
Consensus Gene Co-Expression Network Analysis Identifies Novel Genes Associated with Severity of Fibrotic Lung Disease


 , and
, and
Abstract
:1. Introduction
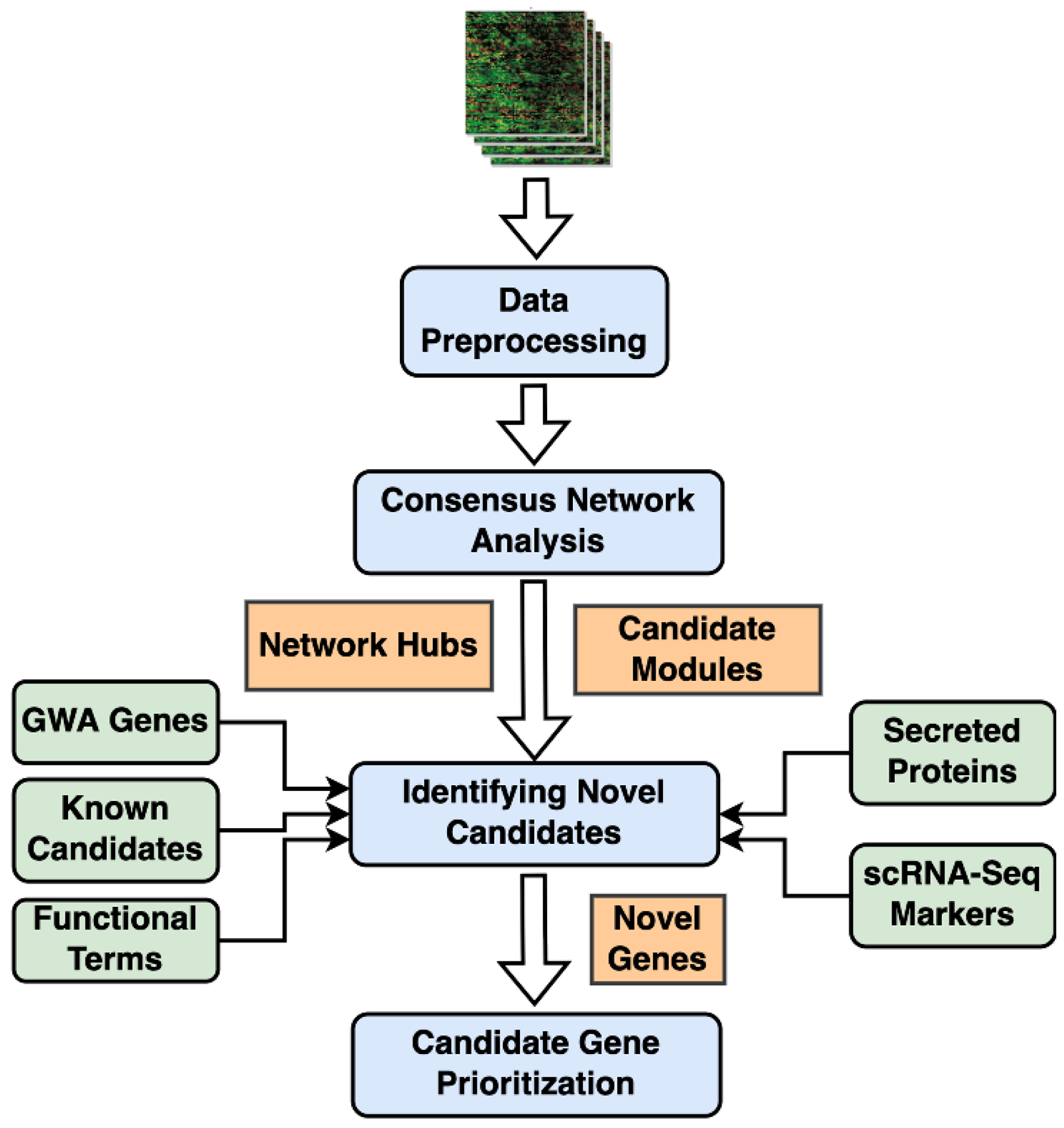
2. Results
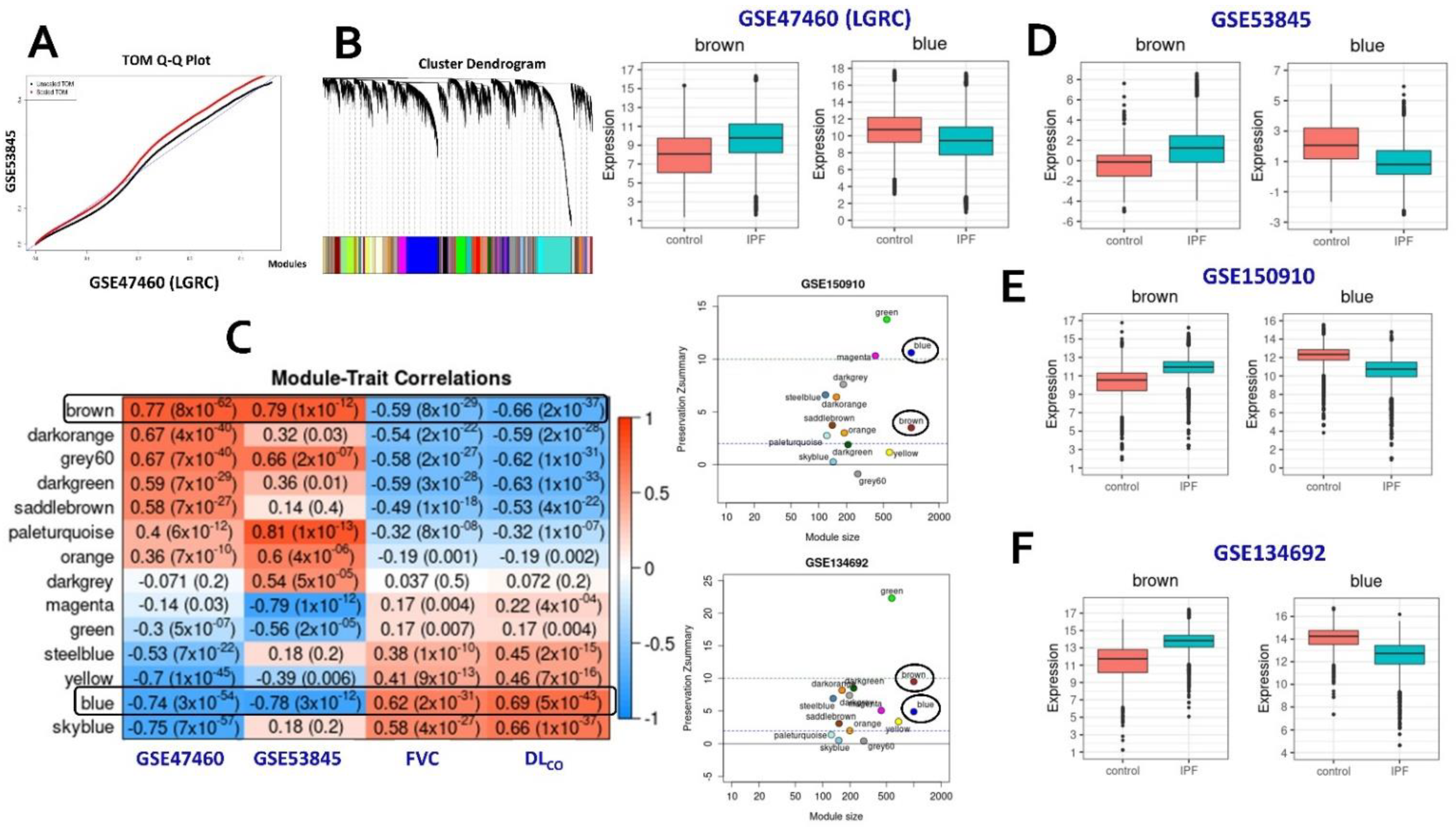
2.1. Consensus Gene Modules
2.2. Identification of Conserved Candidate Modules Correlated with IPF Phenotypic Traits
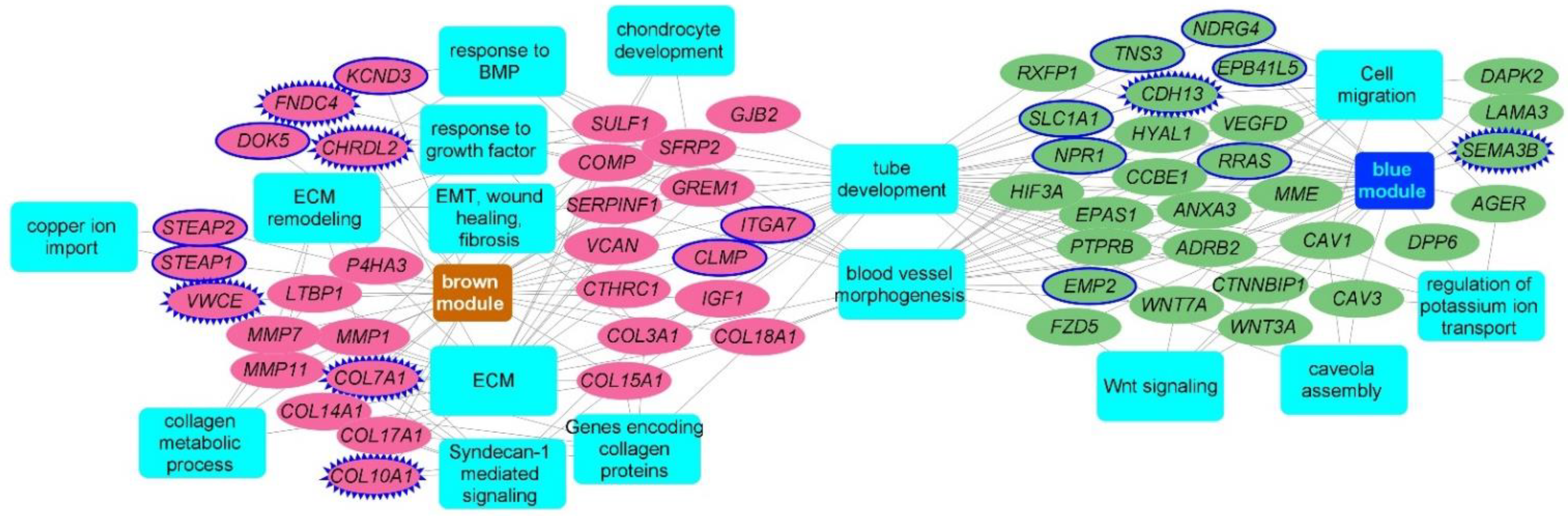
2.3. Consensus Modules-Biological Processes
2.4. Consensus Modules for Specific Cell Types from Normal, and Fibrotic Lung Markers
2.5. Candidate Biomarkers and Novel IPF-Associated Genes
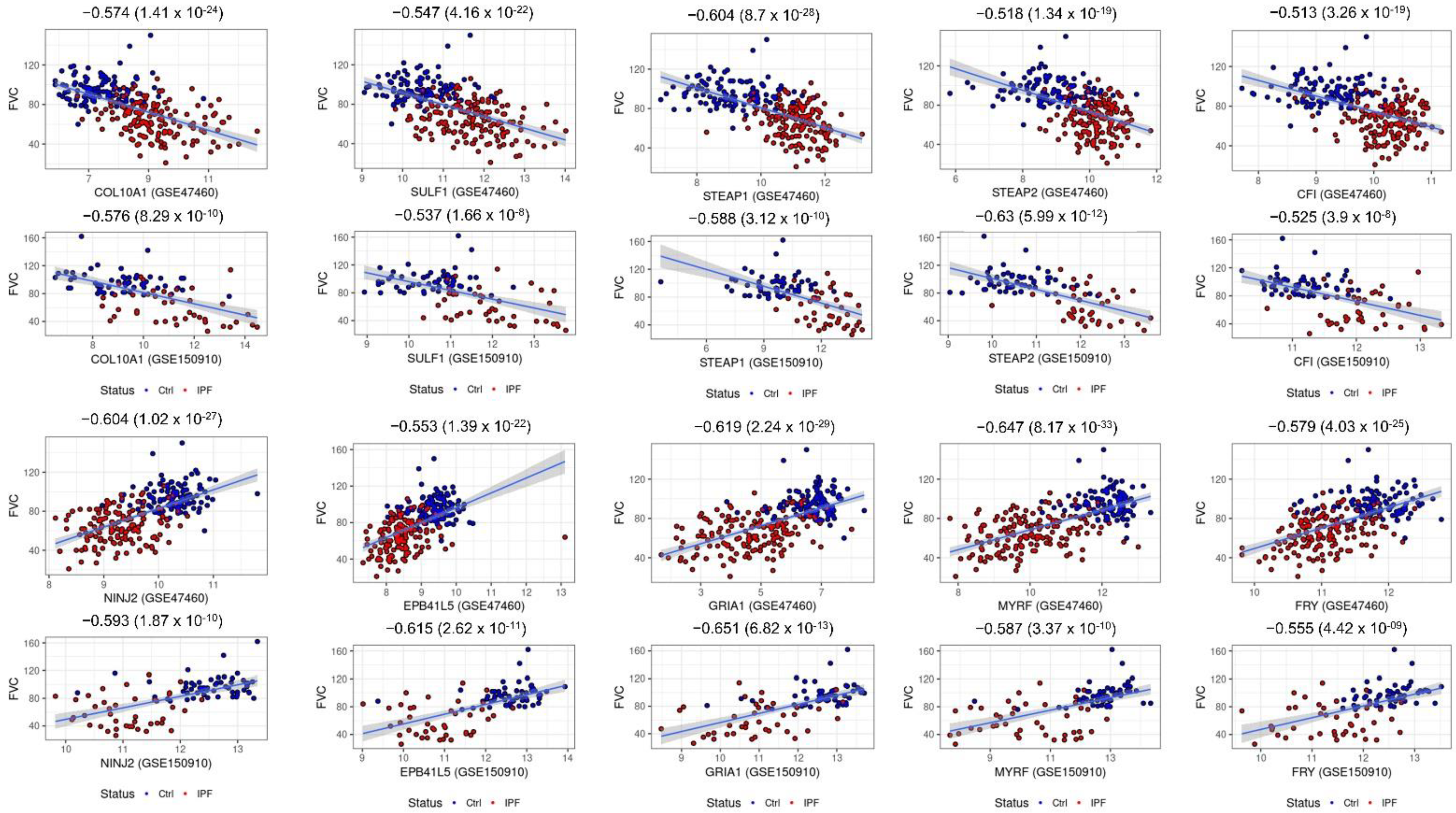
2.6. Consensus Hubs Associated with Lung Function Activity
2.7. Hub Genes Conserved across Different IPF Severities and Acute Exacerbation
2.8. Candidate Genes Categorizing IPF and Other Interstitial Lung Diseases
2.9. Hub Gene Prioritization
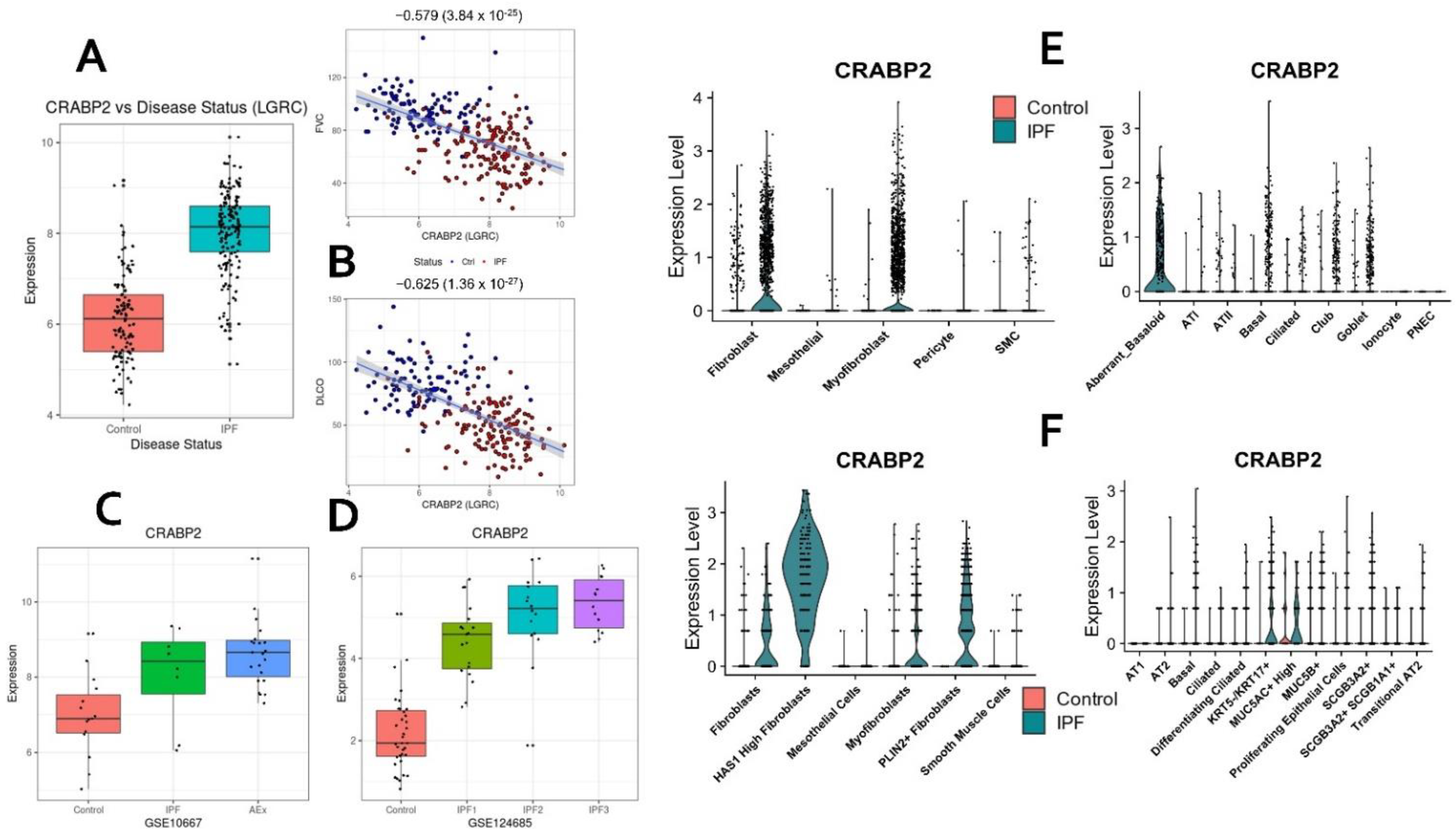
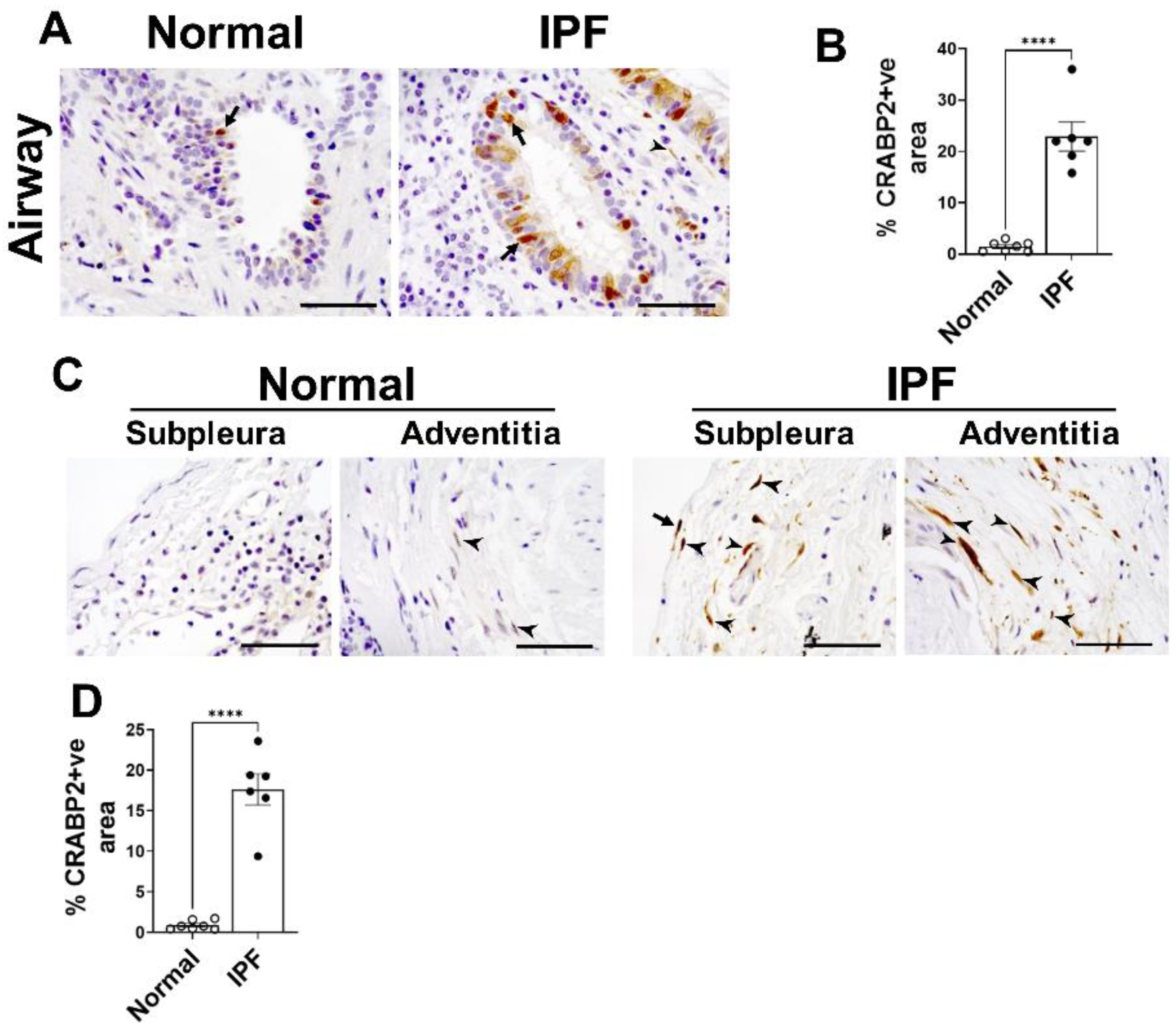
2.10. CRABP2—Novel Candidate Gene and Potential Biomarker of IPF
3. Discussion
4. Materials and Methods
4.1. IPF Transcriptomic Datasets
4.2. Normal and IPF Lung Single-Cell Markers
4.3. Known Pulmonary Fibrosis Genes
4.4. Hub Gene Prioritization
4.5. Lung Function GWA Genes
4.6. Secreted Proteins
4.7. Consensus WGCNA and Candidate Modules
4.8. Immunohistochemistry
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Kaur, A.; Mathai, S.K.; Schwartz, D.A. Genetics in Idiopathic Pulmonary Fibrosis Pathogenesis, Prognosis, and Treatment. Front. Med. 2017, 4, 154. [Google Scholar] [CrossRef] [PubMed]
- Olson, A.L.; Swigris, J.J.; Lezotte, D.C.; Norris, J.M.; Wilson, C.G.; Brown, K.K. Mortality from Pulmonary Fibrosis Increased in the United States from 1992 to 2003. Am. J. Respir. Crit. Care Med. 2007, 176, 277–284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bauer, Y.; Tedrow, J.; De Bernard, S.; Birker-Robaczewska, M.; Gibson, K.F.; Guardela, B.J.; Hess, P.; Klenk, A.; Lindell, K.O.; Poirey, S.; et al. A Novel Genomic Signature with Translational Significance for Human Idiopathic Pulmonary Fibrosis. Am. J. Respir. Cell Mol. Biol. 2015, 52, 217–231. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meltzer, E.B.; Barry, W.T.; A D’Amico, T.; Davis, R.D.; Lin, S.S.; Onaitis, M.W.; Morrison, L.D.; A Sporn, T.; Steele, M.P.; Noble, P.W. Bayesian probit regression model for the diagnosis of pulmonary fibrosis: Proof-of-principle. BMC Med Genom. 2011, 4, 70. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Yella, J.; Chen, J.; McCormack, F.X.; Madala, S.K.; Jegga, A.G. Unsupervised gene expression analyses identify IPF-severity correlated signatures, associated genes and biomarkers. BMC Pulm. Med. 2017, 17, 133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herazo-Maya, J.D.; Noth, I.; Duncan, S.R.; Kim, S.; Ma, S.-F.; Tseng, G.C.; Feingold, E.; Juan-Guardela, B.M.; Richards, T.J.; Lussier, Y.; et al. Peripheral Blood Mononuclear Cell Gene Expression Profiles Predict Poor Outcome in Idiopathic Pulmonary Fibrosis. Sci. Transl. Med. 2013, 5, 205ra136. [Google Scholar] [CrossRef] [Green Version]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef] [Green Version]
