
Heme Oxygenase Modulation Drives Ferroptosis in TNBC Cells

 , , ,
, , ,  , , and
, , and 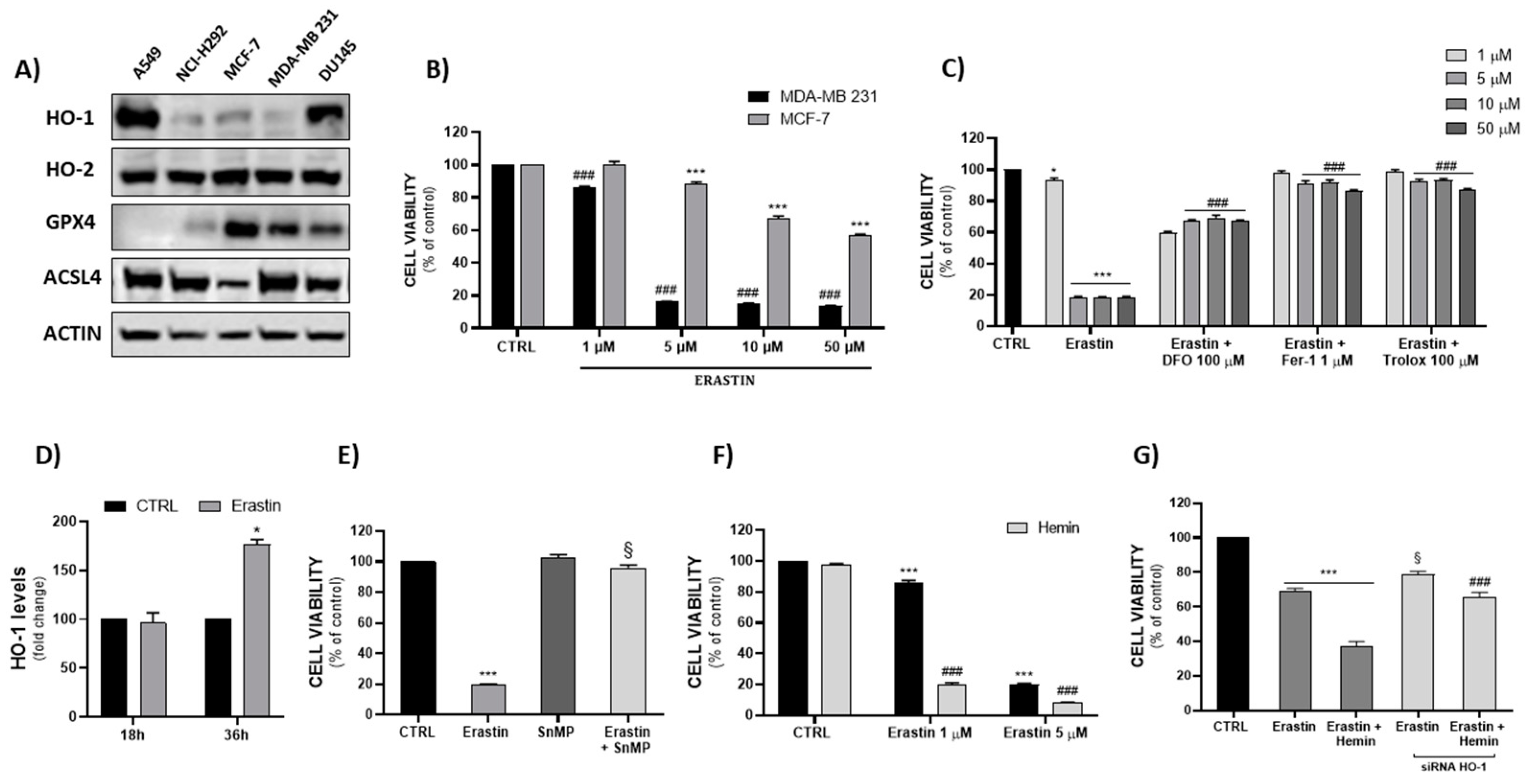{