
Dynamic Phosphorylation of miRNA Biogenesis Factor HYL1 by MPK3 Involving Nuclear–Cytoplasmic Shuttling and Protein Stability in Arabidopsis

 ,
,
Abstract
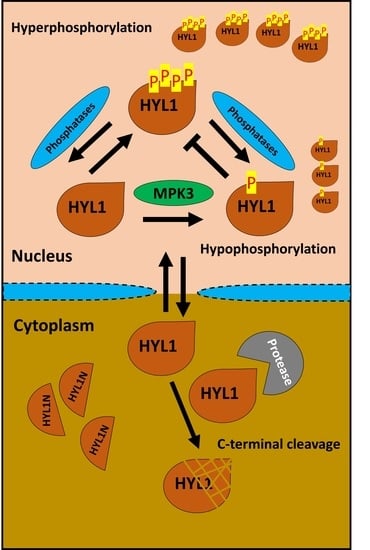
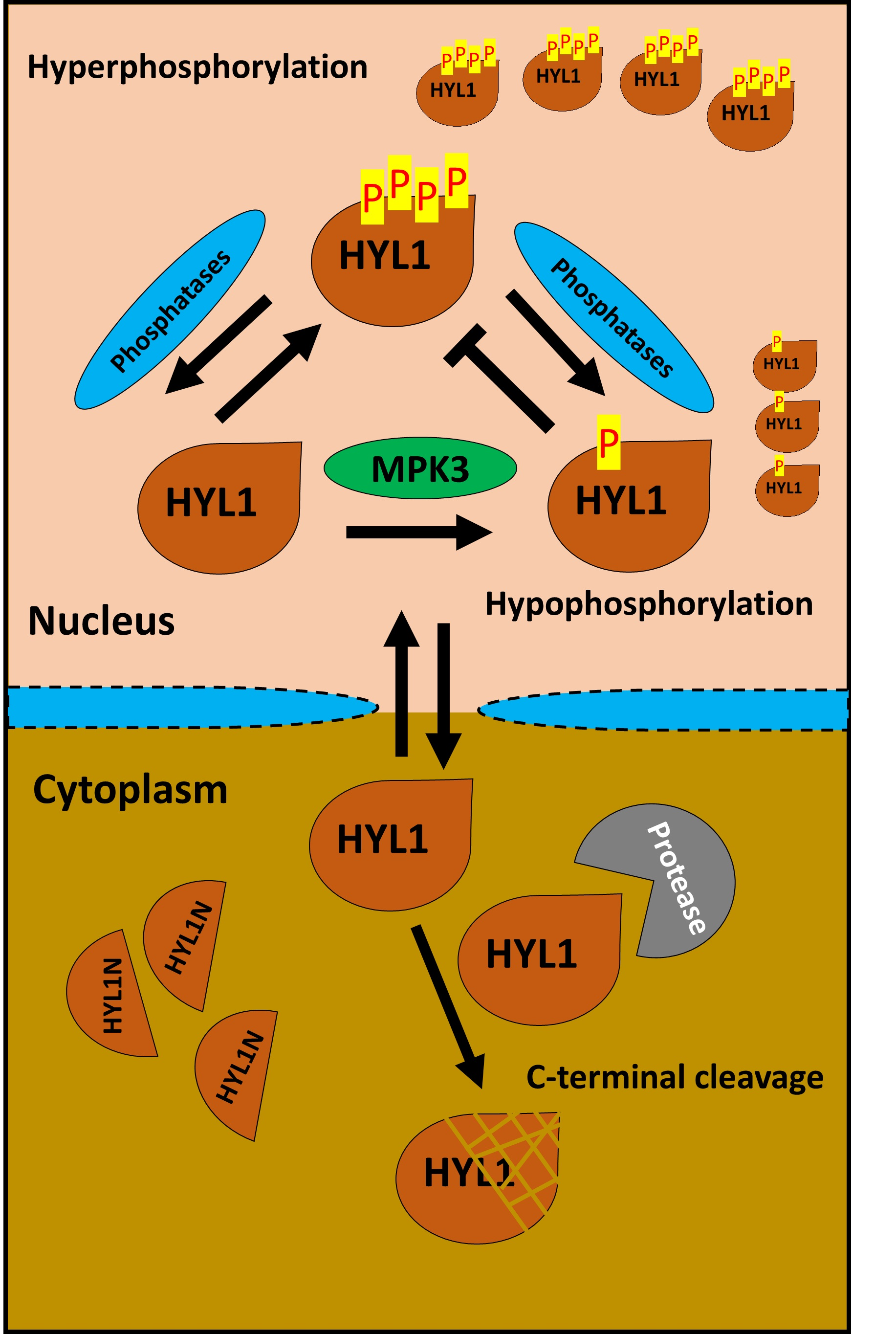
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
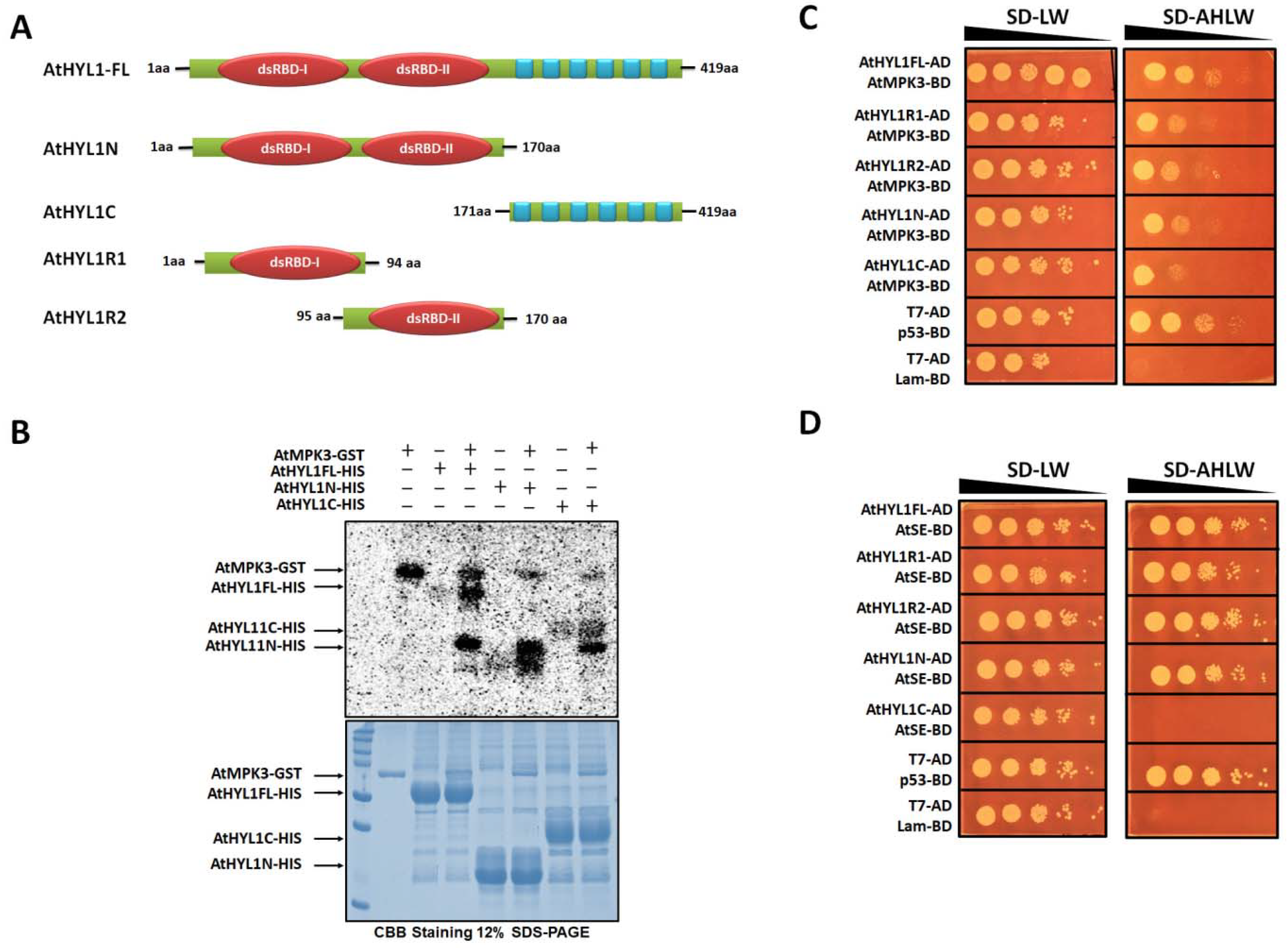
2.1. AtHYL1 Is Phosphorylated by AtMPK3
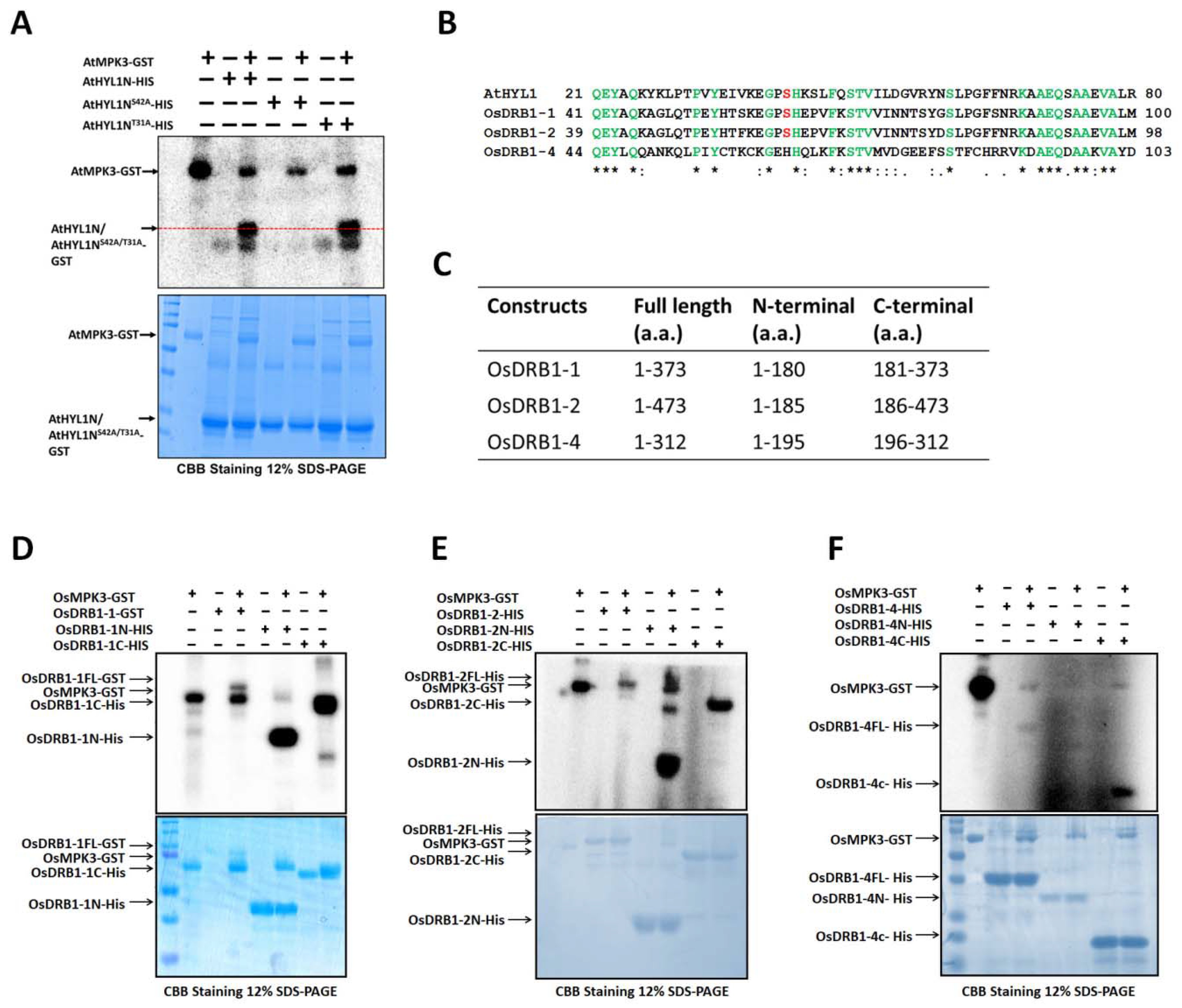
2.2. AtHYL1 Is Phosphorylated by AtMPK3 at Serine-42, a Non-Canonical MAPK Phosphorylation Site
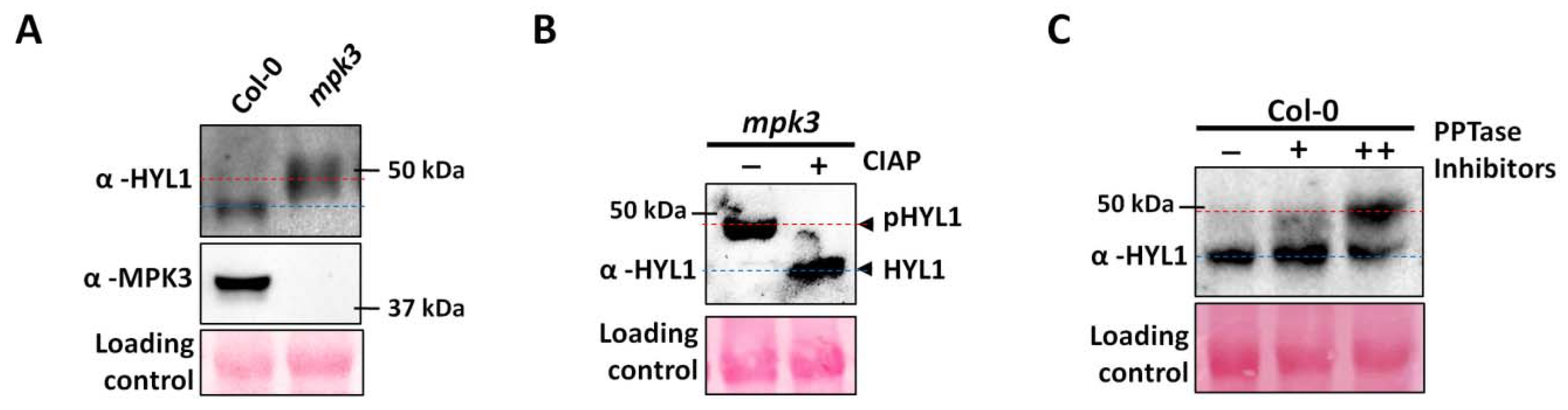
2.3. Mutation in AtMPK3 Leads to the Hyper-Phosphorylation of AtHYL1
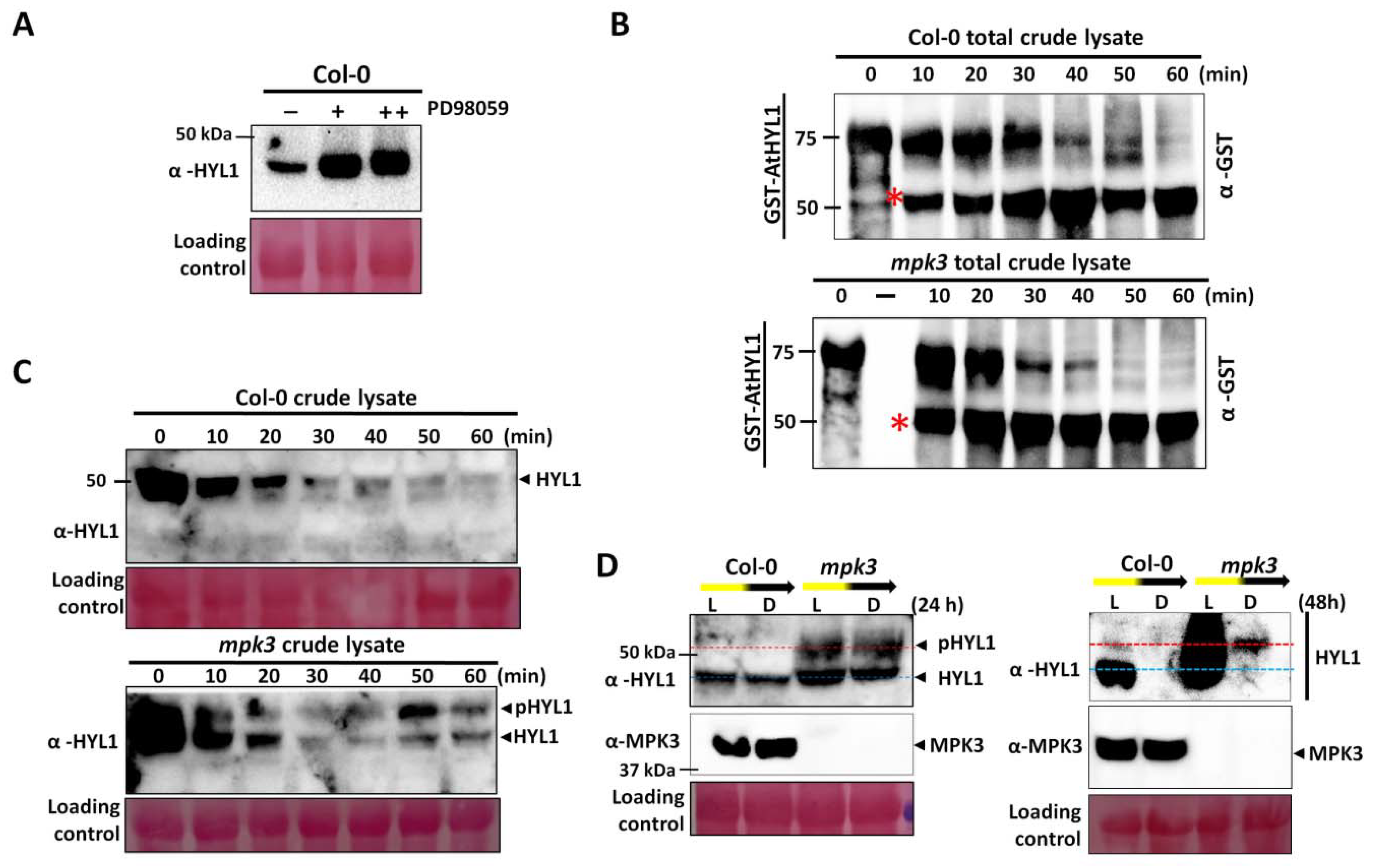
2.4. AtMPK3 Promotes AtHYL1 Protein Degradation
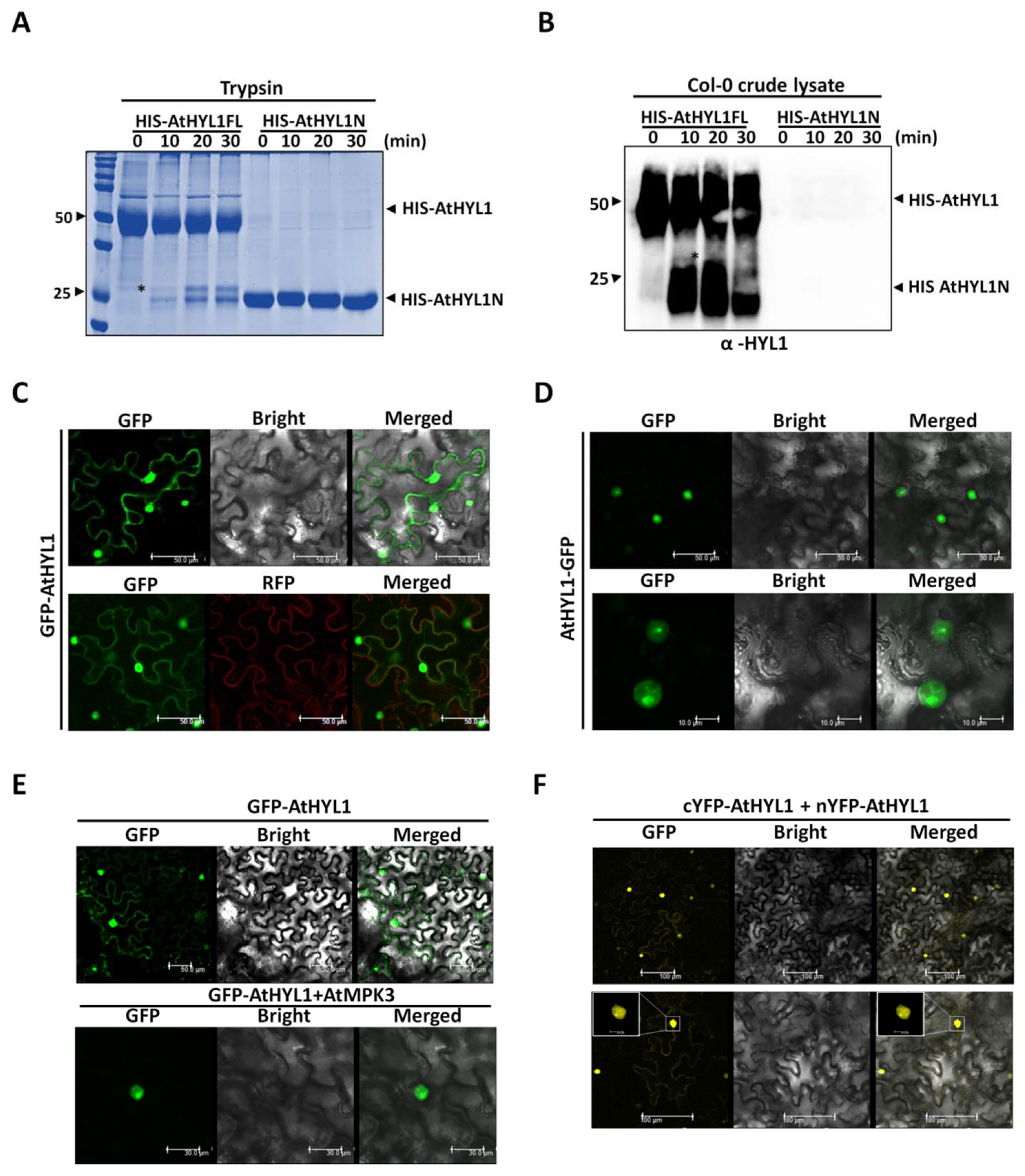
2.5. The Nuclear–Cytosolic Localization of AtHYL1 Is Strictly Regulated by Cytosolic Proteolysis
2.6. Co-Expression of AtMPK3 Regulates AtHYL1 Subcellular Localization
3. Discussion
3.1. AtMPK3 Phosphorylates AtHYL1 at the Serine-42 Position
3.2. AtMPK3 Negatively Regulates AtHYL1 Protein Accumulation
3.3. AtHYL1 Exists in a Hyper-Phosphorylated State in the mpk3 Mutant Background
3.4. Subcellular Localisation of AtHYL1 Is Tightly Governed by Cytoplasmic Proteases
3.5. AtHYL1 Is Localised to Both Nucleus and Cytoplasm
4. Materials and Methods
4.1. Plant Growth Conditions
4.2. Yeast Two-Hybrid Assay
4.3. In Vitro Phosphorylation Assay
4.4. In Vitro Protease Sensitivity Assay