- Langfelder, P.; Mischel, P.S.; Horvath, S. When is hub gene selection better than standard meta-analysis? PLoS ONE 2013, 8, e61505. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.; Zhu, J.; Chen, F.; Ma, L. Weighted Gene Coexpression Network Analysis Identifies Key Genes and Pathways Associated with Idiopathic Pulmonary Fibrosis. Med. Sci. Monit. 2019, 25, 4285–4304. [Google Scholar] [CrossRef]
- E McDonough, J.; Kaminski, N.; Thienpont, B.; Hogg, J.C.; Vanaudenaerde, B.; A Wuyts, W. Gene correlation network analysis to identify regulatory factors in idiopathic pulmonary fibrosis. Thorax 2018, 74, 132–140. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; Gu, L.; Li, W. The Prognostic Value of Integrated Analysis of Inflammation and Hypoxia-Related Genes in Idiopathic Pulmonary Fibrosis. Front. Immunol. 2022, 13, 730186. [Google Scholar] [CrossRef] [PubMed]
- Zhang, N.; Guo, Y.; Wu, C.; Jiang, B.; Wang, Y. Identification of the Molecular Subgroups in Idiopathic Pulmonary Fibrosis by Gene Expression Profiles. Comput. Math. Methods Med. 2021, 2021, 7922594. [Google Scholar] [CrossRef] [PubMed]
- Guillotin, D.; Taylor, A.R.; Platé, M.; Mercer, P.F.; Edwards, L.M.; Haggart, R.; Miele, G.; McAnulty, R.J.; Maher, T.M.; E Hynds, R.; et al. Transcriptome analysis of IPF fibroblastic foci identifies key pathways involved in fibrogenesis. Thorax 2020, 76, 73–82. [Google Scholar] [CrossRef] [PubMed]
- Xia, Y.; Lei, C.; Yang, D.; Luo, H. Identification of key modules and hub genes associated with lung function in idiopathic pulmonary fibrosis. PeerJ 2020, 8, e9848. [Google Scholar] [CrossRef] [PubMed]
- Horvath, S.; Dong, J. Geometric Interpretation of Gene Coexpression Network Analysis. PLoS Comput. Biol. 2008, 4, e1000117. [Google Scholar] [CrossRef] [PubMed]
- Langfelder, P.; Horvath, S. WGCNA: An R package for weighted correlation network analysis. BMC Bioinform. 2008, 9, 559. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, J.; Bardes, E.E.; Aronow, B.J.; Jegga, A.G. ToppGene Suite for gene list enrichment analysis and candidate gene prioritization. Nucleic Acids Res. 2009, 37, W305–W311. [Google Scholar] [CrossRef] [PubMed]
- Adams, T.S.; Schupp, J.C.; Poli, S.; Ayaub, E.A.; Neumark, N.; Ahangari, F.; Chu, S.G.; Raby, B.A.; DeIuliis, G.; Januszyk, M.; et al. Single-cell RNA-seq reveals ectopic and aberrant lung-resident cell populations in idiopathic pulmonary fibrosis. Sci. Adv. 2020, 6, eaba1983. [Google Scholar] [CrossRef]
- Khatib, A.; Solaimuthu, B.; Yosef, M.B.; Rmaileh, A.A.; Tanna, M.; Oren, G.; Frisch, M.S.; Axelrod, J.H.; Lichtenstein, M.; Shaul, Y.D. The glutathione peroxidase 8 (GPX8)/IL-6/STAT3 axis is essential in maintaining an aggressive breast cancer phenotype. Proc. Natl. Acad. Sci. USA 2020, 117, 21420–21431. [Google Scholar] [CrossRef]
- Hill, C.; Jones, M.; Davies, D.; Wang, Y. Epithelial-Mesenchymal Transition Contributes to Pulmonary Fibrosis via Aberrant Epithelial/Fibroblastic Cross-Talk. J. Lung Health Dis. 2019, 3, 31–35. [Google Scholar] [CrossRef] [Green Version]
- Travaglini, K.J.; Nabhan, A.N.; Penland, L.; Sinha, R.; Gillich, A.; Sit, R.V.; Chang, S.; Conley, S.D.; Mori, Y.; Seita, J.; et al. A molecular cell atlas of the human lung from single-cell RNA sequencing. Nature 2020, 587, 619–625. [Google Scholar] [CrossRef] [PubMed]
