
Crosstalk between β-Catenin and CCL2 Drives Migration of Monocytes towards Glioblastoma Cells

 ,
,  , , and
, , and 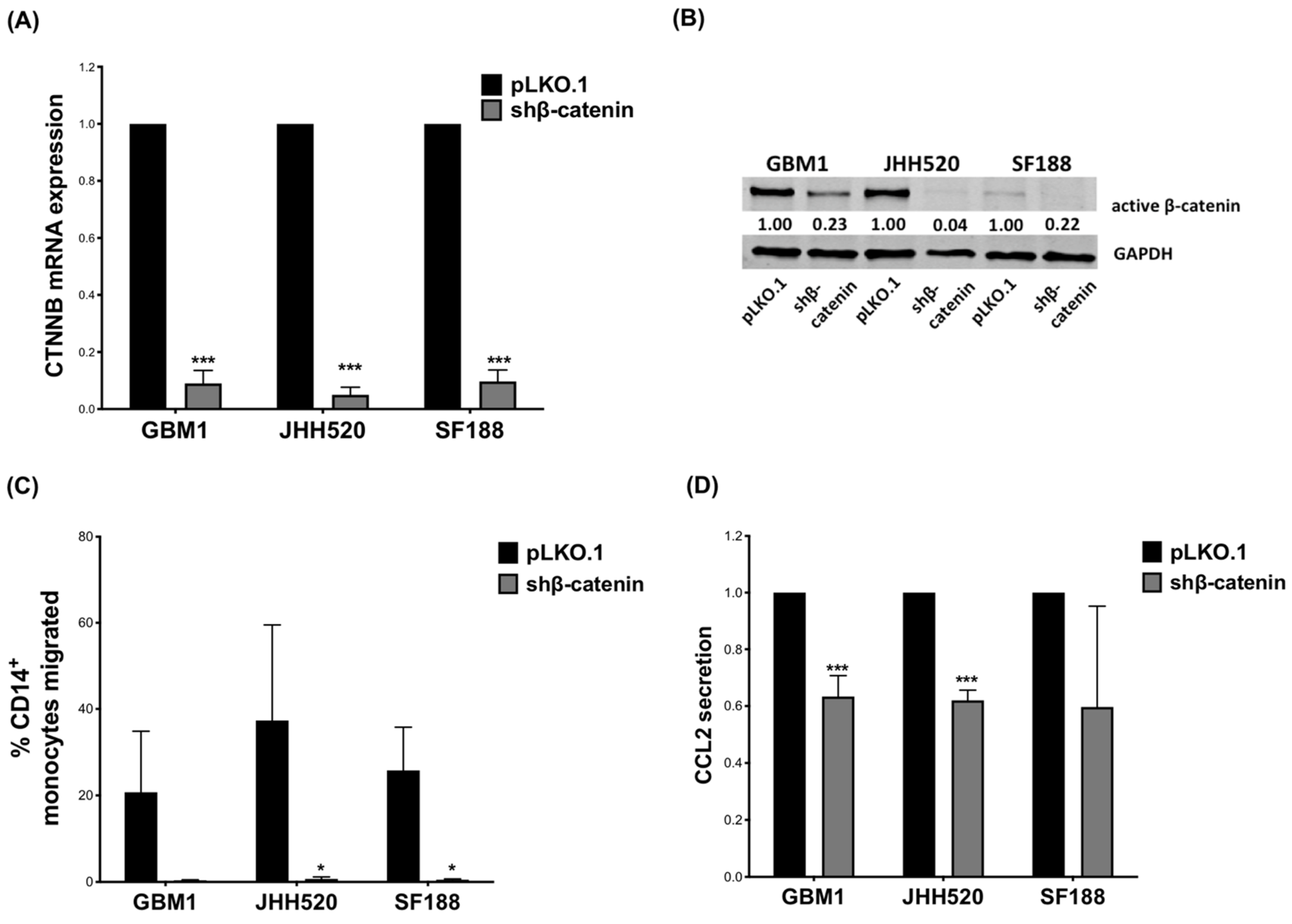{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Material and Methods
2.1. Cell Culture and MSAB Treatment
2.2. Generation of Lentiviral Particles
2.3. Cell Viability and Cell Death Assays
2.4. Clonogenicity Assay
2.5. Quantitative Real Time PCR (RT qPCR)
2.6. Whole Genome Transcriptome Analysis
2.7. Western Blotting
2.8. ELISA
2.9. PBMC Migration Assay
2.10. Luciferase Reporter Assay
2.11. Statistical Analyses
3. Results
3.1. β-Catenin Expression by GBM Cells Impacts Monocyte Migration and CCL2 Secretion
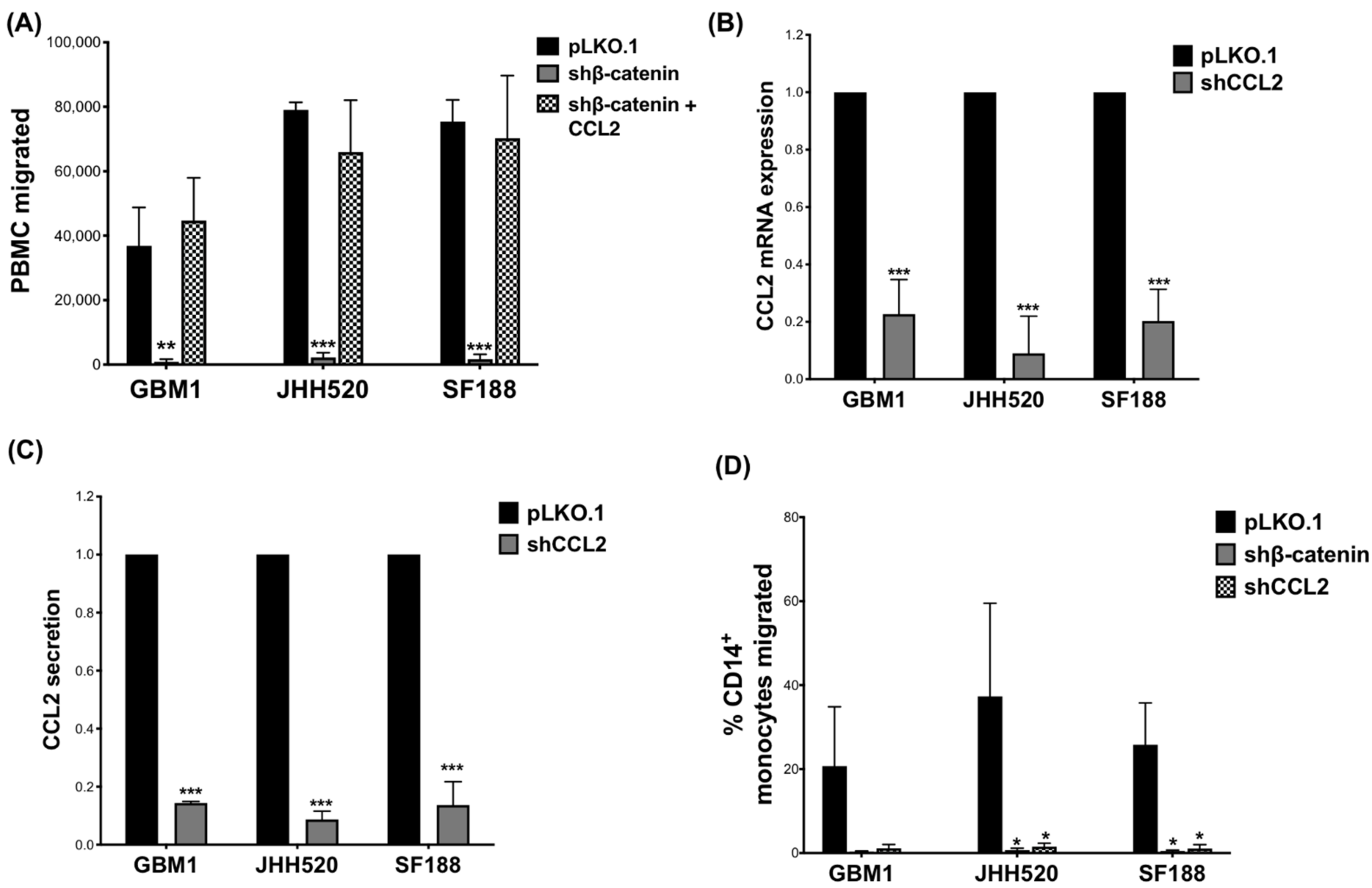
3.2. Recombinant CCL2 Restored PBMC Migration in shβ-Catenin TCM and CCL2 Knockdown Reduced Monocyte Migration
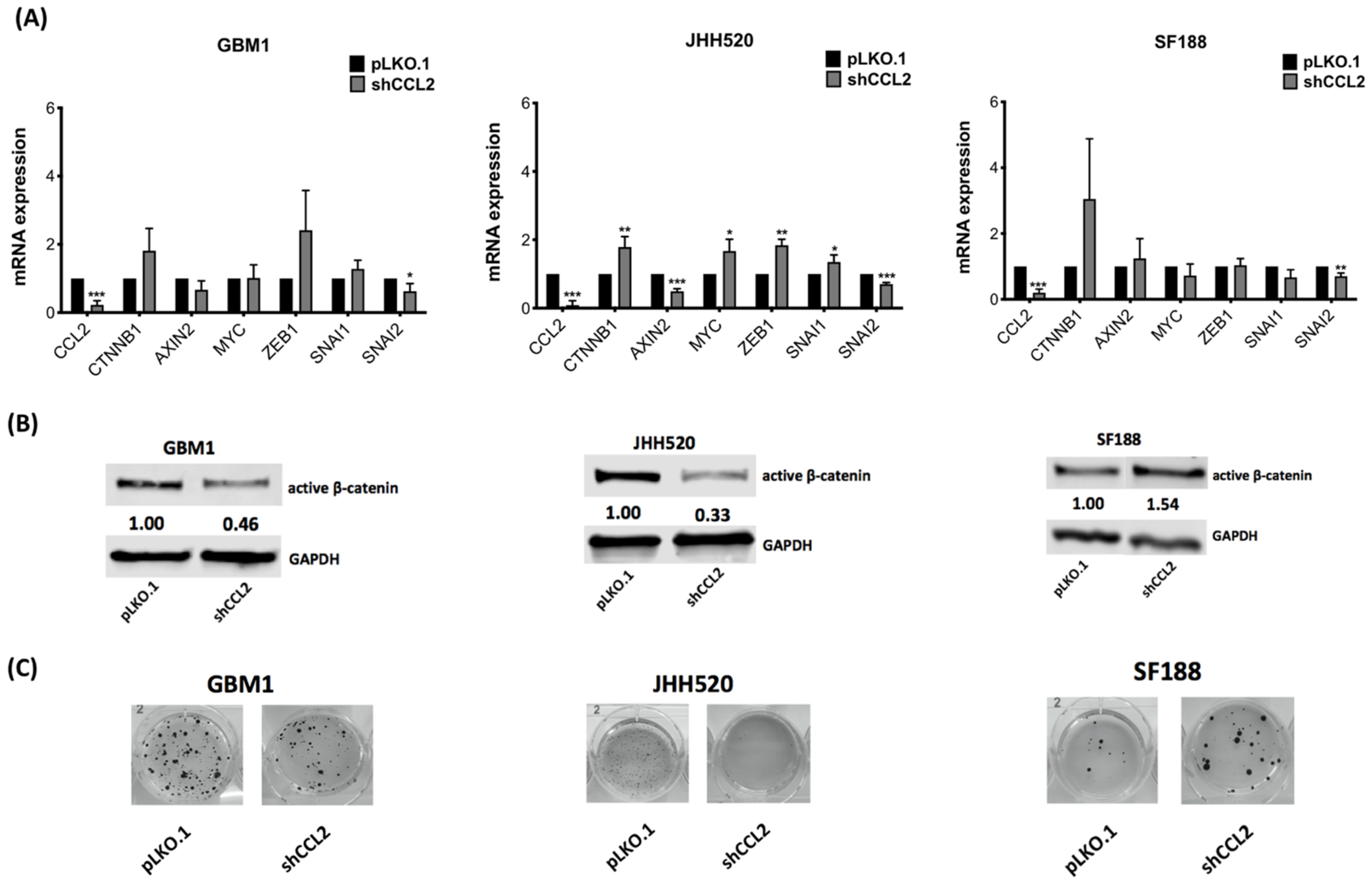
3.3. CCL2 Modulates Components of the Wnt/β-Catenin Pathway and Alters Clonogenicity of GBM Cells
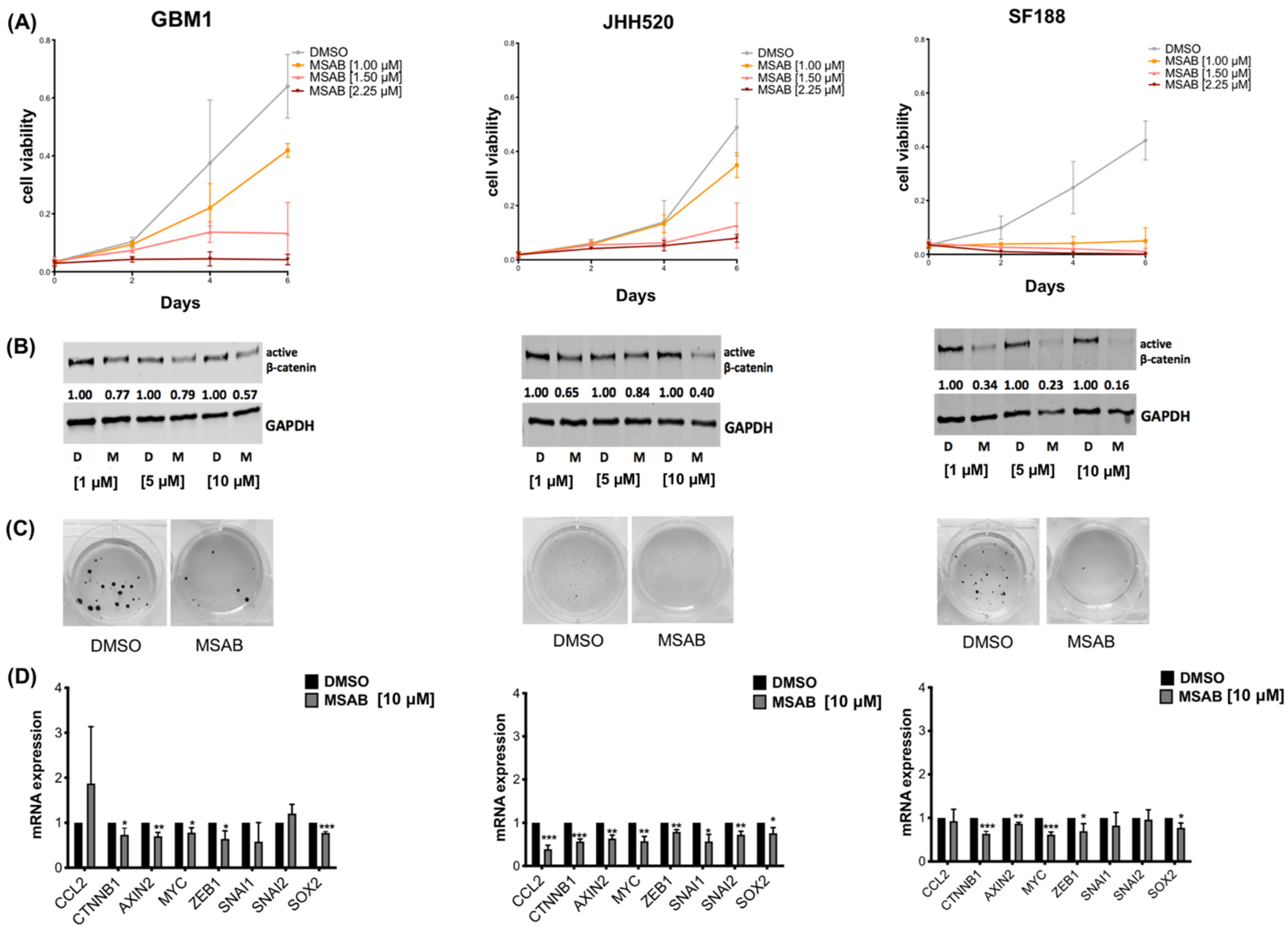
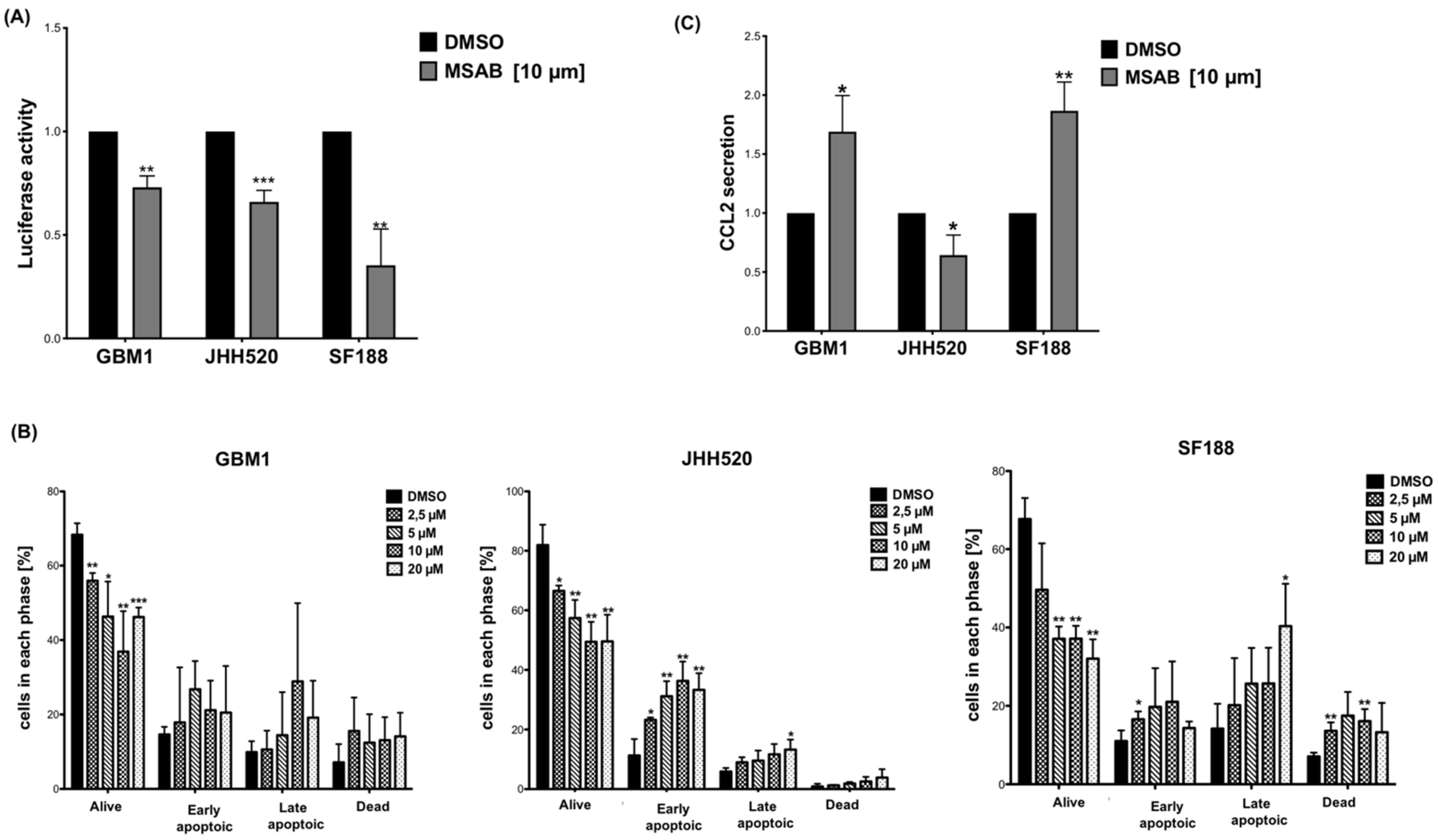
3.4. The β-Catenin Inhibitor MSAB Reduces Viability, Active β-Catenin Levels, Clonogenicity and Expression of β-Catenin Associated Genes in GBM Cells
3.5. MSAB Decreases Wnt/β-Catenin-Activity and Modulates CCL2 Secretion
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Stupp, R.; Mason, W.P.; Van Den Bent, M.J.; Weller, M.; Fisher, B.; Taphoorn, M.J.; Belanger, K.; Brandes, A.A.; Marosi, C.; Bogdahn, U.; et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N. Engl. J. Med. 2005, 352, 987–996. [Google Scholar] [CrossRef] [PubMed]
- Stupp, R.; Hegi, M.E.; Mason, W.P.; Van Den Bent, M.J.; Taphoorn, M.J.; Janzer, R.C.; Ludwin, S.K.; Allgeier, A.; Fisher, B.; Belanger, K.; et al. Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised phase III study: 5-year analysis of the EORTC-NCIC trial. Lancet Oncol. 2009, 10, 459–466. [Google Scholar] [CrossRef]
- Jackson, M.; Hassiotou, F.; Nowak, A. Glioblastoma stem-like cells: At the root of tumor recurrence and a therapeutic target. Carcinogenesis 2015, 36, 177–185. [Google Scholar] [CrossRef] [PubMed]
- Bayin, N.S.; Modrek, A.S.; Placantonakis, D.G. Glioblastoma stem cells: Molecular characteristics and therapeutic implications. World J. Stem Cells 2014, 6, 230–238. [Google Scholar] [CrossRef] [Green Version]
- Bao, S.; Wu, Q.; McLendon, R.E.; Hao, Y.; Shi, Q.; Hjelmeland, A.B.; Dewhirst, M.W.; Bigner, D.D.; Rich, J.N. Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature 2006, 444, 756–760. [Google Scholar] [CrossRef]
- Chen, J.; Li, Y.; Yu, T.S.; McKay, R.M.; Burns, D.K.; Kernie, S.G.; Parada, L.F. A restricted cell population propagates glioblastoma growth after chemotherapy. Nature 2012, 488, 522–526. [Google Scholar] [CrossRef] [Green Version]
- Lathia, J.D.; Mack, S.C.; Mulkearns-Hubert, E.E.; Valentim, C.L.; Rich, J.N. Cancer stem cells in glioblastoma. Genes Dev. 2015, 29, 1203–1217. [Google Scholar] [CrossRef] [Green Version]