4.5. In Vitro Cell-Free Degradation Assay
4.6. Protein Subcellular Localization and Bimolecular Complementation (BiFc) Analysis
4.7. In Vitro Phosphatase Treatment
4.8. Light-to-Dark Transition Assay
4.9. Evolutionary Relationship of AtHYL1 with DRB1 from Other Plants
4.10. Multiple Protein Alignment Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sinha, A.K.; Jaggi, M.; Raghuram, B.; Tuteja, N. Mitogen-activated protein kinase signaling in plants under abiotic stress. Plant Signal Behav. 2011, 6, 196–203. [Google Scholar] [CrossRef] [Green Version]
- Bigeard, J.; Hirt, H. Nuclear Signaling of Plant MAPKs. Front. Plant Sci. 2018, 9, 469. [Google Scholar] [CrossRef] [Green Version]
- Jalmi, S.K.; Sinha, A.K. ROS mediated MAPK signaling in abiotic and biotic stress- striking similarities and differences. Front. Plant Sci. 2015, 6, 769. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.L.; Mei, J.; Ren, G.D. Plant microRNAs: Biogenesis, Homeostasis, and Degradation. Front. Plant Sci. 2019, 10, 360. [Google Scholar] [CrossRef] [Green Version]
- Rogers, K.; Chen, X.M. Biogenesis, Turnover, and Mode of Action of Plant MicroRNAs. Plant Cell 2013, 25, 2383–2399. [Google Scholar] [CrossRef] [Green Version]
- Dexheimer, P.J.; Cochella, L. MicroRNAs: From Mechanism to Organism. Front. Cell Dev. Biol. 2020, 8, 409. [Google Scholar] [CrossRef]