- Habermann, A.C.; Gutierrez, A.J.; Bui, L.T.; Yahn, S.L.; Winters, N.I.; Calvi, C.L.; Peter, L.; Chung, M.-I.; Taylor, C.J.; Jetter, C.; et al. Single-cell RNA sequencing reveals profibrotic roles of distinct epithelial and mesenchymal lineages in pulmonary fibrosis. Sci. Adv. 2020, 6, eaba1972. [Google Scholar] [CrossRef] [PubMed]
- Mayr, C.H.; Simon, L.M.; Leuschner, G.; Ansari, M.; Schniering, J.; E Geyer, P.; Angelidis, I.; Strunz, M.; Singh, P.; Kneidinger, N.; et al. Integrative analysis of cell state changes in lung fibrosis with peripheral protein biomarkers. EMBO Mol. Med. 2021, 13, e12871. [Google Scholar] [CrossRef]
- Furusawa, H.; Cardwell, J.H.; Okamoto, T.; Walts, A.D.; Konigsberg, I.R.; Kurche, J.S.; Bang, T.J.; Schwarz, M.I.; Brown, K.K.; Kropski, J.A.; et al. Chronic Hypersensitivity Pneumonitis, an Interstitial Lung Disease with Distinct Molecular Signatures. Am. J. Respir. Crit. Care Med. 2020, 202, 1430–1444. [Google Scholar] [CrossRef] [PubMed]
- McDonough, J.E.; Ahangari, F.; Li, Q.; Jain, S.; Verleden, S.E.; Herazo-Maya, J.; Vukmirovic, M.; Deiuliis, G.; Tzouvelekis, A.; Tanabe, N.; et al. Transcriptional regulatory model of fibrosis progression in the human lung. JCI Insight 2019, 4, e131597. [Google Scholar] [CrossRef] [PubMed]
- Ritchie, M.E.; Belinda, P.; Wu, D.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. Limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015, 43, e47. [Google Scholar] [CrossRef]
- Konishi, K.; Gibson, K.F.; Lindell, K.O.; Richards, T.J.; Zhang, Y.; Dhir, R.; Bisceglia, M.; Gilbert, S.; Yousem, S.A.; Song, J.W.; et al. Gene Expression Profiles of Acute Exacerbations of Idiopathic Pulmonary Fibrosis. Am. J. Respir. Crit. Care Med. 2009, 180, 167–175. [Google Scholar] [CrossRef]
- Friedman, J.H.; Hastie, T.; Tibshirani, R. Regularization Paths for Generalized Linear Models via Coordinate Descent. J. Stat. Softw. 2010, 33, 1–22. [Google Scholar] [CrossRef] [Green Version]
- Buniello, A.; MacArthur, J.A.L.; Cerezo, M.; Harris, L.W.; Hayhurst, J.; Malangone, C.; McMahon, A.; Morales, J.; Mountjoy, E.; Sollis, E.; et al. The NHGRI-EBI GWAS Catalog of published genome-wide association studies, targeted arrays and summary statistics 2019. Nucleic Acids Res. 2019, 47, D1005–D1012. [Google Scholar] [CrossRef] [Green Version]
- Budhu, A.S.; Noy, N. Direct Channeling of Retinoic Acid between Cellular Retinoic Acid-Binding Protein II and Retinoic Acid Receptor Sensitizes Mammary Carcinoma Cells to Retinoic Acid-Induced Growth Arrest. Mol. Cell. Biol. 2002, 22, 2632–2641. [Google Scholar] [CrossRef] [Green Version]
- Barrett, T.; Wilhite, S.E.; Ledoux, P.; Evangelista, C.; Kim, I.F.; Tomashevsky, M.; Marshall, K.A.; Phillippy, K.H.; Sherman, P.M.; Holko, M.; et al. NCBI GEO: Archive for functional genomics data sets—update. Nucleic Acids Res. 2012, 41, D991–D995. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DePianto, D.J.; Chandriani, S.; Abbas, A.R.; Jia, G.; N’Diaye, E.N.; Caplazi, P.; E Kauder, S.; Biswas, S.; Karnik, S.K.; Ha, C.; et al. Heterogeneous gene expression signatures correspond to distinct lung pathologies and biomarkers of disease severity in idiopathic pulmonary fibrosis. Thorax 2014, 70, 48–56. