K. Sitagliptin attenuated brain damage and cognitive impairment in mice with chronic cerebral hypo-perfusion through suppressing oxidative stress and inflammatory reaction. J. Hypertens. 2015, 33, 1001–1013. [Google Scholar] [CrossRef]
- Flores, G.; Flores-Gomez, G.D.; de Jesus Gomez-Villalobos, M. Neuronal changes after chronic high blood pressure in animal models and its implication for vascular dementia. Synapse 2016, 70, 198–205. [Google Scholar] [CrossRef]
- Rohrer, J.D.; Warren, J.D. Phenotypic signatures of genetic frontotemporal dementia. Curr. Opin. Neurol. 2011, 24, 542–549. [Google Scholar] [CrossRef]
- Weingarten, M.D.; Lockwood, A.H.; Hwo, S.Y.; Kirschner, M.W. A protein factor essential for microtubule assembly. Proc. Natl. Acad. Sci. USA 1975, 72, 1858–1862. [Google Scholar] [CrossRef] [Green Version]
- Drubin, D.G.; Kirschner, M.W. Tau protein function in living cells. J. Cell Biol. 1986, 103, 2739–2746. [Google Scholar] [CrossRef] [Green Version]
- Saxena, A.K.; Abdul-Majeed, S.S.; Gurtu, S.; Mohamed, W.M. Investigation of redox status in chronic cerebral hypoperfusion-induced neurodegeneration in rats. Appl. Transl. Genom. 2015, 5, 30–32. [Google Scholar] [CrossRef] [Green Version]
- Vazquez-Manrique, R.P.; Farina, F.; Cambon, K.; Dolores Sequedo, M.; Parker, A.J.; Millan, J.M.; Weiss, A.; Deglon, N.; Neri, C. AMPK activation protects from neuronal dysfunction and vulnerability across nematode, cellular and mouse models of Huntington’s disease. Hum. Mol. Genet. 2016, 25, 1043–1058. [Google Scholar] [CrossRef]
- Cai, Z.; Yan, L.J.; Li, K.; Quazi, S.H.; Zhao, B. Roles of AMP-activated protein kinase in Alzheimer’s disease. Neuromolecular Med. 2012, 14, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Rui, Y.; Myers, K.R.; Yu, K.; Wise, A.; De Blas, A.L.; Hartzell, H.C.; Zheng, J.Q. Activity-dependent regulation of dendritic growth and maintenance by glycogen synthase kinase 3beta. Nat. Commun. 2013, 4, 2628. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zou, C.; Shi, Y.; Ohli, J.; Schuller, U.; Dorostkar, M.M.; Herms, J. Neuroinflammation impairs adaptive structural plasticity of dendritic spines in a preclinical model of Alzheimer’s disease. Acta Neuropathol. 2016, 131, 235–246. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szabo, C.; Kelemen, O.; Keri, S. Low-grade inflammation disrupts structural plasticity in the human brain. Neuroscience 2014, 275, 81–88. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dahle, C.L.; Jacobs, B.S.; Raz, N. Aging, vascular risk, and cognition: Blood glucose, pulse pressure, and cognitive performance in healthy adults. Psychol. Aging. 2009, 24, 154–162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- VanGuilder, H.D.; Farley, J.A.; Yan, H.; Van Kirk, C.A.; Mitschelen, M.; Sonntag, W.E.; Freeman, W.M. Hippocampal dysregulation of synaptic plasticity-associated proteins with age-related cognitive decline. Neurobiol. Dis. 2011, 43, 201–212. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Foster, T.C. Involvement of hippocampal synaptic plasticity in age-related memory decline. Brain Res. Brain Res. Rev. 1999, 30, 236–249. [Google Scholar] [CrossRef]