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Cellular Screening, Ferroptotic Model Validation and Implications of HO
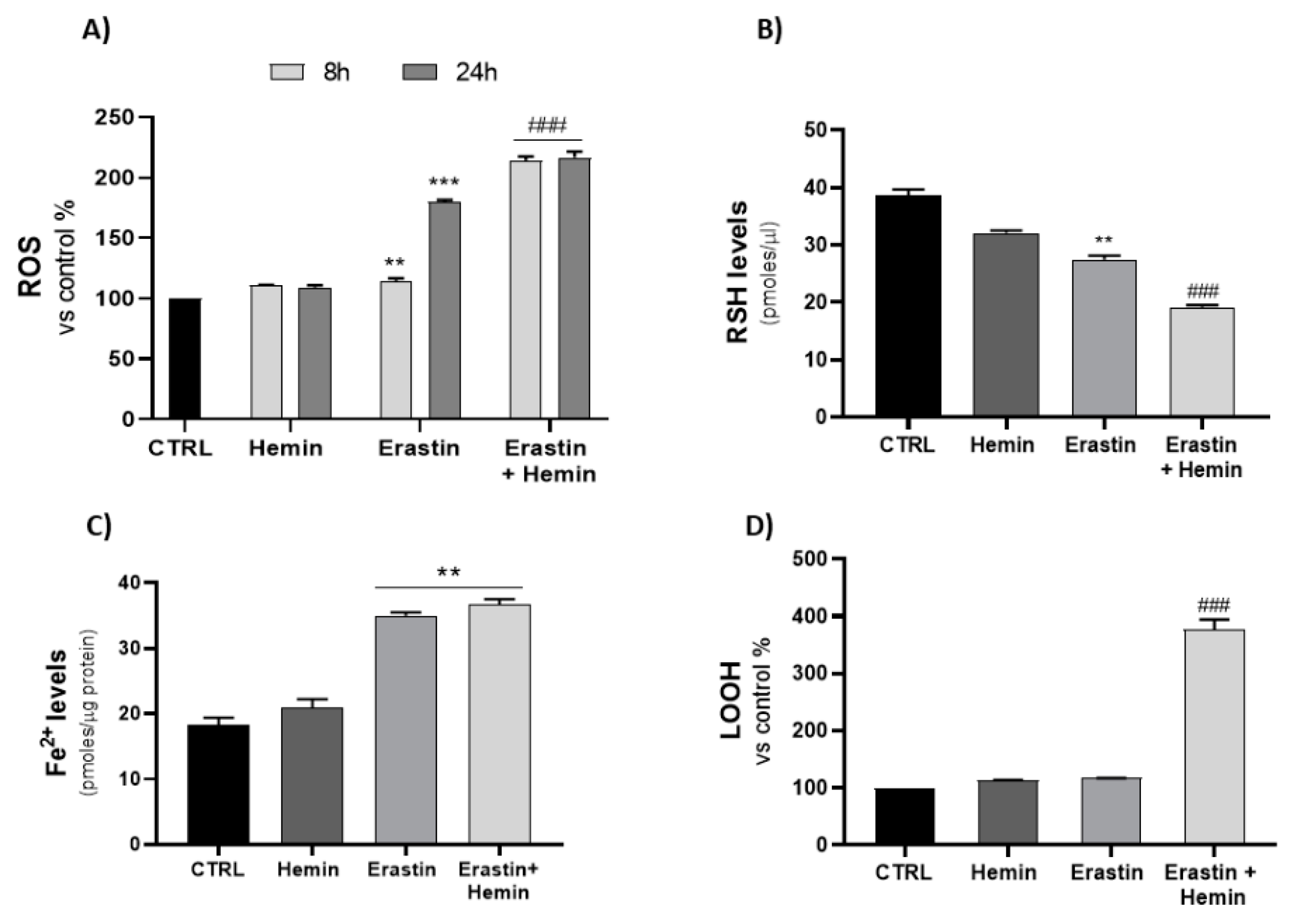