- Kwon, S.C.; Nguyen, T.A.; Choi, Y.G.; Jo, M.H.; Hohng, S.; Kim, V.N.; Woo, J.S. Structure of Human DROSHA. Cell 2016, 164, 81–90. [Google Scholar] [CrossRef] [Green Version]
- Paroo, Z.; Ye, X.; Chen, S.; Liu, Q. Phosphorylation of the Human MicroRNA-Generating Complex Mediates MAPK/Erk Signaling. Cell 2009, 139, 112–122. [Google Scholar] [CrossRef] [Green Version]
- Ledda, B.; Ottaggio, L.; Izzotti, A.; Sukkar, S.G.; Miele, M. Small RNAs in eucaryotes: New clues for amplifying microRNA benefits. Cell Biosci. 2020, 10, 1. [Google Scholar] [CrossRef] [Green Version]
- Cho, S.K.; Ryu, M.Y.; Shah, P.; Poulsen, C.P.; Yang, S.W. Post-Translational Regulation of miRNA Pathway Components, AGO1 and HYL1, in Plants. Mol. Cells 2016, 39, 581–586. [Google Scholar] [CrossRef] [Green Version]
- Siomi, H.; Siomi, M.C. Posttranscriptional Regulation of MicroRNA Biogenesis in Animals. Mol. Cell 2010, 38, 323–332. [Google Scholar] [CrossRef]
- Raghuram, B.; Sheikh, A.H.; Rustagi, Y.; Sinha, A.K. MicroRNA biogenesis factor DRB1 is a phosphorylation target of mitogen activated protein kinase MPK3 in both rice and Arabidopsis. FEBS J. 2015, 282, 521–536. [Google Scholar] [CrossRef]
- Yan, J.; Wang, P.; Wang, B.; Hsu, C.-C.; Tang, K.; Zhang, H.; Hou, Y.-J.; Zhao, Y.; Wang, Q.; Zhao, C.; et al. The SnRK2 kinases modulate miRNA accumulation in Arabidopsis. PLoS Genet. 2017, 13, e1006753. [Google Scholar] [CrossRef] [Green Version]