- Small, G.W. What we need to know about age related memory loss. BMJ 2002, 324, 1502–1505. [Google Scholar] [CrossRef] [Green Version]
- Temido-Ferreira, M.; Coelho, J.E.; Pousinha, P.A.; Lopes, L.V. Novel Players in the Aging Synapse: Impact on Cognition. J. Caffeine Adenosine Res. 2019, 9, 104–127. [Google Scholar] [CrossRef]
- Bettio, L.E.B.; Rajendran, L.; Gil-Mohapel, J. The effects of aging in the hippocampus and cognitive decline. Neurosci. Biobehav. Rev. 2017, 79, 66–86. [Google Scholar] [CrossRef]
- Kumar, A. Calcium Signaling During Brain Aging and Its Influence on the Hippocampal Synaptic Plasticity. Adv. Exp. Med. Biol. 2020, 1131, 985–1012. [Google Scholar] [CrossRef]
- Dahan, L.; Rampon, C.; Florian, C. Age-related memory decline, dysfunction of the hippocampus and therapeutic opportunities. Prog. Neuropsychopharmacol. Biol. Psychiatry 2020, 102, 109943. [Google Scholar] [CrossRef]
- Navakkode, S.; Liu, C.; Soong, T.W. Altered function of neuronal L-type calcium channels in ageing and neuroinflammation: Implications in age-related synaptic dysfunction and cognitive decline. Ageing Res. Rev. 2018, 42, 86–99. [Google Scholar] [CrossRef] [PubMed]
- Diaz, A.; Trevino, S.; Vazquez-Roque, R.; Venegas, B.; Espinosa, B.; Flores, G.; Fernandez, G.J.; Montano, L.F.; Guevara, J. The aminoestrogen prolame increases recognition memory and hippocampal neuronal spine density in aged mice. Synapse 2017, 71, e21987. [Google Scholar] [CrossRef] [PubMed]
- Hatch, R.J.; Leinenga, G.; Gotz, J. Scanning Ultrasound (SUS) Causes No Changes to Neuronal Excitability and Prevents Age-Related Reductions in Hippocampal CA1 Dendritic Structure in Wild-Type Mice. PLoS ONE 2016, 11, e0164278. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Markham, J.A.; McKian, K.P.; Stroup, T.S.; Juraska, J.M. Sexually dimorphic aging of dendritic morphology in CA1 of hippocampus. Hippocampus 2005, 15, 97–103. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
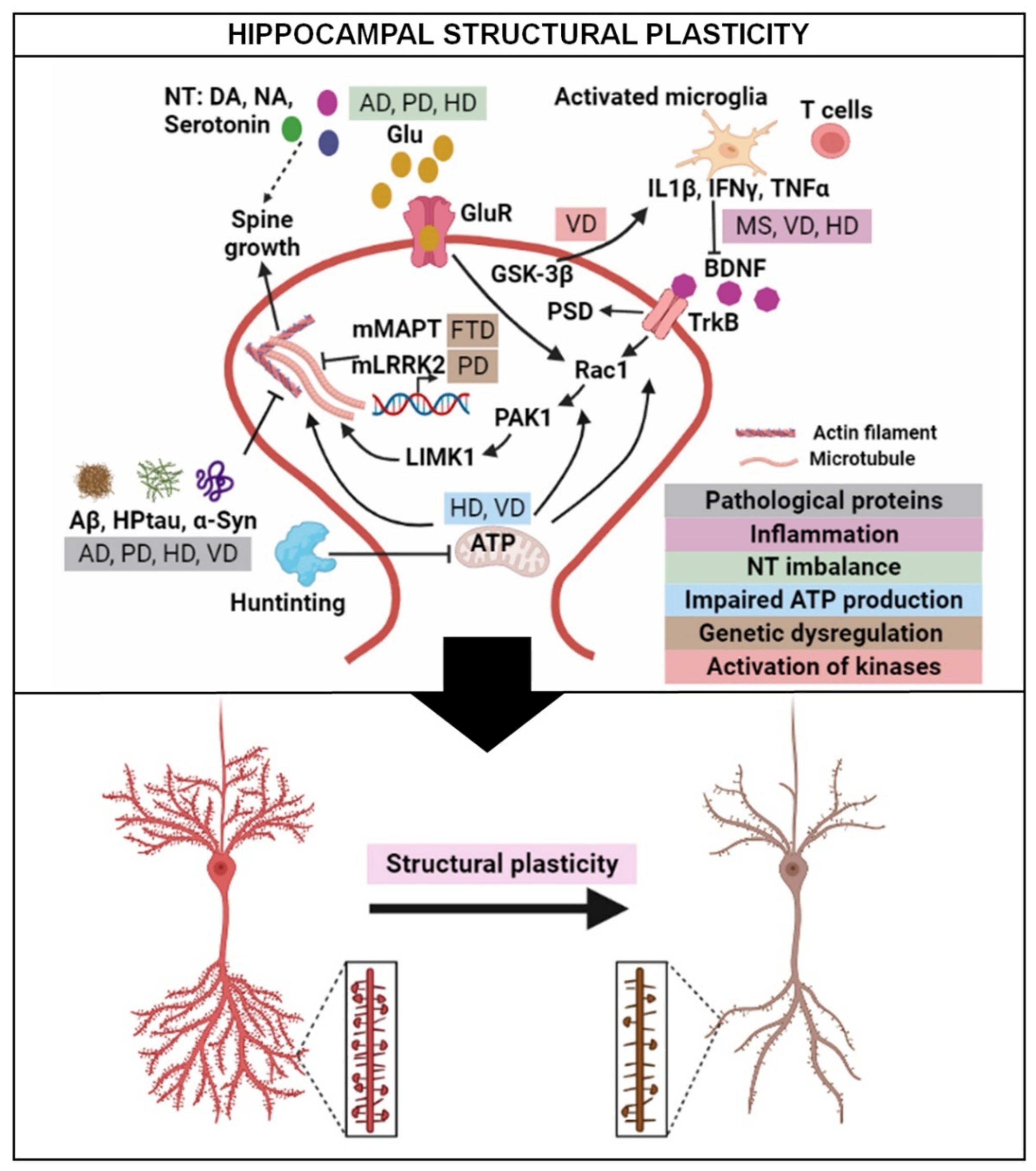{kind=link}
| Model | Dendritic Complexity | Spine Density/ Morphology | Mechanism | Ref. | |
|---|---|---|---|---|---|
| AD | Clinical patients | Decreased | Decreased | [279] | |
| APP/PS1-TG mice | Decreased | Decreased spine density and mushroom | [114] | ||
| APP/PS1-TG mice | Decreased | Fibrillary amyloid deposition | [277] | ||
| APP/PS1-TG mice | Decreased | Fibrillary amyloid deposition | [277] | ||
| APP/PS1-TG mice | Decreased | Decreased | Aβ plaques pathology | [280] | |
| APOE4-TG mice | Decreased | Decreased | [281] | ||
| miR-34c-transfected mouse primary cultured hippocampal neurons | Decreased | Decreased density and filopodia | [282] | ||
| N-tau-TG mice | Increased | Increased | [283] | ||