- Bleau, A.M.; Hambardzumyan, D.; Ozawa, T.; Fomchenko, E.I.; Huse, J.T.; Brennan, C.W.; Holland, E.C. PTEN/PI3K/Akt pathway regulates the side population phenotype and ABCG2 activity in glioma tumor stem-like cells. Cell Stem Cell 2009, 4, 226–235. [Google Scholar] [CrossRef] [Green Version]
- Liu, G.; Yuan, X.; Zeng, Z.; Tunici, P.; Ng, H.; Abdulkadir, I.R.; Lu, L.; Irvin, D.; Black, K.L.; Yu, J.S. Analysis of gene expression and chemoresistance of CD133+ cancer stem cells in glioblastoma. Mol. Cancer 2006, 5, 67. [Google Scholar] [CrossRef] [Green Version]
- Mattei, V.; Santilli, F.; Martellucci, S.; Delle Monache, S.; Fabrizi, J.; Colapietro, A.; Angelucci, A.; Festuccia, C. The importance of tumor stem cells in glioblastoma resistance to therapy. Int. J. Mol. Sci. 2021, 22, 3863. [Google Scholar] [CrossRef]
- Galli, R.; Binda, E.; Orfanelli, U.; Cipelletti, B.; Gritti, A.; De Vitis, S.; Fiocco, R.; Foroni, C.; Dimeco, F.; Vescovi, A. Isolation and characterization of tumorigenic, stem-like neural precursors from human glioblastoma. Cancer Res. 2004, 64, 7011–7021. [Google Scholar] [CrossRef] [Green Version]
- Hemmati, H.D.; Nakano, I.; Lazareff, J.A.; Masterman-Smith, M.; Geschwind, D.H.; Bronner-Fraser, M.; Kornblum, H.I. Cancerous stem cells can arise from pediatric brain tumors. Proc. Natl. Acad. Sci. USA 2003, 100, 15178–15183. [Google Scholar] [CrossRef] [Green Version]
- Ignatova, T.N.; Kukekov, V.G.; Laywell, E.D.; Suslov, O.N.; Vrionis, F.D.; Steindler, D.A. Human cortical glial tumors contain neural stem-like cells expressing astroglial and neuronal markers in vitro. Glia 2002, 39, 193–206. [Google Scholar] [CrossRef]
- Lan, X.; Jörg, D.J.; Cavalli, F.M.; Richards, L.M.; Nguyen, L.V.; Vanner, R.J.; Guilhamon, P.; Lee, L.; Kushida, M.M.; Pellacani, D.; et al. Fate mapping of human glioblastoma reveals an invariant stem cell hierarchy. Nature 2017, 549, 227–232. [Google Scholar] [CrossRef]
- Schonberg, D.L.; Lubelski, D.; Miller, T.E.; Rich, J.N. Brain tumor stem cells: Molecular characteristics and their impact on therapy. Mol. Asp. Med. 2014, 39, 82–101. [Google Scholar] [CrossRef] [Green Version]
- Singh, S.K.; Hawkins, C.; Clarke, I.D.; Squire, J.A.; Bayani, J.; Hide, T.; Henkelman, R.M.; Cusimano, M.D.; Dirks, P.B. Identification of human brain tumour initiating cells. Nature 2004, 432, 396–401. [Google Scholar] [CrossRef]
- Koch, K.; Hartmann, R.; Suwala, A.K.; Rios, D.H.; Kamp, M.A.; Sabel, M.; Steiger, H.J.; Willbold, D.; Sharma, A.; Kahlert, U.D.; et al. Overexpression of cystine/glutamate antiporter xCT correlates with nutrient flexibility and ZEB1 expression in highly clonogenic glioblastoma stem-like cells (GSCs). Cancers 2021, 13, 6001. [Google Scholar] [CrossRef]
- Li, Y.; Sharma, A.; Maciaczyk, J.; Schmidt-Wolf, I.G. Recent development in NKT-based immunotherapy of glioblastoma: From bench to bedside. Int. J. Mol. Sci. 2022, 23, 1311. [Google Scholar] [CrossRef]
- Adamo, A.; Fiore, D.; De Martino, F.; Roscigno, G.; Affinito, A.; Donnarumma, E.; Puoti, I.; Vitiani, L.R.; Pallini, R.; Quintavalle, C.; et al. RYK promotes the stemness of glioblastoma cells via the WNT/β-catenin pathway. Oncotarget 2017, 8, 13476–13487. [Google Scholar] [CrossRef] [Green Version]