- Manavella, P.A.; Hagmann, J.; Ott, F.; Laubinger, S.; Franz, M.; Macek, B.; Weigel, D. Fast-Forward Genetics Identifies Plant CPL Phosphatases as Regulators of miRNA Processing Factor HYL1. Cell 2012, 151, 859–870. [Google Scholar] [CrossRef] [Green Version]
- Su, C.; Li, Z.; Cheng, J.; Li, L.; Zhong, S.; Liu, L.; Zheng, Y.; Zheng, B. The Protein Phosphatase 4 and SMEK1 Complex Dephosphorylates HYL1 to Promotemi RNA Biogenesis by Antagonizing the MAPK Cascade in Arabidopsis. Dev. Cell 2017, 41, 527–539.e5. [Google Scholar] [CrossRef] [Green Version]
- Achkar, N.P.; Cho, S.K.; Poulsen, C.; Arce, A.L.; Re, D.A.; Giudicatti, A.J.; Karayekov, E.; Ryu, M.Y.; Choi, S.W.; Harholt, J.; et al. A Quick HYL1-Dependent Reactivation of MicroRNA Production Is Required for a Proper Developmental Response after Extended Periods of Light Deprivation. Dev. Cell 2018, 46, 236–247.e6. [Google Scholar] [CrossRef] [Green Version]
- Gao, S.; Wang, J.; Jiang, N.; Zhang, S.; Wang, Y.; Zhang, J.; Li, N.; Fang, Y.; Yang, L.; Chen, S.; et al. Hyponastic Leaves 1 protects pri-miRNAs from nuclear exosome attack. Proc. Natl. Acad. Sci. USA 2020, 117, 17429–17437. [Google Scholar] [CrossRef]
- Yang, X.; Dong, W.; Ren, W.; Zhao, Q.; Wu, F.; He, Y. Cytoplasmic HYL1 modulates miRNA-mediated translational repression. Plant Cell 2021, 33, 1980–1996. [Google Scholar] [CrossRef]
- Cho, S.K.; Ben Chaabane, S.; Shah, P.; Poulsen, C.P.; Yang, S.W. COP1 E3 ligase protects HYL1 to retain microRNA biogenesis. Nat. Commun. 2014, 5, 5867. [Google Scholar] [CrossRef] [Green Version]