| TgCRND8 mice | No change | Decreased, thin, and stubby | Aβ plaques pathology Alteration of GluR | [112] | |
| 2576-TG mice | Decreased | Aβ plaques pathology Reactive gliosis | [116] | ||
| Aβ-infused rats | Decreased | [113] | |||
| Aβ-infused rats | Decreased spine density and length Decreased, thin, and filopodia | Glu circulation | [115] | ||
| Aβ-treated rat hippocampal slices | Decreased | NMDAR | [117] | ||
| APP/PS1-TG mice | Decreased spine length Increased neck size | [284] | |||
| APP-knock-in mice | Decreased mushroom | Downregulation of synaptic STIM2–nSOC–CaMKII pathway | [285] | ||
| PD | LRRK2-mutant mouse primary cultured hippocampal neurons | Decreased | LRRK2 regulation | [286] | |
| LRRK2-mutant mice | Decreased | LRRK2 regulation | [287] | ||
| LRRK2-mutant mice | Decreased | Decreased | LRRK2 regulation | [288] | |
| MPTP-lesioned mice | Decreased | [139] | |||
| MPTP-lesioned mice | No change | Decreased | [289] | ||
| PFF-treated primary cultured hippocampal neurons | Decreased mushroom | α-synucleinopathy | [290] | ||
| PFF-treated primary cultured hippocampal neurons | Decreased spine density and mushroom | α-Syn induced dysregulation of the actin-binding protein | [291] | ||
| HD | R6/1-TG mice | Decreased | [292] | ||
| Hip14-deficient mice | Decreased | [293] | |||
| R6/1-TG mice | Decreased | NF-κB signaling | [294] | ||
| Pyk2-deficient mice | Decreased | Pyk2 regulation | [172] | ||
| R6/1-TG mice | No change | [295] | |||
| MS | EAE mice | Decreased | [296] | ||
| EAE mice | Decreased | [297] | |||
| Cuprizone-diet fed mice | No change in total density Increased mushroom | [298] | |||
| VD | BCCAO rats | Decreased | Decreased spine density and mushroom | [65] | |
| BCCAO rats | Decreased | Decreased | APP | [299] | |
| BCCAO rats | Decreased | [300] | |||
| BCCAO rats | Decreased spine density and mushroom | [301] | |||
| BCCAO rats | Decreased | Suppression of AMPK pathway | [302] | ||
| ALS/FTD | TDP-43 overexpressing primary cultured hippocampal neurons | Decreased | RNA-binding function of TDP-43 | [67] | |
| ALS | SOD1G93A TG mice | No change | No change | [303] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Weerasinghe-Mudiyanselage, P.D.E.; Ang, M.J.; Kang, S.; Kim, J.-S.; Moon, C. Structural Plasticity of the Hippocampus in Neurodegenerative Diseases. Int. J. Mol. Sci. 2022, 23, 3349. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23063349
Weerasinghe-Mudiyanselage PDE, Ang MJ, Kang S, Kim J-S, Moon C. Structural Plasticity of the Hippocampus in Neurodegenerative Diseases. International Journal of Molecular Sciences. 2022; 23(6):3349. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23063349
Chicago/Turabian StyleWeerasinghe-Mudiyanselage, Poornima D. E., Mary Jasmin Ang, Sohi Kang, Joong-Sun Kim, and Changjong Moon. 2022. "Structural Plasticity of the Hippocampus in Neurodegenerative Diseases" International Journal of Molecular Sciences 23, no. 6: 3349. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23063349