- Chen, L.; Huang, K.; Han, L.; Shi, Z.; Zhang, K.; Pu, P.; Kang, C. β-catenin/Tcf-4 complex transcriptionally regulates AKT1 in glioma. Int. J. Oncol. 2011, 39, 883–890. [Google Scholar]
- Kahlert, U.D.; Maciaczyk, D.; Doostkam, S.; Orr, B.A.; Simons, B.; Bogiel, T.; Reithmeier, T.; Prinz, M.; Schubert, J.; Niedermann, G.; et al. Activation of canonical WNT/β-catenin signaling enhances in vitro motility of glioblastoma cells by activation of ZEB1 and other activators of epithelial-to-mesenchymal transition. Cancer Lett. 2012, 325, 42–53. [Google Scholar] [CrossRef]
- Kahlert, U.D.; Suwala, A.K.; Koch, K.; Natsumeda, M.; Orr, B.A.; Hayashi, M.; Maciaczyk, J.; Eberhart, C.G. Pharmacologic Wnt inhibition reduces proliferation, survival, and clonogenicity of glioblastoma cells. J. Neuropathol. Exp. Neurol. 2015, 74, 889–900. [Google Scholar] [CrossRef]
- Kaur, N.; Chettiar, S.; Rathod, S.; Rath, P.; Muzumdar, D.; Shaikh, M.L.; Shiras, A. Wnt3a mediated activation of Wnt/β-catenin signaling promotes tumor progression in glioblastoma. Mol. Cell Neurosci. 2013, 54, 44–57. [Google Scholar] [CrossRef]
- Li, Z.; Wang, H.; Eyler, C.E.; Hjelmeland, A.B.; Rich, J.N. Turning cancer stem cells inside out: An exploration of glioma stem cell signaling pathways. J. Biol. Chem. 2009, 284, 16705–16709. [Google Scholar] [CrossRef] [Green Version]
- Liu, C.; Tu, Y.; Sun, X.; Jiang, J.; Jin, X.; Bo, X.; Li, Z.; Bian, A.; Wang, X.; Liu, D.; et al. Wnt/beta-Catenin pathway in human glioma: Expression pattern and clinical/prognostic correlations. Clin. Exp. Med. 2011, 11, 105–112. [Google Scholar] [CrossRef]
- Rheinbay, E.; Suvà, M.L.; Gillespie, S.M.; Wakimoto, H.; Patel, A.P.; Shahid, M.; Oksuz, O.; Rabkin, S.D.; Martuza, R.L.; Rivera, M.N.; et al. An aberrant transcription factor network essential for Wnt signaling and stem cell maintenance in glioblastoma. Cell Rep. 2013, 3, 1567–1579. [Google Scholar] [CrossRef] [Green Version]
- Rossi, M.; Magnoni, L.; Miracco, C.; Mori, E.; Tosi, P.; Pirtoli, L.; Tini, P.; Oliveri, G.; Cosci, E.; Bakker, A. β-catenin and Gli1 are prognostic markers in glioblastoma. Cancer Biol. Ther. 2011, 11, 753–761. [Google Scholar] [CrossRef] [Green Version]
- Schüle, R.; Dictus, C.; Campos, B.; Wan, F.; Felsberg, J.; Ahmadi, R.; Centner, F.S.; Grabe, N.; Reifenberger, G.; Bermejo, J.L.; et al. Potential canonical wnt pathway activation in high-grade astrocytomas. Sci. World J. 2012, 2012, 697313. [Google Scholar] [CrossRef] [Green Version]
- Galluzzi, L.; Spranger, S.; Fuchs, E.; López-Soto, A. WNT Signaling in Cancer Immunosurveillance. Trends Cell Biol. 2019, 29, 44–65. [Google Scholar] [CrossRef]
- Ganesh, S.; Shui, X.; Craig, K.P.; Park, J.; Wang, W.; Brown, B.D.; Abrams, M.T. RNAi-mediated β-catenin inhibition promotes T cell infiltration and antitumor activity in combination with immune checkpoint blockade. Mol. Ther. 2018, 26, 2567–2579. [Google Scholar] [CrossRef]