- Jung, H.J.; Choi, S.W.; Boo, K.H.; Kim, J.E.; Oh, Y.K.; Han, M.K.; Ryu, M.Y.; Lee, C.W.; Moller, C.; Shah, P.; et al. HYL1-CLEAVAGE SUBTILASE 1 (HCS1) suppresses miRNA biogenesis in response to light-to-dark transition. Proc. Natl. Acad. Sci. USA 2022, 119, e2116757119. [Google Scholar] [CrossRef]
- Sacnun, J.M.; Crespo, R.; Palatnik, J.; Rasia, R.; Gonzalez-Schain, N. Dual function of HYPONASTIC LEAVES 1 during early skotomorphogenic growth in Arabidopsis. Plant J. 2020, 102, 977–991. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Guo, X.; Ge, C.; Ma, Z.; Jiang, M.; Li, T.; Koiwa, H.; Yang, S.W.; Zhang, X. KETCH1 imports HYL1 to nucleus for miRNA biogenesis in Arabidopsis. Proc. Natl. Acad. Sci. USA 2017, 114, 4011–4016. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fang, Y.; Spector, D.L. Identification of nuclear dicing bodies containing proteins for microRNA biogenesis in living Arabidopsis plants. Curr. Biol. 2007, 17, 818–823. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vacs, P.; Rasia, R.; Gonzalez-Schain, N. HYPONASTIC LEAVES 1 is required for proper establishment of auxin gradient in apical hooks. Plant Physiol. 2021, 187, 2356–2360. [Google Scholar] [CrossRef]
- Sun, Z.; Li, M.; Zhou, Y.; Guo, T.; Liu, Y.; Zhang, H.; Fang, Y. Coordinated regulation of Arabidopsis microRNA biogenesis and red light signaling through Dicer-like 1 and phytochrome-interacting factor 4. PLoS Genet. 2018, 14, e1007247. [Google Scholar] [CrossRef]
