
Spiro-Oxindole Skeleton Compounds Are Efficient Inhibitors for Indoleamine 2,3-Dioxygenase 1: An Attractive Target for Tumor Immunotherapy

 ,
,
Abstract
:1. Introduction
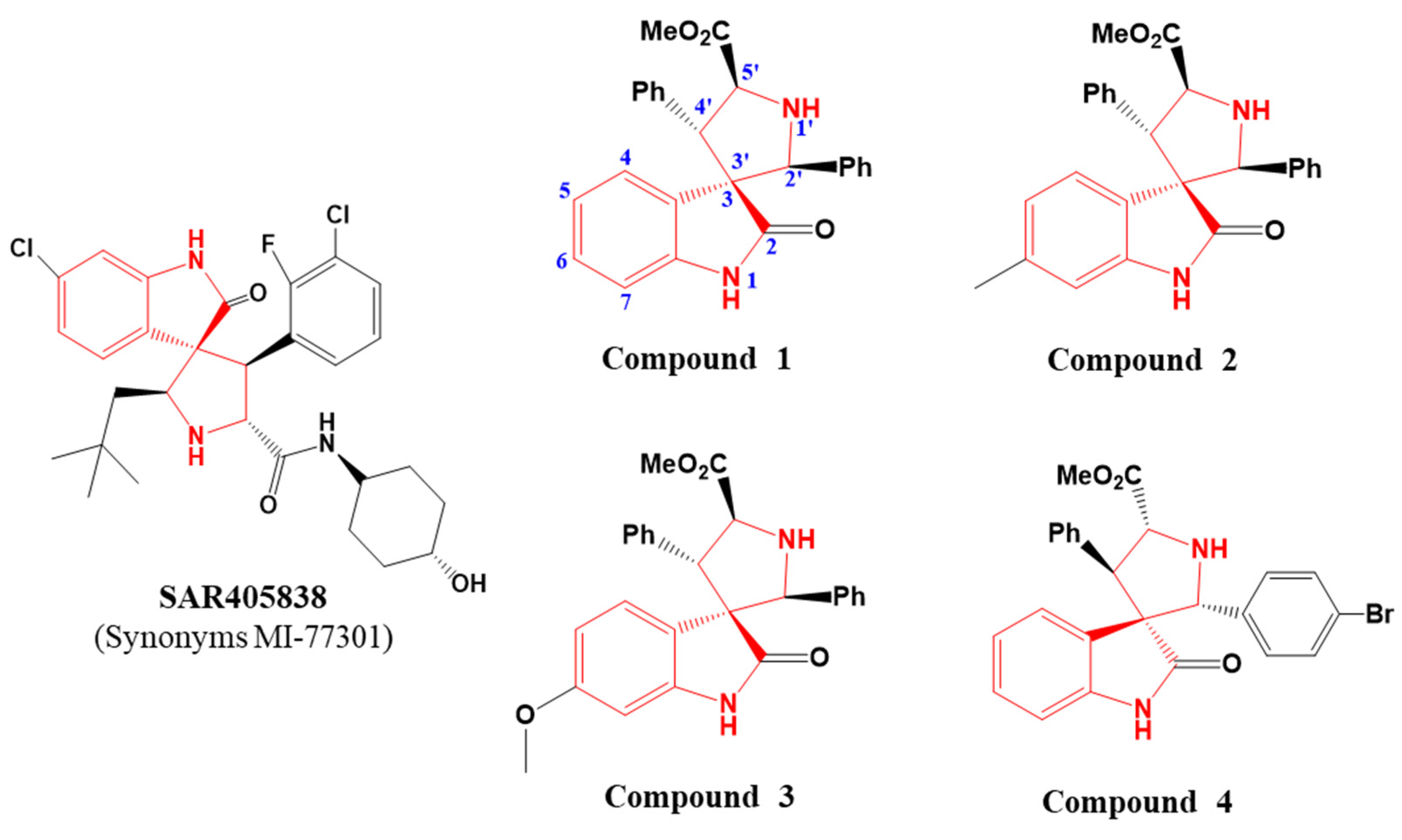
2. Results and Discussion
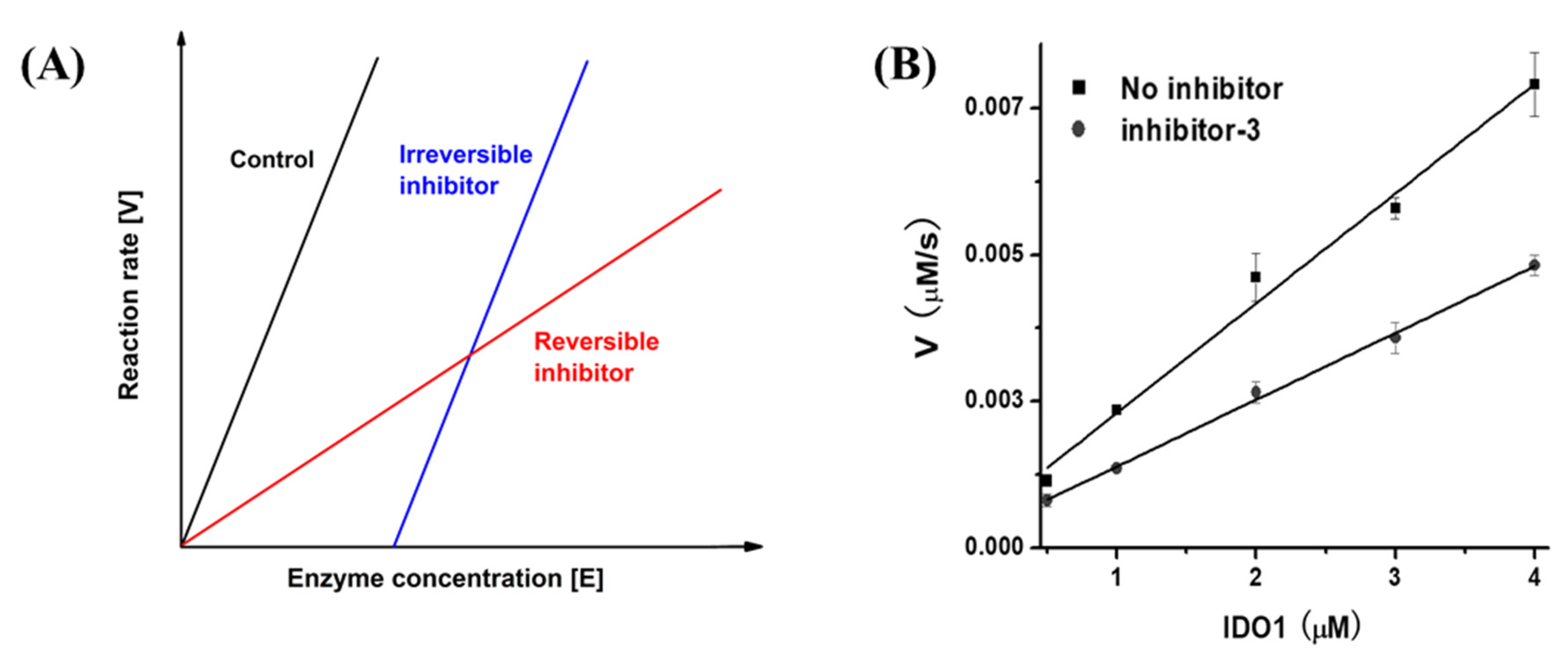
2.1. UV–Vis Spectra of IDO1 and Reversibility Analysis of Inhibitors
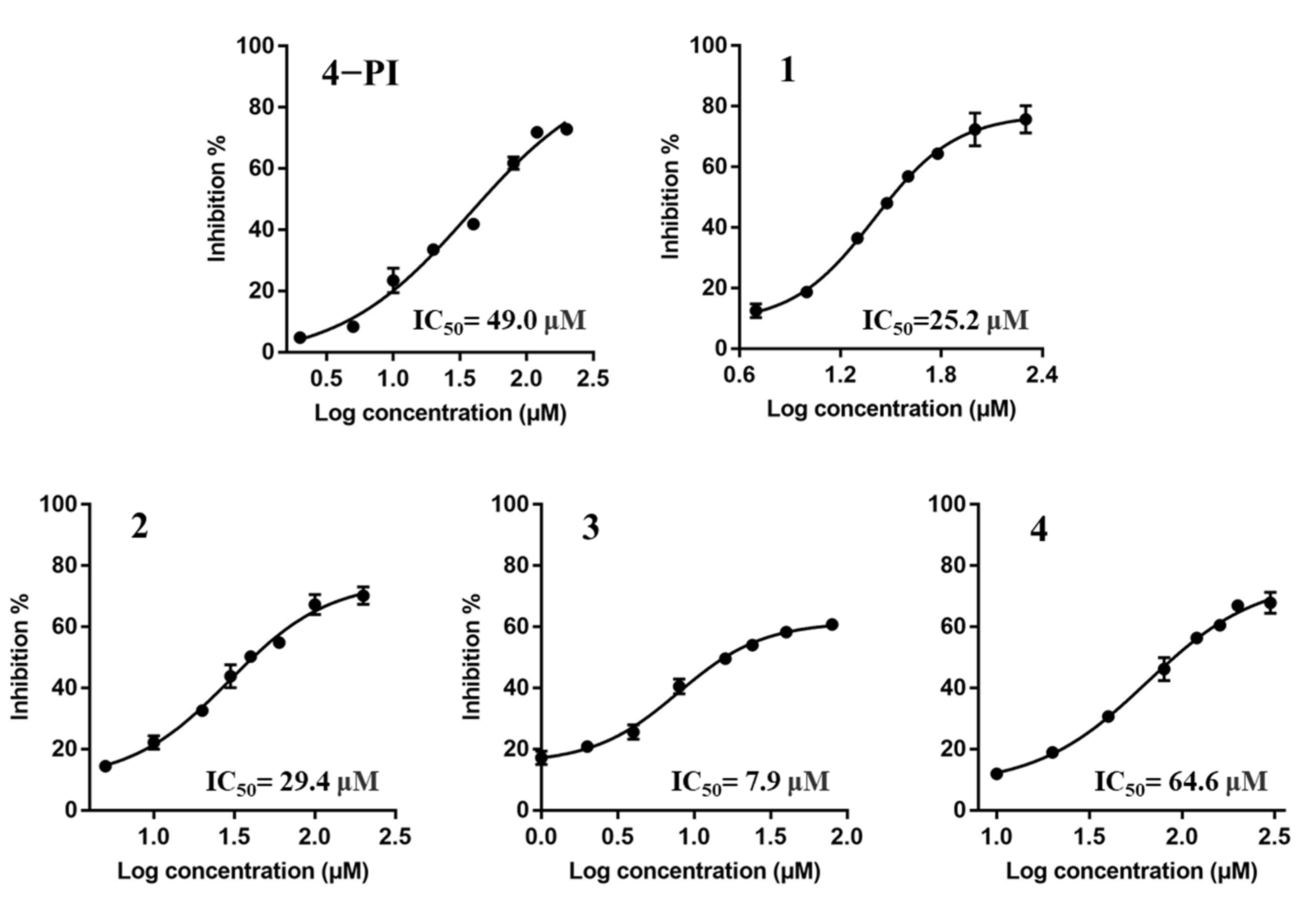
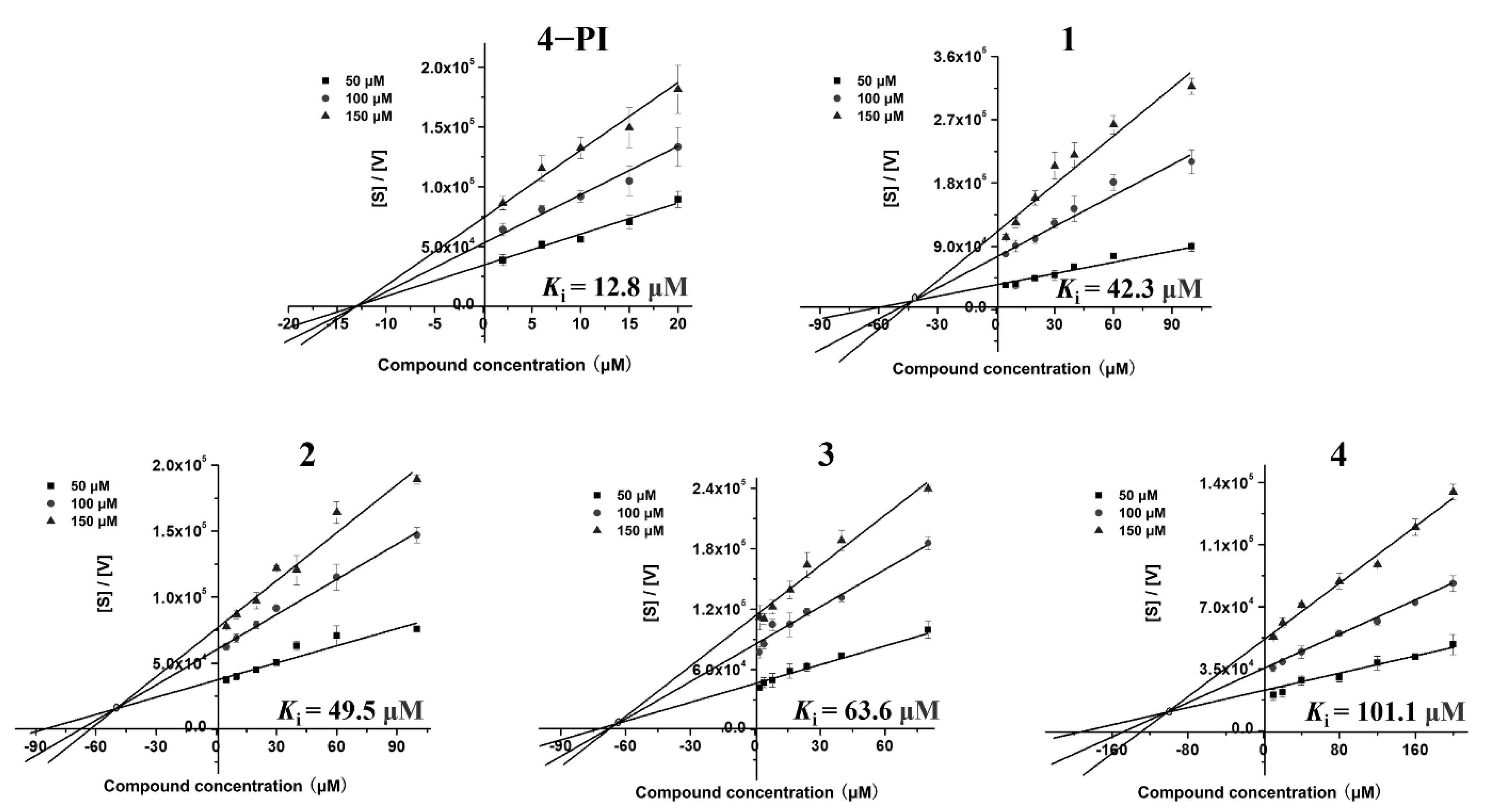
2.2. Enzyme-Based IDO1 Inhibitory Assay to Determine the IC50 and Ki Values
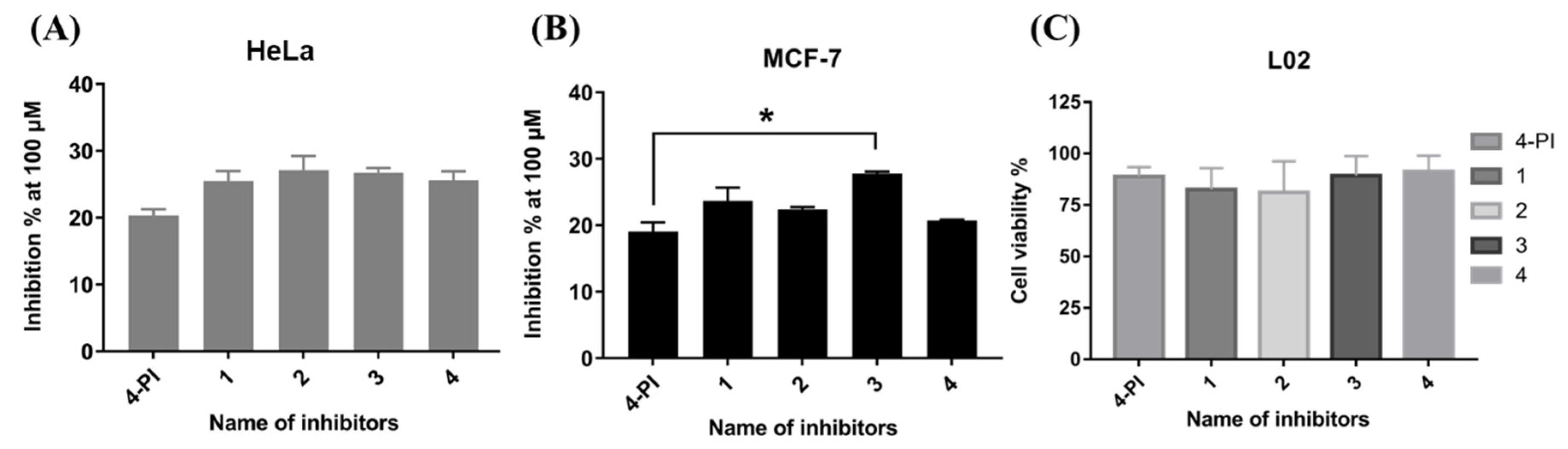
2.3. Cellular IDO1 Inhibitory Activity Assay
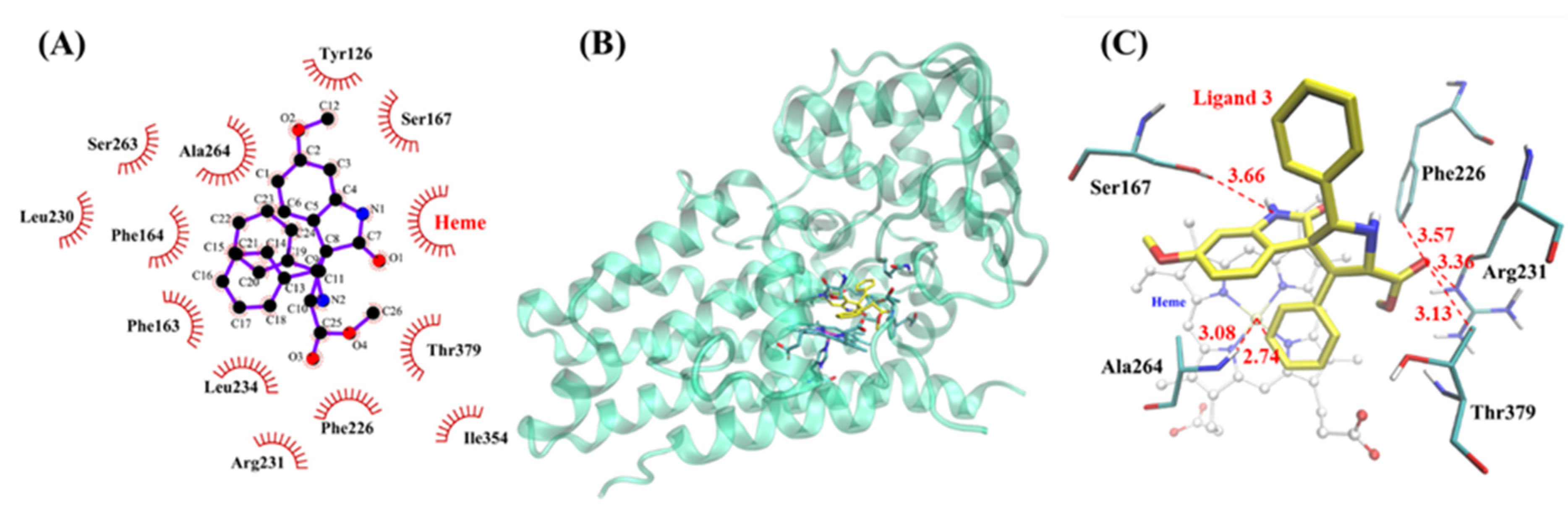
2.4. Molecular Docking Results
3. Conclusions
4. Materials and Methods
4.1. Materials and Reagents
4.2. Expression and Purification of IDO1
4.3. UV–Vis Spectra of IDO1 and IDO1–Complex
4.4. Determination of the Inhibition Types
4.5. Enzyme-Based IDO1 Inhibitory Assay
4.6. IDO1 Inhibitory Assay in Cell Lines
4.7. Molecular Docking Study
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wouatsa, N.A.V. Bioactive Proteins: Source. Synth. Appl. 2015, 676, 79–88. [Google Scholar] [CrossRef]
- Lagasse, H.A.; Alexaki, A.; Simhadri, V.L.; Katagiri, N.H.; Jankowski, W.; Sauna, Z.E.; Kimchi-Sarfaty, C. Recent advances in (therapeutic protein) drug development. F1000Research 2017, 6, 113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Braicu, C.; Buse, M.; Busuioc, C.; Drula, R.; Gulei, D.; Raduly, L.; Rusu, A.; Irimie, A.; Atanasov, A.G.; Slaby, O.; et al. A Comprehensive Review on MAPK: A Promising Therapeutic Target in Cancer. Cancers 2019, 11, 1618. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, M.; Bode, A.M.; Dong, Z.; Lee, M.H. AKT as a Therapeutic Target for Cancer. Cancer Res. 2019, 79, 1019–1031. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, L.; Wang, F.S.; Gershwin, M.E. Human autoimmune diseases: A comprehensive update. J. Intern. Med. 2015, 278, 369–395. [Google Scholar] [CrossRef]