- Luke, J.J.; Bao, R.; Sweis, R.F.; Spranger, S.; Gajewski, T.F. WNT/β-catenin Pathway Activation Correlates with Immune Exclusion across Human Cancers. Clin. Cancer Res. 2019, 25, 3074–3083. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spranger, S.; Bao, R.; Gajewski, T.F. Melanoma-intrinsic β-catenin signalling prevents anti-tumour immunity. Nature 2015, 523, 231–235. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Bi, Y.; Wei, Y.; Liu, J.; Kuerban, K.; Ye, L. Blocking Wnt/β-catenin signal amplifies anti-PD-1 therapeutic efficacy by inhibiting tumor growth, migration, and promoting immune infiltration in glioblastomas. Mol. Cancer Ther. 2021, 20, 1305–1315. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Feng, X.; Herting, C.J.; Garcia, V.A.; Nie, K.; Pong, W.W.; Rasmussen, R.; Dwivedi, B.; Seby, S.; Wolf, S.A.; et al. Cellular and Molecular Identity of Tumor-Associated Macrophages in Glioblastoma. Cancer Res. 2017, 77, 2266–2278. [Google Scholar] [CrossRef] [Green Version]
- Gabrilovich, D.I.; Ostrand-Rosenberg, S.; Bronte, V. Coordinated regulation of myeloid cells by tumours. Nat. Rev. Immunol. 2012, 12, 253–268. [Google Scholar] [CrossRef] [Green Version]
- Mignogna, C.; Signorelli, F.; Vismara, M.F.; Zeppa, P.; Camastra, C.; Barni, T.; Donato, G.; Di Vito, A. A reappraisal of macrophage polarization in glioblastoma: Histopathological and immunohistochemical findings and review of the literature. Pathol. Res. Pract. 2016, 212, 491–499. [Google Scholar] [CrossRef]
- Morantz, R.A.; Wood, G.W.; Foster, M.; Clark, M.; Gollahon, K. Macrophages in experimental and human brain tumors. Part 2: Studies of the macrophage content of human brain tumors. J. Neurosurg. 1979, 50, 305–311. [Google Scholar] [CrossRef]
- Roggendorf, W.; Strupp, S.; Paulus, W. Distribution and characterization of microglia/macrophages in human brain tumors. Acta Neuropathol. 1996, 92, 288–293. [Google Scholar] [CrossRef]
- Rossi, M.L.; Hughes, J.T.; Esiri, M.M.; Coakham, H.B.; Brownell, D.B. Immunohistological study of mononuclear cell infiltrate in malignant gliomas. Acta Neuropathol. 1987, 74, 269–277. [Google Scholar] [CrossRef]
- Biswas, S.K.; Mantovani, A. Macrophage plasticity and interaction with lymphocyte subsets: Cancer as a paradigm. Nat. Immunol. 2010, 11, 889–896. [Google Scholar] [CrossRef]
- Bloch, O.; Crane, C.A.; Kaur, R.; Safaee, M.; Rutkowski, M.J.; Parsa, A.T. Gliomas promote immunosuppression through induction of B7-H1 expression in tumor-associated macrophages. Clin. Cancer Res. 2013, 19, 3165–3175. [Google Scholar] [CrossRef] [Green Version]
- Gabrusiewicz, K.; Ellert-Miklaszewska, A.; Lipko, M.; Sielska, M.; Frankowska, M.; Kaminska, B. Characteristics of the alternative phenotype of microglia/macrophages and its modulation in experimental gliomas. PLoS ONE 2011, 6, e23902. [Google Scholar] [CrossRef] [Green Version]
- Hussain, S.F.; Yang, D.; Suki, D.; Aldape, K.; Grimm, E.; Heimberger, A.B. The role of human glioma-infiltrating microglia/macrophages in mediating antitumor immune responses. Neuro Oncol. 2006, 8, 261–279. [Google Scholar] [CrossRef] [Green Version]