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sivakumar, P.; Thompson, J.R.; Ammar, R.; Porteous, M.; McCoubrey, C.; Cantu, E.; Ravi, K.; Zhang, Y.; Luo, Y.; Streltsov, D.; et al. RNA sequencing of transplant-stage idiopathic pulmonary fibrosis lung reveals unique pathway regula-tion. ERJ Open Res. 2019, 5, 00117–02019. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reyfman, P.A.; Walter, J.M.; Joshi, N.; Anekalla, K.R.; McQuattie-Pimentel, A.C.; Chiu, S.; Fernandez, R.; Akbarpour, M.; Chen, C.-I.; Ren, Z.; et al. Single-Cell Transcriptomic Analysis of Human Lung Provides Insights into the Pathobiology of Pulmonary Fibrosis. Am. J. Respir. Crit. Care Med. 2019, 199, 1517–1536. [Google Scholar] [CrossRef] [PubMed]
- Carvalho-Silva, D.; Pierleoni, A.; Pignatelli, M.; Ong, C.K.; Fumis, L.; Karamanis, N.; Carmona, M.; Faulconbridge, A.; Hercules, A.; McAuley, E.; et al. Open Targets Platform: New developments and updates two years on. Nucleic Acids Res. 2018, 47, D1056–D1065. [Google Scholar] [CrossRef]
- Davis, A.P.; Grondin, C.J.; Johnson, R.J.; Sciaky, D.; McMorran, R.; Wiegers, J.; Wiegers, T.C.; Mattingly, C. The Comparative Toxicogenomics Database: Update 2019. Nucleic Acids Res. 2018, 47, D948–D954. [Google Scholar] [CrossRef]
- Yu, W.; Clyne, M.; Khoury, M.J.; Gwinn, M. Phenopedia and Genopedia: Disease-centered and gene-centered views of the evolving knowledge of human genetic associations. Bioinformatics 2009, 26, 145–146. [Google Scholar] [CrossRef] [Green Version]
- Stelzer, G.; Rosen, N.; Plaschkes, I.; Zimmerman, S.; Twik, M.; Fishilevich, S.; Stein, T.I.; Nudel, R.; Lieder, I.; Mazor, Y.; et al. The GeneCards Suite: From Gene Data Mining to Disease Genome Sequence Analyses. Curr. Protoc. Bioinform. 2016, 54, 1.30.1–1.30.33. [Google Scholar] [CrossRef]
- Uhlén, M.; Fagerberg, L.; Hallström, B.M.; Lindskog, C.; Oksvold, P.; Mardinoglu, A.; Sivertsson, Å.; Kampf, C.; Sjöstedt, E.; Asplund, A.; et al. Tissue-Based Map of the Human Proteome. Science 2015, 347, 1260419. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Horvath, S. A General Framework for Weighted Gene Co-Expression Network Analysis. Stat. Appl. Genet. Mol. Biol. 2005, 4, 17. [Google Scholar] [CrossRef]
- Langfelder, P.; Zhang, B.; Horvath, S. Defining clusters from a hierarchical cluster tree: The Dynamic Tree Cut package for R. Bioinformatics 2008, 24, 719–720. [Google Scholar] [CrossRef] [PubMed]
- Langfelder, P.; Horvath, S. Eigengene networks for studying the relationships between co-expression modules. BMC Syst. Biol. 2007, 1, 54. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Langfelder, P.; Luo, R.; Oldham, M.C.; Horvath, S. Is my network module preserved and reproducible? PLoS Comput. Biol. 2011, 7, e1001057. [Google Scholar] [CrossRef] [PubMed] [Green Version]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | GSE47460 | GSE150910 | ||||||
|---|---|---|---|---|---|---|---|---|
| DLCO | p-Value | FVC | p-Value | DLCO | p-value | FVC | p-Value | |
| Upregulated in IPF | ||||||||
| IGF1 | −0.6 | 7.42 × 10−26 | −0.54 | 5.68 × 10−20 | −0.55 | 6.51 × 10−16 | −0.5 | 4.95 × 10−13 |