2.2. Effects of Hemin and Erastin on Ferroptosis-Associated Oxidative Stress
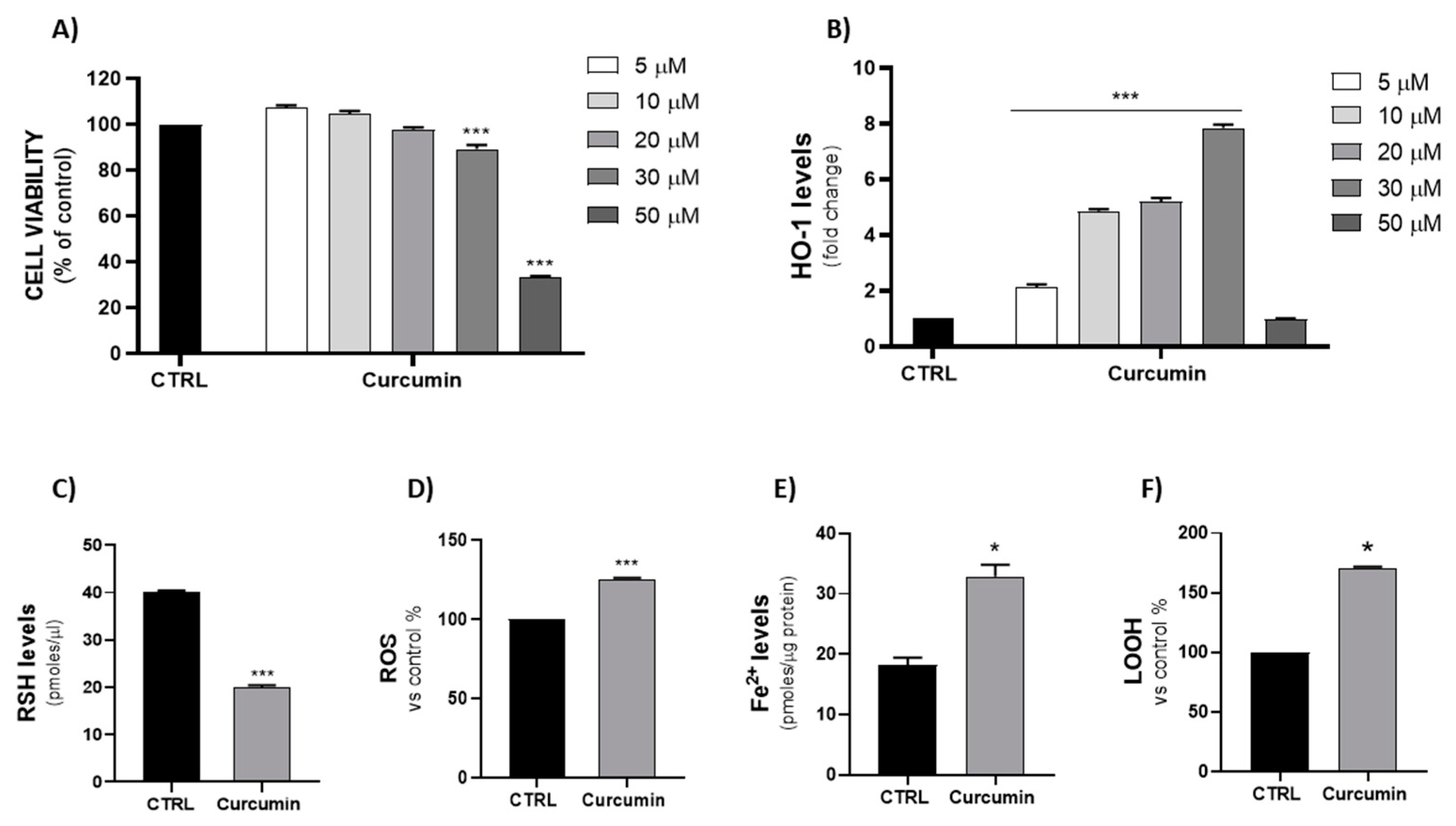
2.3. Effects of Curcumin-Mediated HO-1 Induction