- Mangani, D.; Weller, M.; Roth, P. The network of immunosuppressive pathways in glioblastoma. Biochem. Pharmacol. 2017, 130, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Mantovani, A.; Marchesi, F.; Malesci, A.; Laghi, L.; Allavena, P. Tumour-associated macrophages as treatment targets in oncology. Nat. Rev. Clin. Oncol. 2017, 14, 399–416. [Google Scholar] [CrossRef]
- Wei, J.; Marisetty, A.; Schrand, B.; Gabrusiewicz, K.; Hashimoto, Y.; Ott, M.; Grami, Z.; Kong, L.Y.; Ling, X.; Caruso, H.; et al. Osteopontin mediates glioblastoma-associated macrophage infiltration and is a potential therapeutic target. J. Clin. Investig. 2019, 129, 137–149. [Google Scholar] [CrossRef]
- Chang, A.L.; Miska, J.; Wainwright, D.A.; Dey, M.; Rivetta, C.V.; Yu, D.; Kanojia, D.; Pituch, K.C.; Qiao, J.; Pytel, P.; et al. CCL2 Produced by the Glioma Microenvironment Is Essential for the Recruitment of Regulatory T Cells and Myeloid-Derived Suppressor Cells. Cancer Res. 2016, 76, 5671–5682. [Google Scholar] [CrossRef] [Green Version]
- Bonecchi, R.; Bianchi, G.; Bordignon, P.P.; D’Ambrosio, D.; Lang, R.; Borsatti, A.; Sozzani, S.; Allavena, P.; Gray, P.A.; Mantovani, A.; et al. Differential expression of chemokine receptors and chemotactic responsiveness of type 1 T helper cells (Th1s) and Th2s. J. Exp. Med. 1998, 187, 129–134. [Google Scholar] [CrossRef]
- Gu, L.; Tseng, S.; Horner, R.M.; Tam, C.; Loda, M.; Rollins, B.J. Control of TH2 polarization by the chemokine monocyte chemoattractant protein-1. Nature 2000, 404, 407–411. [Google Scholar] [CrossRef]
- An, Z.; Knobbe-Thomsen, C.B.; Wan, X.; Fan, Q.W.; Reifenberger, G.; Weiss, W.A. EGFR Cooperates with EGFRvIII to Recruit Macrophages in Glioblastoma. Cancer Res. 2018, 78, 6785–6794. [Google Scholar] [CrossRef] [Green Version]
- Karpus, W.J.; Lukacs, N.W.; Kennedy, K.J.; Smith, W.S.; Hurst, S.D.; Barrett, T.A. Differential CC chemokine-induced enhancement of T helper cell cytokine production. J. Immunol. 1997, 158, 4129–4136. [Google Scholar] [PubMed]
- Lesokhin, A.M.; Hohl, T.M.; Kitano, S.; Cortez, C.; Hirschhorn-Cymerman, D.; Avogadri, F.; Rizzuto, G.A.; Lazarus, J.J.; Pamer, E.G.; Houghton, A.N.; et al. Monocytic CCR2(+) myeloid-derived suppressor cells promote immune escape by limiting activated CD8 T-cell infiltration into the tumor microenvironment. Cancer Res. 2012, 72, 876–886. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matsukawa, A.; Lukacs, N.W.; Standiford, T.J.; Chensue, S.W.; Kunkel, S.L. Adenoviral-mediated overexpression of monocyte chemoattractant protein-1 differentially alters the development of Th1 and Th2 type responses in vivo. J. Immunol. 2000, 164, 1699–1704. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goede, V.; Brogelli, L.; Ziche, M.; Augustin, H.G. Induction of inflammatory angiogenesis by monocyte chemoattractant protein-1. Int. J. Cancer 1999, 82, 765–770. [Google Scholar] [CrossRef]
- Low-Marchelli, J.M.; Ardi, V.C.; Vizcarra, E.A.; van Rooijen, N.; Quigley, J.P.; Yang, J. Twist1 induces CCL2 and recruits macrophages to promote angiogenesis. Cancer Res. 2013, 73, 662–671. [Google Scholar] [CrossRef] [Green Version]