- Baranauske, S.; Mickute, M.; Plotnikova, A.; Finke, A.; Venclovas, C.; Klimasauskas, S.; Vilkaitis, G. Functional mapping of the plant small RNA methyltransferase: HEN1 physically interacts with HYL1 and DICER-LIKE 1 proteins. Nucleic Acids Res. 2015, 43, 2802–2812. [Google Scholar] [CrossRef] [Green Version]
- Wu, F.; Yu, L.; Cao, W.; Mao, Y.; Liu, Z.; He, Y. The N-terminal double-stranded RNA binding domains of Arabidopsis HYPONASTIC LEAVES1 are sufficient for pre-microRNA processing. Plant Cell 2007, 19, 914–925. [Google Scholar] [CrossRef] [Green Version]
- Varedi, S.M.; Ventura, A.C.; Merajver, S.D.; Lin, X.N. Multisite Phosphorylation Provides an Effective and Flexible Mechanism for Switch-Like Protein Degradation. PLoS ONE 2010, 5, e14029. [Google Scholar] [CrossRef] [Green Version]
- Yang, S.W.; Chen, H.-Y.; Yang, J.; Machida, S.; Chua, N.-H.; Yuan, Y.A. Structure of Arabidopsis HYPONASTIC LEAVES1 and Its Molecular Implications for miRNA Processing. Structure 2010, 18, 594–605. [Google Scholar] [CrossRef] [Green Version]
- Yamada, Y.; Sato, F. Tyrosine phosphorylation and protein degradation control the transcriptional activity of WRKY involved in benzylisoquinoline alkaloid biosynthesis. Sci. Rep. 2016, 6, 31988. [Google Scholar] [CrossRef] [Green Version]