- Attwood, M.M.; Jonsson, J.; Rask-Andersen, M.; Schioth, H.B. Soluble ligands as drug targets. Nat. Rev. Drug Discov. 2020, 19, 695–710. [Google Scholar] [CrossRef]
- Lin, Y.-W. Biodegradation of aromatic pollutants by metalloenzymes: A structural-functional-environmental perspective. Coord. Chem. Rev. 2021, 434, 213774. [Google Scholar] [CrossRef]
- Chen, S.-F.; Liu, X.-C.; Xu, J.-K.; Li, L.; Lang, J.-J.; Wen, G.-B.; Lin, Y.-W. Conversion of Human Neuroglobin into a Multifunctional Peroxidase by Rational Design. Inorg. Chem. 2021, 60, 2839–2845. [Google Scholar] [CrossRef]
- Poulos, T.L. Heme Enzyme Structure and Function. Chem. Rev. 2014, 114, 3919–3962. [Google Scholar] [CrossRef] [Green Version]
- Luo, J.; Du, K.-J.; Yuan, H.; Wei, C.-W.; Lang, J.-J.; Wen, G.-B.; Wang, Y.-H.; Lin, Y.-W. Rational Design of an Artificial Nuclease by Engineering a Hetero-Dinuclear Center of Mg-Heme in Myoglobin. ACS Catal. 2020, 10, 14359–14365. [Google Scholar] [CrossRef]
- Lin, Y.W. Rational design of heme enzymes for biodegradation of pollutants toward a green future. Biotechnol. Appl. Biochem. 2020, 67, 484–494. [Google Scholar] [CrossRef] [PubMed]
- Nelp, M.T.; Kates, P.A.; Hunt, J.T.; Newitt, J.A.; Balog, A.; Maley, D.; Zhu, X.; Abell, L.; Allentoff, A.; Borzilleri, R. Immune-modulating enzyme indoleamine 2,3-dioxygenase is effectively inhibited by targeting its apo-form. Proc. Natl. Acad. Sci. USA 2018, 115, 3249–3254. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fang, K.; Dong, G.; Li, Y.; He, S.; Wu, Y.; Wu, S.; Wang, W.; Sheng, C. Discovery of Novel Indoleamine 2,3-Dioxygenase 1 (IDO1) and Histone Deacetylase (HDAC) Dual Inhibitors. ACS Med. Chem. Lett. 2018, 9, 312–317. [Google Scholar] [CrossRef] [PubMed]
- Yan, D.; Lin, Y.W.; Tan, X. Heme-containing enzymes and inhibitors for tryptophan metabolism. Metallomics 2017, 9, 1230–1240. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.; Li, X.; Hu, F.; Li, Y.; Yang, Y.; Yan, J.; Kuang, C.; Yang, Q. Discovery of Tryptanthrin Derivatives as Potent Inhibitors of Indoleamine 2,3-Dioxygenase with Therapeutic Activity in Lewis Lung Cancer (LLC) Tumor-Bearing Mice. J. Med. Chem. 2013, 56, 8321–8331. [Google Scholar] [CrossRef] [PubMed]