- Mehrad, B.; Keane, M.P.; Strieter, R.M. Chemokines as mediators of angiogenesis. Thromb. Haemost. 2007, 97, 755–762. [Google Scholar] [CrossRef] [Green Version]
- Salcedo, R.; Ponce, M.L.; Young, H.A.; Wasserman, K.; Ward, J.M.; Kleinman, H.K.; Oppenheim, J.J.; Murphy, W.J. Human endothelial cells express CCR2 and respond to MCP-1: Direct role of MCP-1 in angiogenesis and tumor progression. Blood 2000, 96, 34–40. [Google Scholar] [CrossRef]
- Stamatovic, S.M.; Keep, R.F.; Mostarica-Stojkovic, M.; Andjelkovic, A.V. CCL2 regulates angiogenesis via activation of Ets-1 transcription factor. J. Immunol. 2006, 177, 2651–2661. [Google Scholar] [CrossRef] [Green Version]
- Sandhu, S.K.; Papadopoulos, K.; Fong, P.C.; Patnaik, A.; Messiou, C.; Olmos, D.; Wang, G.; Tromp, B.J.; Puchalski, T.A.; Balkwill, F.; et al. A first-in-human, first-in-class, phase I study of carlumab (CNTO 888), a human monoclonal antibody against CC-chemokine ligand 2 in patients with solid tumors. Cancer Chemother. Pharmacol. 2013, 71, 1041–1050. [Google Scholar] [CrossRef]
- Kuratsu, J.; Yoshizato, K.; Yoshimura, T.; Leonard, E.J.; Takeshima, H.; Ushio, Y. Quantitative study of monocyte chemoattractant protein-1 (MCP-1) in cerebrospinal fluid and cyst fluid from patients with malignant glioma. J. Natl. Cancer Inst. 1993, 85, 1836–1839. [Google Scholar] [CrossRef]
- Moogooei, M.; Shamaei, M.; Khorramdelazad, H.; Fattahpour, S.; Seyedmehdi, S.M.; Hassanshahi, G.; Kalantari Khandani, B. The Intricate Expression of CC Chemokines in Glial Tumors: Evidence for Involvement of CCL2 and CCL5 but Not CCL11. Acta Med. Iran. 2015, 53, 770–777. [Google Scholar]
- Yi, L.; Xiao, H.; Xu, M.; Ye, X.; Hu, J.; Li, F.; Li, M.; Luo, C.; Yu, S.; Bian, X.; et al. Glioma-initiating cells: A predominant role in microglia/macrophages tropism to glioma. J. Neuroimmunol. 2011, 232, 75–82. [Google Scholar] [CrossRef]
- Ye, X.Z.; Xu, S.L.; Xin, Y.H.; Yu, S.C.; Ping, Y.F.; Chen, L.; Xiao, H.L.; Wang, B.; Yi, L.; Wang, Q.L.; et al. Tumor-associated microglia/macrophages enhance the invasion of glioma stem-like cells via TGF-β1 signaling pathway. J. Immunol. 2012, 189, 444–453. [Google Scholar] [CrossRef] [Green Version]
- Zhang, F.; Li, P.; Liu, S.; Yang, M.; Zeng, S.; Deng, J.; Chen, D.; Yi, Y.; Liu, H. β-Catenin-CCL2 feedback loop mediates crosstalk between cancer cells and macrophages that regulates breast cancer stem cells. Oncogene 2021, 40, 5854–5865. [Google Scholar] [CrossRef]
- Hwang, S.Y.; Deng, X.; Byun, S.; Lee, C.; Lee, S.J.; Suh, H.; Zhang, J.; Kang, Q.; Zhang, T.; Westover, K.D.; et al. Direct Targeting of β-Catenin by a Small Molecule Stimulates Proteasomal Degradation and Suppresses Oncogenic Wnt/β-Catenin Signaling. Cell Rep. 2016, 16, 28–36. [Google Scholar] [CrossRef] [Green Version]
- Moffat, J.; Grueneberg, D.A.; Yang, X.; Kim, S.Y.; Kloepfer, A.M.; Hinkle, G.; Piqani, B.; Eisenhaure, T.M.; Luo, B.; Grenier, J.K.; et al. A lentiviral RNAi library for human and mouse genes applied to an arrayed viral high-content screen. Cell 2006, 124, 1283–1298. [Google Scholar] [CrossRef] [Green Version]