| LTBP1 | −0.61 | 7.23 × 10−27 | −0.55 | 8.62 × 10−21 | −0.33 | 4.91 × 10−6 | −0.31 | 2.32 × 10−5 |
| SULF1 | −0.61 | 7.23 × 10−27 | −0.55 | 8.62 × 10−21 | −0.54 | 1.34 × 10−15 | −0.54 | 4.64 × 10−15 |
| COL15A1 | −0.59 | 7.59 × 10−25 | −0.55 | 8.62 × 10−21 | −0.33 | 9.12 × 10−6 | −0.18 | 0.0249 |
| SERPINF1 | −0.65 | 2.85 × 10−31 | −0.59 | 2.91 × 10−24 | −0.57 | 2.46 × 10−17 | −0.47 | 1.60 × 10−11 |
| PDIA4 | −0.65 | 2.85 × 10−31 | −0.57 | 1.77 × 10−22 | −0.49 | 1.57 × 10−12 | −0.46 | 4.54 × 10−11 |
| COL10A1 | −0.61 | 7.23 × 10−27 | −0.57 | 1.77 × 10−22 | −0.49 | 1.15 × 10−12 | −0.58 | 1.41 × 10−17 |
| COL14A1 | −0.64 | 4.43 × 10−30 | −0.54 | 5.68 × 10−20 | −0.55 | 4.21 × 10−16 | −0.5 | 4.72 × 10−13 |
| COL18A1 | −0.61 | 7.23 × 10−27 | −0.58 | 2.27 × 10−23 | −0.45 | 2.46 × 10−10 | −0.4 | 3.38 × 10−8 |
| SCRG1 | −0.62 | 6.88 × 10−28 | −0.56 | 1.28 × 10−21 | −0.41 | 6.28 × 10−9 | −0.35 | 1.67 × 10−6 |
| GPX8 | −0.59 | 7.59 × 10−25 | −0.56 | 1.28 × 10−21 | −0.3 | 3.65 × 10−5 | −0.31 | 2.97 × 10−5 |
| COL3A1 | −0.58 | 6.83 × 10−24 | −0.55 | 8.62 × 10−21 | −0.48 | 3.25 × 10−12 | −0.51 | 1.75 × 10−13 |
| STEAP2 | −0.59 | 7.59 × 10−25 | −0.52 | 2.07 × 10−18 | −0.68 | 7.19 × 10−27 | −0.63 | 1.24 × 10−21 |
| STEAP1 | −0.65 | 2.85 × 10−31 | −0.6 | 3.15 × 10−25 | −0.64 | 4.73 × 10−23 | −0.59 | 2.23 × 10−18 |
| TTC39C | −0.66 | 2.13 × 10−32 | −0.57 | 1.77 × 10−22 | −0.47 | 2.14 × 10−11 | −0.41 | 1.42 × 10−8 |
| ITGA7 | −0.59 | 7.59 × 10−25 | −0.51 | 1.17 × 10−17 | −0.46 | 3.61 × 10−11 | −0.41 | 1.67 × 10−8 |
| ZNF385D | −0.61 | 7.23 × 10−27 | −0.53 | 3.50 × 10−19 | −0.53 | 1.51 × 10−14 | −0.44 | 7.71 × 10−10 |
| DCLK1 | −0.61 | 7.23 × 10−27 | −0.53 | 3.50 × 10−19 | −0.5 | 4.62 × 10−13 | −0.48 | 6.39 × 10−12 |
| CFI | −0.61 | 7.23 × 10−27 | −0.51 | 1.17 × 10−17 | −0.62 | 2.30 × 10−21 | −0.53 | 2.43 × 10−14 |
| Downregulated in IPF | ||||||||
| ADRB2 | 0.67 | 1.24 × 10−33 | 0.6 | 3.15 × 10−25 | 0.69 | 5.22 × 10−28 | 0.63 | 5.79 × 10−22 |
| WNT7A | 0.64 | 4.43 × 10−30 | 0.6 | 3.15 × 10−25 | 0.59 | 8.33 × 10−19 | 0.56 | 1.33 × 10−16 |
| AFF3 | 0.61 | 7.23 × 10−27 | 0.52 | 2.07 × 10−18 | 0.65 | 6.70 × 10−24 | 0.51 | 1.10 ×10−13 |
| AGER | 0.7 | 1.50 × 10−37 | 0.64 | 2.81 × 10−29 | 0.7 | 1.36 × 10−28 | 0.66 | 1.80 × 10−24 |
| MATN3 | 0.61 | 7.23 × 10−27 | 0.52 | 2.07 × 10−18 | 0.25 | 0.0007 | 0.2 | 0.0105 |
| NINJ2 | 0.65 | 2.85 × 10−31 | 0.6 | 3.15 × 10−25 | 0.67 | 2.67 × 10−25 | 0.59 | 8.19 × 10−19 |
| FRY | 0.66 | 2.13 × 10−32 | 0.58 | 2.27 × 10−23 | 0.61 | 1.42 × 10−20 | 0.55 | 3.61 × 10−16 |
| ARHGAP31 | 0.64 | 4.43 × 10−30 | 0.55 | 8.62 × 10−21 | 0.57 | 1.24 × 10−17 | 0.5 | 1.03 × 10−12 |
| ARHGEF26 | 0.61 | 7.23 × 10−27 | 0.55 | 8.62 × 10−21 | 0.63 | 8.08 × 10−22 | 0.53 | 1.25 × 10−14 |
| ANKS1A | 0.6 | 7.42 × 10−26 | 0.5 | 6.32 × 10−17 | 0.2 | 0.0104 | 0.21 | 0.0061 |
| CCBE1 | 0.62 | 6.88 × 10−28 | 0.56 | 1.28 × 10−21 | 0.59 | 1.75 × 10−18 | 0.44 | 5.15 × 10−10 |
| NCKAP5 | 0.65 | 2.85 × 10−31 | 0.62 | 3.05 × 10−27 | 0.63 | 7.77 × 10−22 | 0.61 | 4.24 × 10−20 |
| EPB41L5 | 0.61 | 7.23 × 10−27 | 0.55 | 8.62 × 10−21 | 0.66 | 3.84 × 10−25 | 0.62 | 1.89 × 10−20 |
| ANXA3 | 0.66 | 2.13 × 10−32 | 0.59 | 2.91 × 10−24 | 0.65 | 5.04 × 10−24 | 0.61 | 3.39 × 10−20 |
| EMP2 | 0.68 | 7.32 × 10−35 | 0.63 | 3.15 × 10−28 | 0.3 | 4.57 × 10−5 | 0.21 | 0.0056 |