- Dolui, E.; Larrieu, P.; Blanc, S.; Sapunaric, F.; Pouyez, J.; Moineaux, L.; Colette, D.; Stroobant, V.; Pilotte, L.; Colau, D. Discovery and preliminary SARs of keto-indoles as novel indoleamine 2,3-dioxygenase (IDO) inhibitors. Eur. J. Med. Chem. 2011, 46, 3058–3065. [Google Scholar]
- Kumar, S.; Waldo, J.P.; Jaipuri, F.A.; Marcinowicz, A.; Van Allen, C.; Adams, J.; Kesharwani, T. Discovery of Clinical Candidate (1R,4r)-4-((R)-2-((S)-6-Fluoro-5H-imidazo[5,1-a]isoindol-5-yl)-1-hydroxyethyl)cyclohexan-1-ol (Navoximod), a Potent and Selective Inhibitor of Indoleamine 2,3-Dioxygenase 1. J. Med. Chem. 2019, 62, 6705–6733. [Google Scholar] [CrossRef]
- Mondanelli, G.; Ugel, S.; Grohmann, U.; Bronte, V. The immune regulation in cancer by the amino acid metabolizing enzymes ARG and IDO. Curr. Opin. Pharmacol. 2017, 35, 30–39. [Google Scholar] [CrossRef]
- Pantouris, G.; Loudon-Griffiths, J.; Mowat, C.G. Insights into the mechanism of inhibition of tryptophan 2,3-dioxygenase by isatin derivatives. J. Enzyme Inhib. Med. Chem. 2016, 31, 70–78. [Google Scholar] [CrossRef] [Green Version]
- Gollner, A.; Rudolph, D.; Arnhof, H.; Bauer, M.; Blake, S.M.; Boehmelt, G.; Cockroft, X.L.; Dahmann, G.; Ettmayer, P.; Gerstberger, T.; et al. Discovery of Novel Spiro[3H-indole-3,2’-pyrrolidin]-2(1H)-one Compounds as Chemically Stable and Orally Active Inhibitors of the MDM2-p53 Interaction. J. Med. Chem. 2016, 59, 10147–10162. [Google Scholar] [CrossRef]
- Sosin, A.M.; Burger, A.M.; Siddiqi, A.; Abrams, J.; Mohammad, R.M.; Al-Katib, A.M. HDM2 antagonist MI-219 (spiro-oxindole), but not Nutlin-3 (cis-imidazoline), regulates p53 through enhanced HDM2 autoubiquitination and degradation in human malignant B-cell lymphomas. J. Hematol. Oncol. 2012, 5, 57. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shangary, S.; Qin, D.; McEachern, D.; Liu, M.; Miller, R.S.; Qiu, S.; Nikolovska-Coleska, Z.; Ding, K.; Wang, G.; Chen, J.; et al. Temporal activation of p53 by a specific MDM2 inhibitor is selectively toxic to tumors and leads to complete tumor growth inhibition. Proc. Natl. Acad. Sci. USA 2008, 105, 3933–3938. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, S.; Sun, W.; Zhao, Y.; McEachern, D.; Meaux, I.; Barrière, C.; Stuckey, J.A.; Meagher, J.L.; Bai, L.; Liu, L.; et al. SAR405838: An optimized inhibitor of MDM2-p53 interaction that induces complete and durable tumor regression. Cancer Res. 2014, 74, 5855–5865. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Weger, V.A.; De Jonge, M.; Langenberg, M.H.G.; Schellens, J.H.M.; Lolkema, M.; Varga, A.; Demers, B.; Thomas, K.; Hsu, K.; Tuffal, G. A phase I study of the HDM2 antagonist SAR405838 combined with the MEK inhibitor pimasertib in patients with advanced solid tumours. Br. J. Cancer 2019, 120, 286–293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Röhrig, U.F.; Majjigapu, S.R.; Vogel, P.; Zoete, V.; Michielin, O. Challenges in the Discovery of Indoleamine 2,3-Dioxygenase 1 (IDO1) Inhibitors. J. Med. Chem. 2015, 58, 9421–9437. [Google Scholar] [CrossRef]
- Platten, M.; Nollen, E.A.A.; Rohrig, U.F.; Fallarino, F.; Opitz, C.A. Tryptophan metabolism as a common therapeutic target in cancer, neurodegeneration and beyond. Nat. Rev. Drug Discov. 2019, 18, 379–401. [Google Scholar] [CrossRef]
- Zakharia, Y.; McWilliams, R.R.; Rixe, O.; Drabick, J.; Shaheen, M.F.; Grossmann, K.F.; Kolhe, R.; Pacholczyk, R.; Sadek, R.; Tennant, L.L.; et al. Phase II trial of the IDO pathway inhibitor indoximod plus pembrolizumab for the treatment of patients with advanced melanoma. J. Immunother. Cancer 2021, 9, e002057. [Google Scholar] [CrossRef]
- Fung, S.P.S.; Wang, H.; Tomek, P.; Squire, C.J.; Flanagan, J.U.; Palmer, B.D.; Bridewell, D.J.A.; Tijono, S.M.; Jamie, J.F.; Ching, L.M. Discovery and characterisation of hydrazines as inhibitors of the immune suppressive enzyme, indoleamine 2,3-dioxygenase 1 (IDO1). Bioorg. Med. Chem. 2013, 21, 7595–7603. [Google Scholar] [CrossRef]
