
Dynamics of Connexin 43 Down Modulation in Human Articular Chondrocytes Stimulated by Tumor Necrosis Factor Alpha

 , , ,
, , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
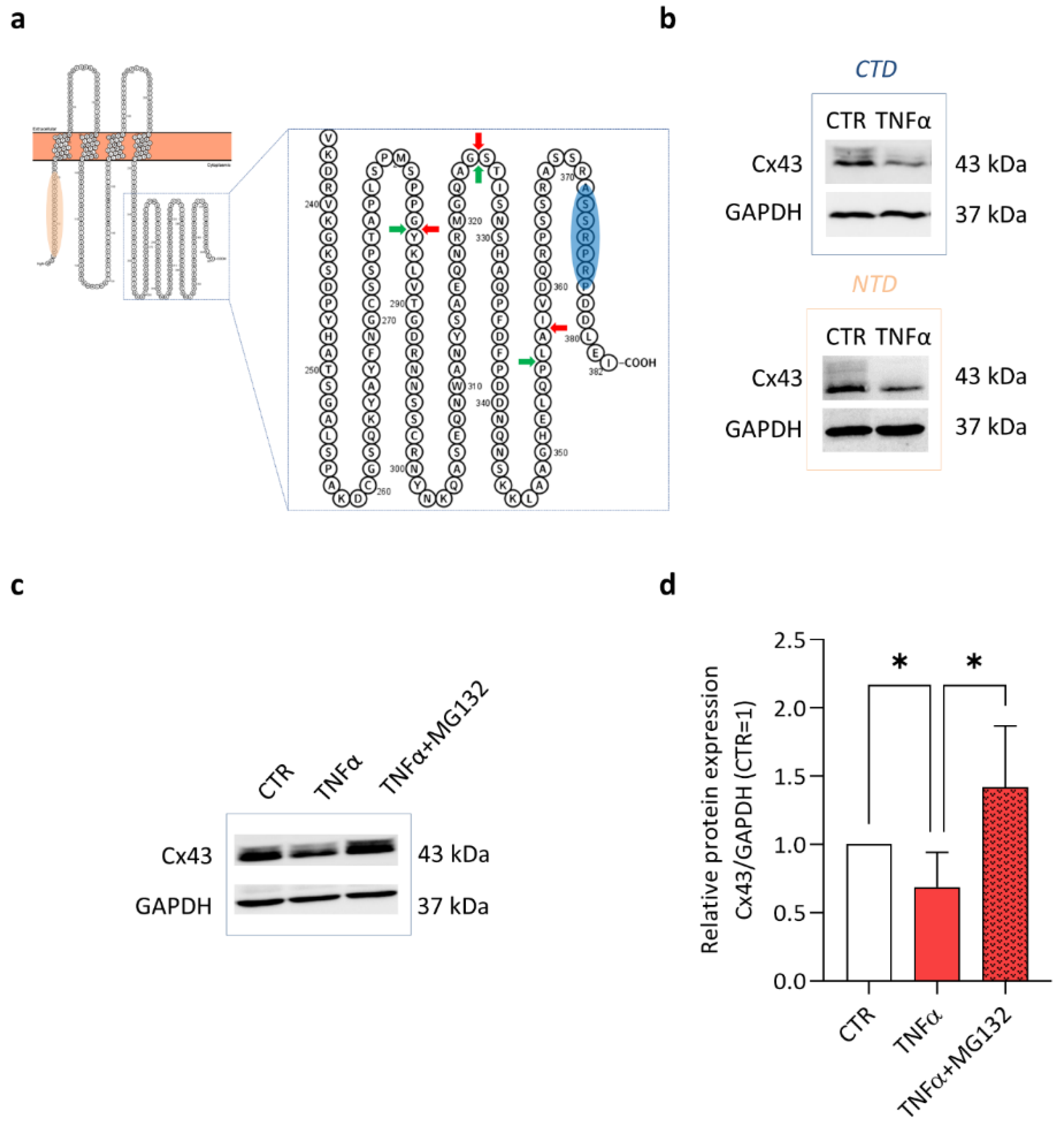
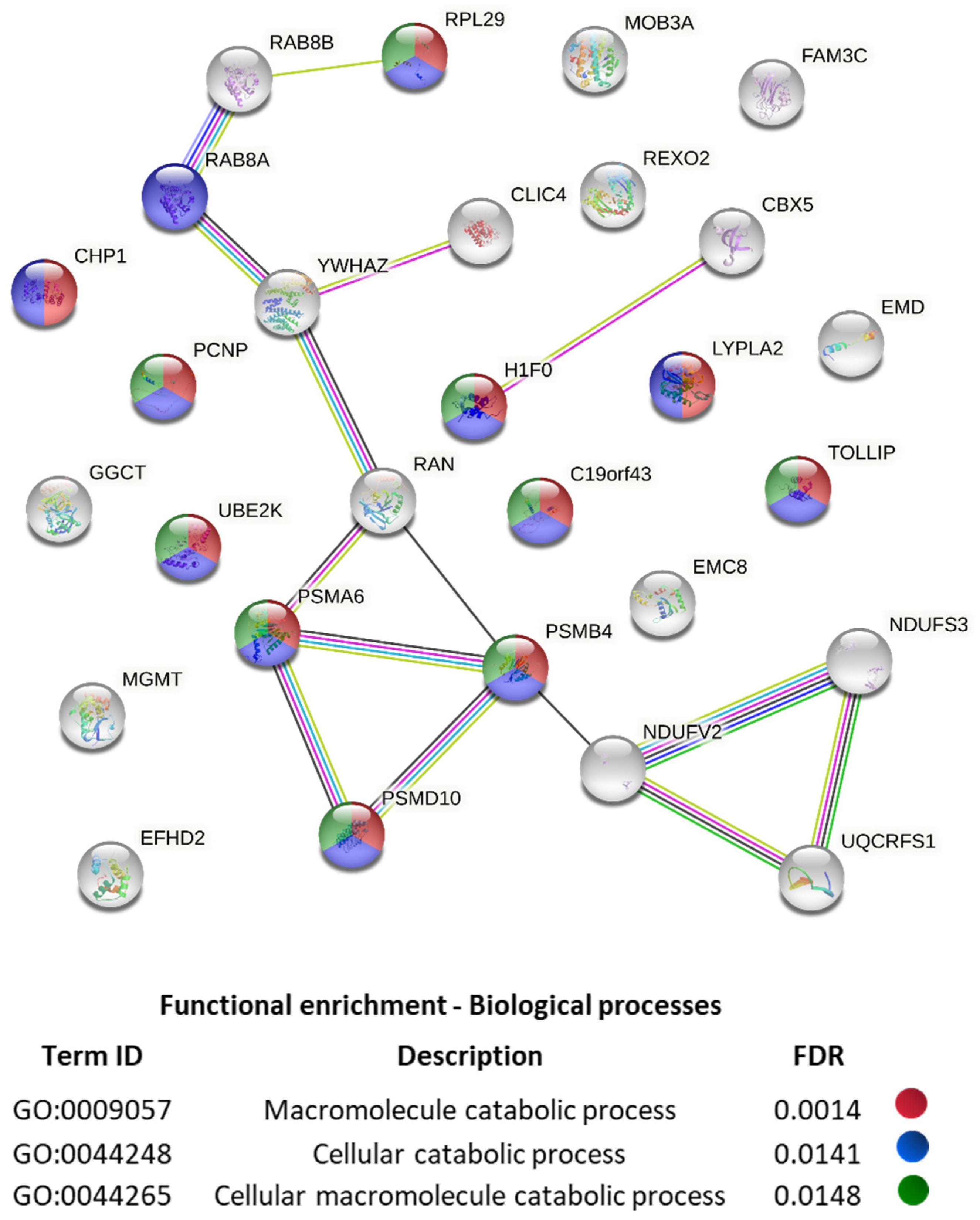
2. Results and Discussion
3. Materials and Methods
3.1. Cell Cultures
3.2. Treatments
3.3. Cell Lysates
3.4. Subcellular Fractionation
3.5. Western Blotting
3.6. Gene Expression Analysis
3.7. Scrape Loading Assay
3.8. Immunofluorescence
3.9. nLC-MS/MS Analysis for Protein Identification
3.10. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Donahue, H.J.; Qu, R.W.; Genetos, D.C. JoInt diseases: From connexins to gap junctions. Nat. Rev. Rheumatol. 2017, 14, 42–51. [Google Scholar] [CrossRef] [PubMed]
- Kotini, M.; Barriga, E.H.; Leslie, J.; Gentzel, M.; Rauschenberger, V.; Schambony, A.; Mayor, R. Gap junction protein Connexin-43 is a direct transcriptional regulator of N-cadherin in vivo. Nat. Commun. 2018, 9, 3846. [Google Scholar] [CrossRef] [PubMed]
- Leithe, E.; Mesnil, M.; Aasen, T. The Connexin 43 C-terminus: A tail of many tales. Biochim. Biophys. Acta Biomembr. 2018, 1860, 48–64. [Google Scholar] [CrossRef] [PubMed]
- Mayan, M.D.; Gago-Fuentes, R.; Carpintero-Fernandez, P.; Fernandez-Puente, P.; Filgueira-Fernandez, P.; Goyanes, N.; Valiunas, V.; Brink, P.R.; Goldberg, G.S.; Blanco, F.J. Articular chondrocyte network mediated by gap junctions: Role in metabolic cartilage homeostasis. Ann. Rheum. Dis. 2015, 74, 275–284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gago-Fuentes, R.; Bechberger, J.F.; Varela-Eirin, M.; Varela-Vazquez, A.; Acea, B.; Fonseca, E.; Naus, C.C.; Mayan, M.D. The C-terminal domain of connexin43 modulates cartilage structure via chondrocyte phenotypic changes. Oncotarget 2016, 7, 73055–73067. [Google Scholar] [CrossRef] [Green Version]
- Niada, S.; Giannasi, C.; Gomarasca, M.; Stanco, D.; Casati, S.; Brini, A.T. Adipose-derived stromal cell secretome reduces TNFalpha-induced hypertrophy and catabolic markers in primary human articular chondrocytes. Stem Cell Res. 2019, 38, 101463. [Google Scholar] [CrossRef]