- Niyazi, M.; Niyazi, I.; Belka, C. Counting colonies of clonogenic assays by using densitometric software. Radiat. Oncol. 2007, 2, 4. [Google Scholar] [CrossRef] [Green Version]
- Gschwandtner, M.; Derler, R.; Midwood, K.S. More Than Just Attractive: How CCL2 Influences Myeloid Cell Behavior Beyond Chemotaxis. Front. Immunol. 2019, 10, 2759. [Google Scholar] [CrossRef] [Green Version]
- Mestdagt, M.; Polette, M.; Buttice, G.; Noël, A.; Ueda, A.; Foidart, J.M.; Gilles, C. Transactivation of MCP-1/CCL2 by beta-catenin/TCF-4 in human breast cancer cells. Int. J. Cancer 2006, 118, 35–42. [Google Scholar] [CrossRef] [Green Version]
- Zhu, X.; Fujita, M.; Snyder, L.A.; Okada, H. Systemic delivery of neutralizing antibody targeting CCL2 for glioma therapy. J. Neuro-Oncol. 2011, 104, 83–92. [Google Scholar] [CrossRef] [Green Version]
- Leung, S.Y.; Wong, M.P.; Chung, L.P.; Chan, A.S.; Yuen, S.T. Monocyte chemoattractant protein-1 expression and macrophage infiltration in gliomas. Acta Neuropathol. 1997, 93, 518–527. [Google Scholar] [CrossRef]
- Kielian, T.; van Rooijen, N.; Hickey, W.F. MCP-1 expression in CNS-1 astrocytoma cells: Implications for macrophage infiltration into tumors in vivo. J. Neurooncol. 2002, 56, 1–12. [Google Scholar] [CrossRef]
- Ou, B.; Cheng, X.; Xu, Z.; Chen, C.; Shen, X.; Zhao, J.; Lu, A. A positive feedback loop of β-catenin/CCR2 axis promotes regorafenib resistance in colorectal cancer. Cell Death Dis. 2019, 10, 643. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, S.; Yang, L.; Dong, H.; Guo, H. Human telomerase reverse transcriptase recruits the β-catenin/TCF-4 complex to transactivate chemokine (C-C motif) ligand 2 expression in colorectal cancer. Biomed. Pharmacother. 2019, 112, 108700. [Google Scholar] [CrossRef] [PubMed]
- Lévy, L.; Neuveut, C.; Renard, C.A.; Charneau, P.; Branchereau, S.; Gauthier, F.; Van Nhieu, J.T.; Cherqui, D.; Petit-Bertron, A.F.; Mathieu, D.; et al. Transcriptional activation of interleukin-8 by beta-catenin-Tcf4. J. Biol. Chem. 2002, 277, 42386–42393. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guan, R.; Zhang, X.; Guo, M. Glioblastoma stem cells and Wnt signaling pathway: Molecular mechanisms and therapeutic targets. Chin. Neurosurg. J. 2020, 6, 25. [Google Scholar] [CrossRef]
- Lee, Y.; Lee, J.K.; Ahn, S.H.; Lee, J.; Nam, D.H. WNT signaling in glioblastoma and therapeutic opportunities. Lab. Investig. J. Tech. Methods Pathol. 2016, 96, 137–150. [Google Scholar] [CrossRef] [Green Version]
- Wu, A.; Wei, J.; Kong, L.Y.; Wang, Y.; Priebe, W.; Qiao, W.; Sawaya, R.; Heimberger, A.B. Glioma cancer stem cells induce immunosuppressive macrophages/microglia. Neuro Oncol. 2010, 12, 1113–1125. [Google Scholar] [CrossRef]
- Zhou, W.; Ke, S.Q.; Huang, Z.; Flavahan, W.; Fang, X.; Paul, J.; Wu, L.; Sloan, A.E.; McLendon, R.E.; Li, X.; et al. Periostin secreted by glioblastoma stem cells recruits M2 tumour-associated macrophages and promotes malignant growth. Nat. Cell Biol. 2015, 17, 170–182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ding, P.; Wang, W.; Wang, J.; Yang, Z.; Xue, L. Expression of tumor-associated macrophage in progression of human glioma. Cell Biochem. Biophys. 2014, 70, 1625–1631. [Google Scholar] [CrossRef]