- Sha, Z.; Peth, A.; Goldberg, A.L. Keeping proteasomes under control-a role for phosphorylation in the nucleus. Proc. Natl. Acad. Sci. USA 2011, 108, 18573–18574. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kurihara, Y.; Takashi, Y.; Watanabe, Y. The interaction between DCL1 and HYL1 is important for efficient and precise processing of pri-miRNA in plant microRNA biogenesis. RNA 2006, 12, 206–212. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dong, Z.; Han, M.-H.; Fedoroff, N. The RNA-binding proteins HYL1 and SE promote accurate in vitro processing of pri-miRNA by DCL1. Proc. Natl. Acad. Sci. USA 2008, 105, 9970–9975. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, X.; Ren, W.; Zhao, Q.; Zhang, P.; Wu, F.; He, Y. Homodimerization of HYL1 ensures the correct selection of cleavage sites in primary miRNA. Nucleic Acids Res. 2014, 42, 12224–12236. [Google Scholar] [CrossRef] [Green Version]
- Yamamoto, S.; Subedi, G.P.; Hanashima, S.; Satoh, T.; Otaka, M.; Wakui, H.; Sawada, K.I.; Yokota, S.I.; Yamaguchi, Y.; Kubota, H.; et al. ATPase Activity and ATP-dependent Conformational Change in the Co-chaperone HSP70/HSP90-organizing Protein (HOP)*. J. Biol. Chem. 2014, 289, 9880–9886. [Google Scholar] [CrossRef] [Green Version]
- Singh, P.; Sinha, A.K. A Positive Feedback Loop Governed by SUB1A1 Interaction with MITOGEN-ACTIVATED PROTEIN KINASE3 Imparts Submergence Tolerance in Rice. Plant Cell 2016, 28, 1127–1143. [Google Scholar] [CrossRef] [Green Version]