- Giannasi, C.; Niada, S.; Magagnotti, C.; Ragni, E.; Andolfo, A.; Brini, A.T. Comparison of two ASC-derived therapeutics in an in vitro OA model: Secretome versus extracellular vesicles. Stem Cell Res. Ther. 2020, 11, 521. [Google Scholar] [CrossRef]
- Chisari, E.; Yaghmour, K.M.; Khan, W.S. The effects of TNF-alpha inhibition on cartilage: A systematic review of preclinical studies. Osteoarthr. Cartil. 2020, 28, 708–718. [Google Scholar] [CrossRef]
- van der Kraan, P.M.; Blaney Davidson, E.N.; Blom, A.; van den Berg, W.B. TGF-beta signaling in chondrocyte terminal differentiation and osteoarthritis: Modulation and integration of signaling pathways through receptor-Smads. Osteoarthr. Cartil. 2009, 17, 1539–1545. [Google Scholar] [CrossRef] [Green Version]
- Phull, A.R.; Eo, S.H.; Abbas, Q.; Ahmed, M.; Kim, S.J. Applications of Chondrocyte-Based Cartilage Engineering: An Overview. Biomed. Res. Int. 2016, 2016, 1879837. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, M.C.; Ha, C.W.; Elmallah, R.K.; Cherian, J.J.; Cho, J.J.; Kim, T.W.; Bin, S.I.; Mont, M.A. A placebo-controlled randomised trial to assess the effect of TGF-ß1-expressing chondrocytes in patients with arthritis of the knee. Bone Jt. J. 2015, 97-B, 924–932. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Zhou, C.; Zhang, D.; Zou, J.; Liu, W.; Cai, L.; Cui, Y.; Lai, W.; Xie, J. The involvement of the ERK-MAPK pathway in TGF-β1-mediated connexin43-gap junction formation in chondrocytes. Connect. Tissue Res. 2019, 60, 477–486. [Google Scholar] [CrossRef]
- Duan, M.; Liu, Y.; Guo, D.; Kan, S.; Niu, Z.; Pu, X.; Bai, M.; Zhang, D.; Du, W.; Xie, J. TGF-β2 increases cell-cell communication in chondrocytes via p-Smad3 signalling. Biochim. Biophys. Acta Mol. Cell Res. 2021, 1869, 119175. [Google Scholar] [CrossRef] [PubMed]
- Spreafico, A.; Chellini, F.; Frediani, B.; Bernardini, G.; Niccolini, S.; Serchi, T.; Collodel, G.; Paffetti, A.; Fossombroni, V.; Galeazzi, M.; et al. Biochemical investigation of the effects of human platelet releasates on human articular chondrocytes. J. Cell Biochem. 2009, 108, 1153–1165. [Google Scholar] [CrossRef]