| RTKN2 | 0.66 | 2.13 × 10−32 | 0.61 | 3.18 × 10−26 | 0.7 | 8.68 × 10−29 | 0.66 | 1.72 × 10−24 |
| SPRYD7 | 0.66 | 2.13 × 10−32 | 0.61 | 3.18 × 10−26 | 0.52 | 3.90 × 10−14 | 0.47 | 2.00 × 10−11 |
| SLC44A2 | 0.58 | 6.83 × 10−24 | 0.53 | 3.50 × 10−19 | 0.56 | 7.94 × 10−17 | 0.55 | 5.85 × 10−16 |
| KCNMB4 | 0.62 | 6.88 × 10−28 | 0.53 | 3.50 × 10−19 | 0.53 | 5.99 × 10−15 | 0.48 | 8.93 × 10−12 |
| FAM167A | 0.67 | 1.24 × 10−33 | 0.59 | 2.91 × 10−24 | 0.6 | 1.84 × 10−19 | 0.49 | 1.45 × 10−12 |
| OLFML2A | 0.68 | 7.32 × 10−35 | 0.61 | 3.18 × 10−26 | 0.67 | 4.13 × 10−26 | 0.67 | 3.79 × 10−25 |
| ECHDC3 | 0.57 | 5.91 × 10−23 | 0.54 | 5.68 × 10−20 | 0.2 | 0.0086 | 0.092 | 0.2773 |
| SEMA3B | 0.66 | 2.13 × 10−32 | 0.62 | 3.05 × 10−27 | 0.48 | 5.53 × 10−12 | 0.52 | 5.33 × 10−14 |
| LAMA3 | 0.64 | 4.42 × 10−30 | 0.57 | 1.77 × 10−22 | 0.62 | 1.62 × 10−21 | 0.6 | 1.34 × 10−19 |
| PCYOX1 | 0.65 | 2.85 × 10−31 | 0.57 | 1.77 × 10−22 | 0.17 | 0.03216 | 0.17 | 0.03719 |
| RNF144B | 0.6 | 7.42 × 10−26 | 0.54 | 5.68 × 10−20 | 0.5 | 5.28 × 10−13 | 0.42 | 3.36 × 10−9 |
| HYAL1 | 0.58 | 6.83 × 10−24 | 0.53 | 3.50 × 10−19 | 0.54 | 9.30 × 10−16 | 0.47 | 1.55 × 10−11 |
| CDH13 | 0.62 | 6.88 × 10−28 | 0.59 | 2.91 × 10−24 | 0.44 | 6.94 × 10−10 | 0.41 | 1.45 × 10−8 |
| CTNND2 | 0.71 | 4.57 × 10−39 | 0.64 | 2.81 × 10−29 | 0.63 | 3.33 × 10−22 | 0.57 | 6.84 × 10−17 |
| DPP6 | 0.69 | 3.50 × 10−36 | 0.62 | 3.05 × 10−27 | 0.65 | 3.78 × 10−24 | 0.61 | 7.69 × 10−20 |
| GRIA1 | 0.67 | 1.24 × 10−33 | 0.62 | 3.05 × 10−27 | 0.67 | 6.26 × 10−26 | 0.65 | 1.80 × 10−23 |
| DENND3 | 0.64 | 4.43 × 10−30 | 0.57 | 1.77 × 10−22 | 0.46 | 4.49 × 10−11 | 0.38 | 1.25 × 10−7 |
| Hub Gene | Name | IPF1—Early logFC | IPF2—Progressive logFC | IPF3—Advanced logFC |
|---|---|---|---|---|
| Brown Module | ||||
| CDH3 | Cadherin 3 | 2.16 | 2.53 | 2.59 |
| CFI | Complement factor I | 1.01 | 0.7 | 0.95 |
| CHRDL2 | Chordin like 2 | 1.03 | 2.04 | 1.8 |
| COL10A1 | Collagen type X alpha 1 chain | 2.78 | 2.84 | 2.91 |
| CRABP2 | Cellular retinoic acid binding protein 2 | 2.45 | 3 | 3.25 |
| DCLK1 | Doublecortin-like kinase 1 | 0.86 | 0.94 | 1.04 |
| DOK5 | Docking protein 5 | 1.16 | 1.37 | 1.23 |
| FNDC4 | Fibronectin type III domain containing 4 | 1.04 | 1.19 | 1.29 |
| GPX8 | Glutathione peroxidase 8 (putative) | 1.08 | 0.98 | 1.16 |
| SCRG1 | Stimulator of chondrogenesis 1 | 1.23 | 1.29 | 1.48 |
| SPRR1A | Small proline rich protein 1A | 2.51 | 3.19 | 3.66 |
| STEAP1 | STEAP family member 1 | 1.3 | 1.57 | 1.74 |
| STEAP2 | STEAP2 metalloreductase | 1.03 | 1.35 | 1.34 |
| TDO2 | Tryptophan 2,3-dioxygenase | 2.34 | 2.92 | 2.8 |
| Blue Module | ||||
| AATK | Apoptosis associated tyrosine kinase | −0.94 | −0.96 | −0.99 |
| AFF3 | AF4/FMR2 family member 3 | −1.22 | −1.43 | −1.44 |
| ARHGEF26 | Rho guanine nucleotide exchange factor 26 | −0.91 | −1.14 | −1.27 |
| BTNL9 | Butyrophilin like 9 | −3.39 | −4 | −3.69 |
| C1orf115 | Chromosome 1 open reading frame 115 | −0.85 | −1.14 | −1.26 |
| CDH13 | Cadherin 13 | −0.69 | −0.84 | −0.88 |
| CRTAC1 | Cartilage acidic protein 1 | −1.57 | −2 | −2.33 |
| DENND3 | DENN domain containing 3 | −1.17 | −1.16 | −1.33 |
| EMP2 | Epithelial membrane protein 2 | −0.74 | −1.4 | −1.53 |