- Sørensen, M.D.; Dahlrot, R.H.; Boldt, H.B.; Hansen, S.; Kristensen, B.W. Tumour-associated microglia/macrophages predict poor prognosis in high-grade gliomas and correlate with an aggressive tumour subtype. Neuropathol. Appl. Neurobiol. 2018, 44, 185–206. [Google Scholar] [CrossRef] [Green Version]
- Komohara, Y.; Horlad, H.; Ohnishi, K.; Fujiwara, Y.; Bai, B.; Nakagawa, T.; Suzu, S.; Nakamura, H.; Kuratsu, J.; Takeya, M. Importance of direct macrophage-tumor cell interaction on progression of human glioma. Cancer Sci. 2012, 103, 2165–2172. [Google Scholar] [CrossRef]
- Lu-Emerson, C.; Snuderl, M.; Kirkpatrick, N.D.; Goveia, J.; Davidson, C.; Huang, Y.; Riedemann, L.; Taylor, J.; Ivy, P.; Duda, D.G.; et al. Increase in tumor-associated macrophages after antiangiogenic therapy is associated with poor survival among patients with recurrent glioblastoma. Neuro Oncol. 2013, 15, 1079–1087. [Google Scholar] [CrossRef]
- Lim, S.Y.; Yuzhalin, A.E.; Gordon-Weeks, A.N.; Muschel, R.J. Targeting the CCL2-CCR2 signaling axis in cancer metastasis. Oncotarget 2016, 7, 28697–28710. [Google Scholar] [CrossRef] [Green Version]
- Charo, I.F.; Myers, S.J.; Herman, A.; Franci, C.; Connolly, A.J.; Coughlin, S.R. Molecular cloning and functional expression of two monocyte chemoattractant protein 1 receptors reveals alternative splicing of the carboxyl-terminal tails. Proc. Natl. Acad. Sci. USA 1994, 91, 2752–2756. [Google Scholar] [CrossRef] [Green Version]
- Sharma, A.; Reutter, H.; Ellinger, J. DNA Methylation and Bladder Cancer: Where Genotype does not Predict Phenotype. Curr. Genom. 2020, 21, 34–36. [Google Scholar] [CrossRef]
- Brennan, C.W.; Verhaak, R.G.; McKenna, A.; Campos, B.; Noushmehr, H.; Salama, S.R.; Zheng, S.; Chakravarty, D.; Sanborn, J.Z.; Berman, S.H.; et al. The somatic genomic landscape of glioblastoma. Cell 2013, 155, 462–477. [Google Scholar] [CrossRef]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Aretz, P.; Maciaczyk, D.; Yusuf, S.; Sorg, R.V.; Hänggi, D.; Liu, H.; Liu, H.; Dakal, T.C.; Sharma, A.; Bethanabatla, R.; et al. Crosstalk between β-Catenin and CCL2 Drives Migration of Monocytes towards Glioblastoma Cells. Int. J. Mol. Sci. 2022, 23, 4562. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23094562
Aretz P, Maciaczyk D, Yusuf S, Sorg RV, Hänggi D, Liu H, Liu H, Dakal TC, Sharma A, Bethanabatla R, et al. Crosstalk between β-Catenin and CCL2 Drives Migration of Monocytes towards Glioblastoma Cells. International Journal of Molecular Sciences. 2022; 23(9):4562. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23094562
Chicago/Turabian StyleAretz, Philippe, Donata Maciaczyk, Suad Yusuf, Rüdiger V. Sorg, Daniel Hänggi, Hongjia Liu, Hongde Liu, Tikam Chand Dakal, Amit Sharma, Ramakrishna Bethanabatla, and et al. 2022. "Crosstalk between β-Catenin and CCL2 Drives Migration of Monocytes towards Glioblastoma Cells" International Journal of Molecular Sciences 23, no. 9: 4562. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23094562