- Mayan, M.D.; Carpintero-Fernandez, P.; Gago-Fuentes, R.; Martinez-de-Ilarduya, O.; Wang, H.Z.; Valiunas, V.; Brink, P.; Blanco, F.J. Human articular chondrocytes express multiple gap junction proteins: Differential expression of connexins in normal and osteoarthritic cartilage. Am. J. Pathol. 2013, 182, 1337–1346. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tacheau, C.; Laboureau, J.; Mauviel, A.; Verrecchia, F. TNF-alpha represses connexin43 expression in HaCat keratinocytes via activation of JNK signaling. J. Cell Physiol. 2008, 216, 438–444. [Google Scholar] [CrossRef]
- Kimura, K.; Nishida, T. Role of the ubiquitin-proteasome pathway in downregulation of the gap-junction protein Connexin43 by TNF-{alpha} in human corneal fibroblasts. Investig. Opthalmol. Vis. Sci. 2010, 51, 1943–1947. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kimura, K.; Orita, T.; Morishige, N.; Nishida, T.; Sonoda, K.H. Role of the JNK signaling pathway in downregulation of connexin43 by TNF-alpha in human corneal fibroblasts. Curr. Eye Res. 2013, 38, 926–932. [Google Scholar] [CrossRef] [PubMed]
- Morioka, N.; Zhang, F.F.; Nakamura, Y.; Kitamura, T.; Hisaoka-Nakashima, K.; Nakata, Y. Tumor necrosis factor-mediated downregulation of spinal astrocytic connexin43 leads to increased glutamatergic neurotransmission and neuropathic pain in mice. Brain Behav. Immun. 2015, 49, 293–310. [Google Scholar] [CrossRef]
- Zhang, F.F.; Morioka, N.; Kitamura, T.; Hisaoka-Nakashima, K.; Nakata, Y. Proinflammatory cytokines downregulate Connexin 43-gap junctions via the ubiquitin-proteasome system in rat spinal astrocytes. Biochem. Biophys. Res. Commun. 2015, 464, 1202–1208. [Google Scholar] [CrossRef]
- Kabátková, M.; Svobodová, J.; Pěnčíková, K.; Mohatad, D.S.; Šmerdová, L.; Kozubík, A.; Machala, M.; Vondráček, J. Interactive effects of inflammatory cytokine and abundant low-molecular-weight PAHs on inhibition of gap junctional intercellular communication, disruption of cell proliferation control, and the AhR-dependent transcription. Toxicol. Lett. 2015, 232, 113–121. [Google Scholar] [CrossRef] [PubMed]