- Zhang, J.X.; Wang, H.Y.; Jin, Q.W.; Zheng, C.W.; Zhao, G.; Shang, Y.J. Thiourea-Quaternary Ammonium Salt Catalyzed Asymmetric 1,3-Dipolar Cycloaddition of Imino Esters to Construct Spiro[pyrrolidin-3,3′-oxindoles. Org. Lett. 2016, 18, 4774–4777. [Google Scholar] [CrossRef]
- Wang, G.; Liu, X.; Huang, T.; Kuang, Y.; Lin, L.; Feng, X. Asymmetric catalytic 1,3-dipolar cycloaddition reaction of nitrile imines for the synthesis of chiral spiro-pyrazoline-oxindoles. Org. Lett. 2013, 15, 76–79. [Google Scholar] [CrossRef]
- Xu, X.; Ren, J.; Ma, Y.; Liu, H.; Rong, Q.; Feng, Y.; Wang, Y.; Cheng, Y.; Ge, R.; Li, Z. Discovery of cyanopyridine scaffold as novel indoleamine-2,3-dioxygenase 1 (IDO1) inhibitors through virtual screening and preliminary hit optimisation. J. Enzyme Inhib. Med. Chem. 2019, 34, 250–263. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Jaller, D.; Patel, B.; Lalonde, J.M.; Duhadaway, J.B.; Malachowski, W.P.; Prendergast, G.C.; Muller, A.J. Structure based development of phenylimidazole-derived inhibitors of indoleamine 2,3-dioxygenase. J. Med. Chem. 2008, 51, 4968. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pérez-Cruz, F.; Serra, S.; Delogu, G.; Lapier, M.; Maya, J.D.; Olea-Azar, C.; Santana, L.; Uriarte, E. Antitrypanosomal and antioxidant properties of 4-hydroxycoumarins derivatives. Bioorg. Med. Chem. Lett. 2012, 22, 5569–5573. [Google Scholar] [CrossRef] [PubMed]
- Copeland, A.R. Reversible Modes of Inhibitor Interactions with Enzymes. In Evaluation of Enzyme Inhibitors in Drug Discovery; John and Wiley and Sons: Hoboken, NJ, USA, 2013; pp. 57–121. [Google Scholar]
- Dougall, I.G.; Unitt, J. Chapter 2-Evaluation of the Biological Activity of Compounds: Techniques and Mechanism of Action Studies. In The Practice of Medicinal Chemistry, 4th ed.; Wermuth, C.G., Aldous, D., Raboisson, P., Rognan, D., Eds.; Academic Press: San Diego, CA, USA, 2015; pp. 15–43. [Google Scholar]