| EPB41L5 | Erythrocyte membrane protein band 4.1 like 5 | −0.81 | −1.04 | −1.1 |
| GALNT18 | Polypeptide N-acetylgalactosaminyltransferase 18 | −1.33 | −1.6 | −1.71 |
| GRIA1 | Glutamate ionotropic receptor AMPA type subunit 1 | −0.71 | −1.1 | −1.09 |
| HPCAL1 | Hippocalcin like 1 | −0.86 | −1.34 | −1.71 |
| ITLN2 | Intelectin 2 | −2.29 | −3.52 | −4.18 |
| KANK3 | KN motif and ankyrin repeat domains 3 | −0.92 | −1.08 | −1.16 |
| KCNMB4 | Potassium calcium-activated channel subfamily M regulatory beta subunit 4 | −1.02 | −1.36 | −1.41 |
| MATN3 | Matrilin 3 | −0.92 | −1.4 | −1.61 |
| MYRF | Myelin regulatory factor | −1.43 | −1.59 | −2.13 |
| NDRG4 | NDRG family member 4 | −1.59 | −2.24 | −2.61 |
| NPR1 | Natriuretic peptide receptor 1 | −0.79 | −1.22 | −1.27 |
| OLFML2A | Olfactomedin like 2A | −0.85 | −0.94 | −0.9 |
| PAPSS2 | 3′-phosphoadenosine 5′-phosphosulfate synthase 2 | −0.66 | −1.01 | −1.3 |
| PLLP | Plasmolipin | −0.86 | −1.49 | −1.7 |
| RNF144B | Ring finger protein 144B | −0.65 | −0.91 | −1.17 |
| RS1 | Retinoschisin 1 | −0.71 | −1.61 | −1.79 |
| SERTM1 | Serine rich and transmembrane domain containing 1 | −1.11 | −1.8 | −2.02 |
| STARD8 | StAR related lipid transfer domain containing 8 | −0.95 | −1.08 | −1.29 |
| STXBP6 | Syntaxin binding protein 6 | −0.76 | −1.71 | −2.17 |
| VIPR1 | Vasoactive intestinal peptide receptor 1 | −1.51 | −2.41 | −2.72 |
| VSIG10 | V-set and immunoglobulin domain containing 10 | −0.75 | −1.09 | −0.96 |
| Symbol | Gene ID | Description |
|---|---|---|
| Brown Module | ||
| CHEK2 | 11200 | Checkpoint kinase 2 |
| CRABP2 | 1382 | Cellular retinoic acid binding protein 2 |
| TSHZ2 | 128553 | Teashirt zinc finger homeobox 2 |
| COL7A1 | 1294 | Collagen type VII alpha 1 chain |
| SEC24D | 9871 | SEC24 homolog D, COPII coat complex component |
| REEP2 | 51308 | Receptor accessory protein 2 |
| COL10A1 | 1300 | Collagen type X alpha 1 chain |
| TTC39C | 125488 | Tetratricopeptide repeat domain 39C |
| STEAP1 | 26872 | STEAP family member 1 |
| EFNA4 | 1945 | Ephrin A4 |
| CLMP | 79827 | CXADR-like membrane protein |
| CDH3 | 1001 | Cadherin 3 |
| NPM3 | 10360 | Nucleophosmin/nucleoplasmin 3 |
| VWCE | 220001 | von Willebrand factor C and EGF domains |
| PLEKHA4 | 57664 | Pleckstrin homology domain containing A4 |
| CFI | 3426 | Complement factor I |
| TDO2 | 6999 | Tryptophan 2,3-dioxygenase |
| TMEM229A | 730130 | Transmembrane protein 229A |
| Blue Module | ||
| MATN3 | 4148 | Matrilin 3 |
| FRY | 10129 | FRY microtubule binding protein |
| CTNND2 | 1501 | Catenin delta 2 |
| RADIL | 55698 | Rap associating with DIL domain |
| ECHDC3 | 79746 | Enoyl-CoA hydratase domain containing 3 |
| KANK3 | 256949 | KN motif and ankyrin repeat domains 3 |
| SPRING1 | 79794 | SREBF pathway regulator in golgi 1 |
| ANKS1A | 23294 | Ankyrin repeat and sterile alpha motif domain containing 1A |
| SLC44A2 | 57153 | Solute carrier family 44 member 2 |
| TNS3 | 64759 | Tensin 3 |
| ST6GALNAC5 | 81849 | ST6 N-acetylgalactosaminide alpha-2,6-sialyltransferase 5 |
| C5orf38 | 153571 | Chromosome 5 open reading frame 38 |
| AFF3 | 3899 | AF4/FMR2 family member 3 |
| RNF182 | 221687 | Ring finger protein 182 |
| CRTAC1 | 55118 | Cartilage acidic protein 1 |