- Moon, M.H.; Jeong, J.K.; Lee, Y.J.; Seol, J.W.; Jackson, C.J.; Park, S.Y. SIRT1, a class III histone deacetylase, regulates TNF-α-induced inflammation in human chondrocytes. Osteoarthr. Cartil. 2013, 21, 470–480. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Bock, M.; Wang, N.; Decrock, E.; Bultynck, G.; Leybaert, L. Intracellular Cleavage of the Cx43 C-Terminal Domain by Matrix-Metalloproteases: A Novel Contributor to Inflammation? Mediat. Inflamm. 2015, 2015, 257471. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szklarczyk, D.; Morris, J.H.; Cook, H.; Kuhn, M.; Wyder, S.; Simonovic, M.; Santos, A.; Doncheva, N.T.; Roth, A.; Bork, P.; et al. The STRING database in 2017: Quality-controlled protein-protein association networks, made broadly accessible. Nucleic Acids Res. 2017, 45, D362–D368. [Google Scholar] [CrossRef]
- Quan, R.; Huang, Z.; Yue, Z.; Xin, D.; Yang, D.; Pan, J.; Zhang, L. Effects of a proteasome inhibitor on the NF-κB signalling pathway in experimental osteoarthritis. Scand. J. Rheumatol. 2013, 42, 400–407. [Google Scholar] [CrossRef]
- Radwan, M.; Wilkinson, D.J.; Hui, W.; Destrument, A.P.; Charlton, S.H.; Barter, M.J.; Gibson, B.; Coulombe, J.; Gray, D.A.; Rowan, A.D.; et al. Protection against murine osteoarthritis by inhibition of the 26S proteasome and lysine-48 linked ubiquitination. Ann. Rheum. Dis. 2015, 74, 1580–1587. [Google Scholar] [CrossRef] [Green Version]
- Mathy-Hartert, M.; Hogge, L.; Sanchez, C.; Deby-Dupont, G.; Crielaard, J.M.; Henrotin, Y. Interleukin-1beta and interleukin-6 disturb the antioxidant enzyme system in bovine chondrocytes: A possible explanation for oxidative stress generation. Osteoarthr. Cartil. 2008, 16, 756–763. [Google Scholar] [CrossRef] [Green Version]
- Zhan, J.; Yan, Z.; Kong, X.; Liu, J.; Lin, Z.; Qi, W.; Wu, Y.; Lin, J.; Pan, X.; Xue, X. Lycopene inhibits IL-1β-induced inflammation in mouse chondrocytes and mediates murine osteoarthritis. J. Cell. Mol. Med. 2021, 25, 3573–3584. [Google Scholar] [CrossRef]
- Yao, J.; Ke, J.; Zhou, Z.; Tan, G.; Yin, Y.; Liu, M.; Chen, J.; Wu, W. Combination of HGF and IGF-1 promotes Connexin 43 expression and improves ventricular arrhythmia after myocardial infarction through activating the MAPK/ERK and MAPK/p38 signaling pathways in a rat model. Cardiovasc. Diagn. Ther. 2019, 9, 346–354. [Google Scholar] [CrossRef]