- Cornish-Bowden, A. A simple graphical method for determining the inhibition constants of mixed, uncompetitive and non-competitive inhibitors (Short Communication). Biochem. Pharmacol. 1974, 137, 143–144. [Google Scholar] [CrossRef] [PubMed]
- Nitsche, C.; Passioura, T.; Varava, P.; Mahawaththa, M.C.; Leuthold, M.M.; Klein, C.D.; Suga, H.; Otting, G. De Novo Discovery of Nonstandard Macrocyclic Peptides as Noncompetitive Inhibitors of the Zika Virus NS2B-NS3 Protease. ACS Med. Chem. Lett. 2019, 10, 168–174. [Google Scholar] [CrossRef] [PubMed]
- García-García, A.; Rojas, S. A gliclazide complex based on palladium towards Alzheimer’s disease: Promising protective activity against Aβ-induced toxicity in C. elegans. Chem. Commun. 2022, 58, 1514–1517. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Y.-C.; Prusoff, W.H. Relationship between the inhibition constant (KI) and the concentration of inhibitor which causes 50 per cent inhibition (I50) of an enzymatic reaction. Biochem. Pharmacol. 1973, 22, 3099–3108. [Google Scholar] [PubMed]
- Sono, M.; Cady, S.G. Enzyme kinetic and spectroscopic studies of inhibitor and effector interactions with indoleamine 2,3-dioxygenase. 1. Norharman and 4-phenylimidazole binding to the enzyme as inhibitors and heme ligands. Biochemistry 1989, 28, 5392–5399. [Google Scholar] [CrossRef]
- Sugimoto, H.; Oda, S.; Otsuki, T.; Hino, T.; Yoshida, T.; Shiro, Y. Crystal structure of human indoleamine 2,3-dioxygenase: Catalytic mechanism of O2 incorporation by a heme-containing dioxygenase. Proc. Natl. Acad. Sci. USA 2006, 103, 2611–2616. [Google Scholar] [CrossRef] [Green Version]
- Meininger, D.; Zalameda, L.; Liu, Y.; Stepan, L.P.; Borges, L.; Mccarter, J.D.; Sutherland, C.L. Purification and kinetic characterization of human indoleamine 2,3-dioxygenases 1 and 2 (IDO1 and IDO2) and discovery of selective IDO1 inhibitors. Biochim. Biophys. Acta 2011, 1814, 1947–1954. [Google Scholar] [CrossRef]