| PLLP | 51090 | Plasmolipin |
| NINJ2 | 4815 | Ninjurin 2 |
| KCNMB4 | 27345 | Potassium calcium-activated channel subfamily M regulatory beta subunit 4 |
| VSIG10 | 54621 | V-set and immunoglobulin domain containing 10 |
| PDZD2 | 23037 | PDZ domain containing 2 |
| BTNL9 | 153579 | Butyrophilin like 9 |
| VIPR1 | 7433 | Vasoactive intestinal peptide receptor 1 |
| DENND3 | 22898 | DENN domain containing 3 |
| FAM189A1 | 23359 | Family with sequence similarity 189 member A1 |
| Rank | Upregulated Hub Genes | Description | Downregulated Hub Genes | Description |
|---|---|---|---|---|
| 1 | CHEK2 | Checkpoint kinase 2 | CTNND2 | Catenin delta 2 |
| 2 | CDH3 | Cadherin 3 | CDH13 | Cadherin 13 |
| 3 | COL7A1 | Collagen type VII alpha 1 chain | SELENBP1 | Selenium binding protein 1 |
| 4 | CFI | Complement factor I | ARHGAP31 | Rho GTPase activating protein 31 |
| 5 | KCND3 | Potassium voltage-gated channel subfamily D member 3 | CAVIN2 | Caveolae associated protein 2 |
| 6 | CRABP2 | Cellular retinoic acid binding protein 2 | DENND3 | DENN domain containing 3 |
| 7 | ZNF469 | Zinc finger protein 469 | SLC1A1 | Solute carrier family 1 member 1 |
| 8 | STEAP2 | STEAP2 metalloreductase | EMP2 | Epithelial membrane protein 2 |
| 9 | TDO2 | Tryptophan 2,3-dioxygenase | PAPSS2 | 3′-phosphoadenosine 5′-phosphosulfate synthase 2 |
| 10 | SEC24D | SEC24 homolog D, COPII coat complex component | SLC44A2 | Solute carrier family 44 member 2 |
| 11 | CLMP | CXADR like membrane protein | N4BP1 | NEDD4 binding protein 1 |
| 12 | DCLK1 | Doublecortin like kinase 1 | GPM6A | Glycoprotein M6A |
| 13 | MAGED4B | MAGE family member D4B | NINJ2 | Ninjurin 2 |
| 14 | PDIA4 | Protein disulfide isomerase family A member 4 | RRAS | RAS related |
| 15 | ITGA7 | Integrin subunit alpha 7 | HPCAL1 | Hippocalcin like 1 |
| 16 | NPM3 | Nucleophosmin/nucleoplasmin 3 | AFF3 | AF4/FMR2 family member 3 |
| 17 | COL10A1 | Collagen type X alpha 1 chain | NDRG4 | NDRG family member 4 |
| 18 | GPX8 | Glutathione peroxidase 8 (putative) | VIPR1 | Vasoactive intestinal peptide receptor 1 |
| 19 | EFNA4 | Ephrin A4 | MYRF | Myelin regulatory factor |
| 20 | DOK5 | Docking protein 5 | RS1 | Retinoschisin 1 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ghandikota, S.; Sharma, M.; Ediga, H.H.; Madala, S.K.; Jegga, A.G. Consensus Gene Co-Expression Network Analysis Identifies Novel Genes Associated with Severity of Fibrotic Lung Disease. Int. J. Mol. Sci. 2022, 23, 5447. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23105447
Ghandikota S, Sharma M, Ediga HH, Madala SK, Jegga AG. Consensus Gene Co-Expression Network Analysis Identifies Novel Genes Associated with Severity of Fibrotic Lung Disease. International Journal of Molecular Sciences. 2022; 23(10):5447. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23105447
Chicago/Turabian StyleGhandikota, Sudhir, Mihika Sharma, Harshavardhana H. Ediga, Satish K. Madala, and Anil G. Jegga. 2022. "Consensus Gene Co-Expression Network Analysis Identifies Novel Genes Associated with Severity of Fibrotic Lung Disease" International Journal of Molecular Sciences 23, no. 10: 5447. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23105447