- Cheng, J.C.; Chang, H.M.; Fang, L.; Sun, Y.P.; Leung, P.C. TGF-β1 up-regulates connexin43 expression: A potential mechanism for human trophoblast cell differentiation. J. Cell. Physiol. 2015, 230, 1558–1566. [Google Scholar] [CrossRef]
- Li, X.; Guo, L.; Yang, X.; Wang, J.; Hou, Y.; Zhu, S.; Du, J.; Feng, J.; Xie, Y.; Zhuang, L.; et al. TGF-β1-induced connexin43 promotes scar formation via the Erk/MMP-1/collagen III pathway. J. Oral Rehabil. 2020, 47 (Suppl. 1), 99–106. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Liu, W.; Wei, J.; Cui, Y.; Zhang, D.; Xie, J. Transforming growth factor-β1-induced N-cadherin drives cell-cell communication through connexin43 in osteoblast lineage. Int. J. Oral Sci. 2021, 13, 15. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Duan, M.; Guo, D.; Kan, S.; Zhang, L.; Aili, M.; Zhang, D.; Du, W.; Xie, J. PDGF-AA promotes cell-to-cell communication in osteocytes through PI3K/Akt signaling pathway. Acta Biochim. Biophys. Sin. 2021, 53, 1640–1649. [Google Scholar] [CrossRef] [PubMed]
- Babica, P.; Sovadinová, I.; Upham, B.L. Scrape Loading/Dye Transfer Assay. Methods Mol. Biol. 2016, 1437, 133–144. [Google Scholar] [CrossRef]
- Schrobback, K.; Klein, T.J.; Woodfield, T.B. The importance of connexin hemichannels during chondroprogenitor cell differentiation in hydrogel versus microtissue culture models. Tissue Eng. Part A 2015, 21, 1785–1794. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.; Kong, X.; Zhuang, W.; Teng, B.; Yu, X.; Hua, S.; Wang, S.; Liang, F.; Ma, D.; Zhang, S.; et al. Dynamic changes in protein interaction between AKAP95 and Cx43 during cell cycle progression of A549 cells. Sci. Rep. 2016, 6, 21224. [Google Scholar] [CrossRef] [Green Version]
- Gago-Fuentes, R.; Fernández-Puente, P.; Megias, D.; Carpintero-Fernández, P.; Mateos, J.; Acea, B.; Fonseca, E.; Blanco, F.J.; Mayan, M.D. Proteomic Analysis of Connexin 43 Reveals Novel Interactors Related to Osteoarthritis. Mol. Cell. Proteom. 2015, 14, 1831–1845. [Google Scholar] [CrossRef] [Green Version]