- Panda, S.; Roy, A.; Deka, S.J.; Trivedi, V.; Manna, D. Fused Heterocyclic Compounds as Potent Indoleamine-2,3-dioxygenase 1 Inhibitors. ACS Med. Chem. Lett. 2016, 7, 1167–1172. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Booth, E.S.; Basran, J.; Lee, M.; Handa, S.; Raven, E.L. Substrate Oxidation by Indoleamine 2,3-Dioxygenase: Evidence for a common reaction mechanism. J. Biol. Chem. 2015, 290, 30924–30930. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, Q.; Zheng, M.; Yang, S.; Kuang, C.; Yu, C.; Yang, Q. Structure–activity relationship and enzyme kinetic studies on 4-aryl-1H-1,2,3-triazoles as indoleamine 2,3-dioxygenase (IDO) inhibitors. Eur. J. Med. Chem. 2011, 46, 5680–5687. [Google Scholar] [CrossRef]
- Crosignani, S.; Bingham, P.; Bottemanne, P.; Cannelle, H.; Cauwenberghs, S.; Cordonnier, M.; Dalvie, D.; Deroose, F.; Feng, J.L.; Gomes, B. Discovery of a Novel and Selective Indoleamine 2,3-Dioxygenase (IDO-1) Inhibitor 3-(5-Fluoro-1 H -indol-3-yl)pyrrolidine-2,5-dione (EOS200271/PF-06840003) and Its Characterization as a Potential Clinical Candidate. J. Med. Chem. 2017, 60, 9617–9629. [Google Scholar] [CrossRef]
- Lewis-Ballester, A.; Pham, K.N.; Batabyal, D.; Karkashon, S.; Bonanno, J.B.; Poulos, T.L.; Yeh, S.R. Structural insights into substrate and inhibitor binding sites in human indoleamine 2,3-dioxygenase 1. Nat. Commun. 2017, 8, 1693. [Google Scholar] [CrossRef] [PubMed]
- Schüttelkopf, A.W.; Aalten, D.M.F.V. PRODRG: A tool for high-throughput crystallography of protein–ligand complexes. Acta Crystallogr. D Biol. Crystallogr. 2004, 60, 1355–1363. [Google Scholar] [CrossRef] [PubMed] [Green Version]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compounds | Type of Inhibition | IC50 (μM) | Ki (μM) |
|---|---|---|---|
| 4−PI | Noncompetitive [40] | 49.0 ± 0.7 | 12.8 ± 2.1 |
| 1 | Uncompetitive | 25.2 ± 3.6 | 42.3 ± 0.8 |
| 2 | Uncompetitive | 29.4 ± 1.3 | 49.5 ± 6.0 |