- Reddy, M.S.; Guhan, N.; Muniyappa, K. Characterization of single-stranded DNA-binding proteins from mycobacteria-The carboxyl-terminal domain of SSB is essential for stable association with its cognate RecA protein. J. Biol. Chem. 2001, 276, 45959–45968. [Google Scholar] [CrossRef] [Green Version]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bhagat, P.K.; Verma, D.; Singh, K.; Badmi, R.; Sharma, D.; Sinha, A.K. Dynamic Phosphorylation of miRNA Biogenesis Factor HYL1 by MPK3 Involving Nuclear–Cytoplasmic Shuttling and Protein Stability in Arabidopsis. Int. J. Mol. Sci. 2022, 23, 3787. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23073787
Bhagat PK, Verma D, Singh K, Badmi R, Sharma D, Sinha AK. Dynamic Phosphorylation of miRNA Biogenesis Factor HYL1 by MPK3 Involving Nuclear–Cytoplasmic Shuttling and Protein Stability in Arabidopsis. International Journal of Molecular Sciences. 2022; 23(7):3787. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23073787
Chicago/Turabian StyleBhagat, Prakash Kumar, Deepanjali Verma, Kirti Singh, Raghuram Badmi, Deepika Sharma, and Alok Krishna Sinha. 2022. "Dynamic Phosphorylation of miRNA Biogenesis Factor HYL1 by MPK3 Involving Nuclear–Cytoplasmic Shuttling and Protein Stability in Arabidopsis" International Journal of Molecular Sciences 23, no. 7: 3787. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23073787