- Swärd, P.; Frobell, R.; Englund, M.; Roos, H.; Struglics, A. Cartilage and bone markers and inflammatory cytokines are increased in synovial fluid in the acute phase of knee injury (hemarthrosis)—A cross-sectional analysis. Osteoarthr. Cartil. 2012, 20, 1302–1308. [Google Scholar] [CrossRef] [Green Version]
- Struglics, A.; Larsson, S.; Kumahashi, N.; Frobell, R.; Lohmander, L.S. Changes in Cytokines and Aggrecan ARGS Neoepitope in Synovial Fluid and Serum and in C-Terminal Crosslinking Telopeptide of Type II Collagen and N-Terminal Crosslinking Telopeptide of Type I Collagen in Urine Over Five Years After Anterior Cruciate Ligament Rupture: An Exploratory Analysis in the Knee Anterior Cruciate Ligament, Nonsurgical Versus Surgical Treatment Trial. Arthritis Rheumatol. 2015, 67, 1816–1825. [Google Scholar] [CrossRef]
- Roemer, F.W.; Englund, M.; Turkiewicz, A.; Struglics, A.; Guermazi, A.; Lohmander, L.S.; Larsson, S.; Frobell, R. Molecular and Structural Biomarkers of Inflammation at Two Years After Acute Anterior Cruciate Ligament Injury Do Not Predict Structural Knee Osteoarthritis at Five Years. Arthritis Rheumatol. 2019, 71, 238–243. [Google Scholar] [CrossRef] [Green Version]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Della Morte, E.; Niada, S.; Giannasi, C.; Zagra, L.; Brini, A.T. Dynamics of Connexin 43 Down Modulation in Human Articular Chondrocytes Stimulated by Tumor Necrosis Factor Alpha. Int. J. Mol. Sci. 2022, 23, 5575. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23105575
Della Morte E, Niada S, Giannasi C, Zagra L, Brini AT. Dynamics of Connexin 43 Down Modulation in Human Articular Chondrocytes Stimulated by Tumor Necrosis Factor Alpha. International Journal of Molecular Sciences. 2022; 23(10):5575. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23105575
Chicago/Turabian StyleDella Morte, Elena, Stefania Niada, Chiara Giannasi, Luigi Zagra, and Anna Teresa Brini. 2022. "Dynamics of Connexin 43 Down Modulation in Human Articular Chondrocytes Stimulated by Tumor Necrosis Factor Alpha" International Journal of Molecular Sciences 23, no. 10: 5575. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23105575