| 3 | Uncompetitive | 7.9 ± 1.7 | 63.6 ± 3.5 |
| 4 | Uncompetitive | 64.6 ± 3.9 | 101.1 ± 4.4 |
| Compounds | HeLa Cell | MCF-7 Cell | CT26 Cell | 4T1 Cell |
|---|---|---|---|---|
| 4−PI | 20.3 ± 0.9 | 19.1 ± 1.4 | 5.8 ± 0.5 | 5.7 ± 0.5 |
| 1 | 25.5 ± 1.5 | 23.7 ± 2.0 | 3.7 ± 0.8 | 2.6 ± 0.6 |
| 2 | 27.1 ± 2.1 | 22.4 ± 0.4 | 4.2 ± 0.5 | 4.2 ± 1.0 |
| 3 | 26.7 ± 0.7 | 27.8 ± 0.3 | 6.4 ± 0.8 | 4.3 ± 0.1 |
| 4 | 25.6 ± 1.3 | 20.7 ± 0.1 | 1.7 ± 0.5 | 2.8 ± 0.3 |
| Model | Ebinding a (kcal/mol) | Einter-mol b (kcal/mol) | Evdw c (kcal/mol) | Eelec d (kcal/mol) |
|---|---|---|---|---|
| 1 | −5.88 | −7.08 | −2.7 | −0.25 |
| 2 | −5.40 | −6.59 | −3.09 | −0.28 |
| 3 | −5.01 | −6.20 | −2.71 | −0.19 |
| 4 | −4.83 | −6.03 | −2.92 | −0.22 |
| 5 | −4.61 | −5.80 | −2.45 | −0.12 |
| 6 | −4.49 | −5.68 | −2.27 | −0.26 |
| 7 | −4.49 | −5.68 | −2.13 | −0.18 |
| 8 | −4.41 | −5.61 | −2.88 | −0.24 |
| 9 | −4.40 | −5.59 | −2.03 | −0.18 |
| 10 | −4.40 | −5.60 | −2.58 | −0.10 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yan, D.; Xu, J.; Wang, X.; Zhang, J.; Zhao, G.; Lin, Y.; Tan, X. Spiro-Oxindole Skeleton Compounds Are Efficient Inhibitors for Indoleamine 2,3-Dioxygenase 1: An Attractive Target for Tumor Immunotherapy. Int. J. Mol. Sci. 2022, 23, 4668. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23094668
Yan D, Xu J, Wang X, Zhang J, Zhao G, Lin Y, Tan X. Spiro-Oxindole Skeleton Compounds Are Efficient Inhibitors for Indoleamine 2,3-Dioxygenase 1: An Attractive Target for Tumor Immunotherapy. International Journal of Molecular Sciences. 2022; 23(9):4668. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23094668
Chicago/Turabian StyleYan, Daojing, Jiakun Xu, Xiang Wang, Jiaxing Zhang, Gang Zhao, Yingwu Lin, and Xiangshi Tan. 2022. "Spiro-Oxindole Skeleton Compounds Are Efficient Inhibitors for Indoleamine 2,3-Dioxygenase 1: An Attractive Target for Tumor Immunotherapy" International Journal of Molecular Sciences 23, no. 9: 4668. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23094668