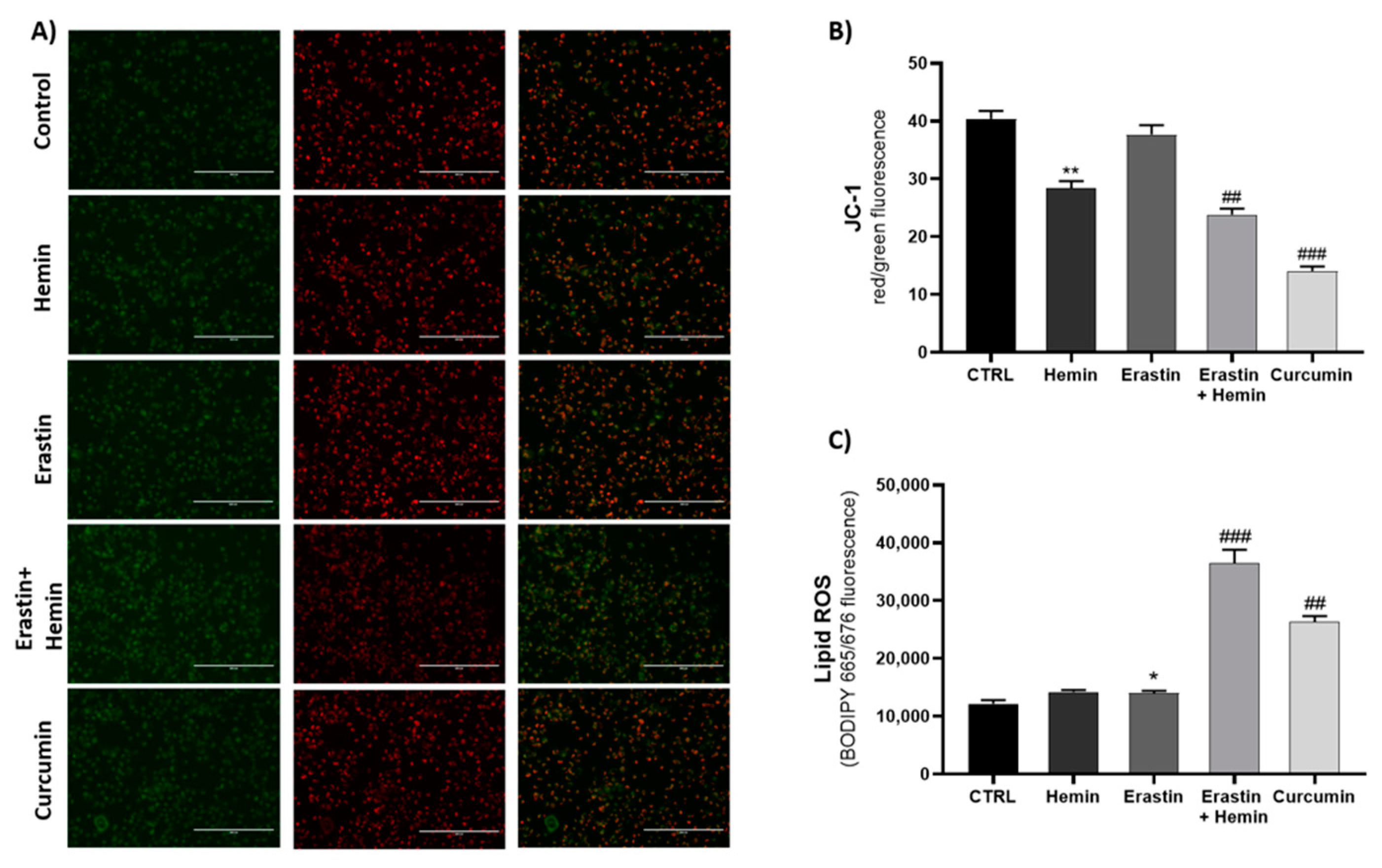
2.4. Effect of Ferroptosis Inducers on Mitochondrial Dysfunction and Lipid ROS Accumulation
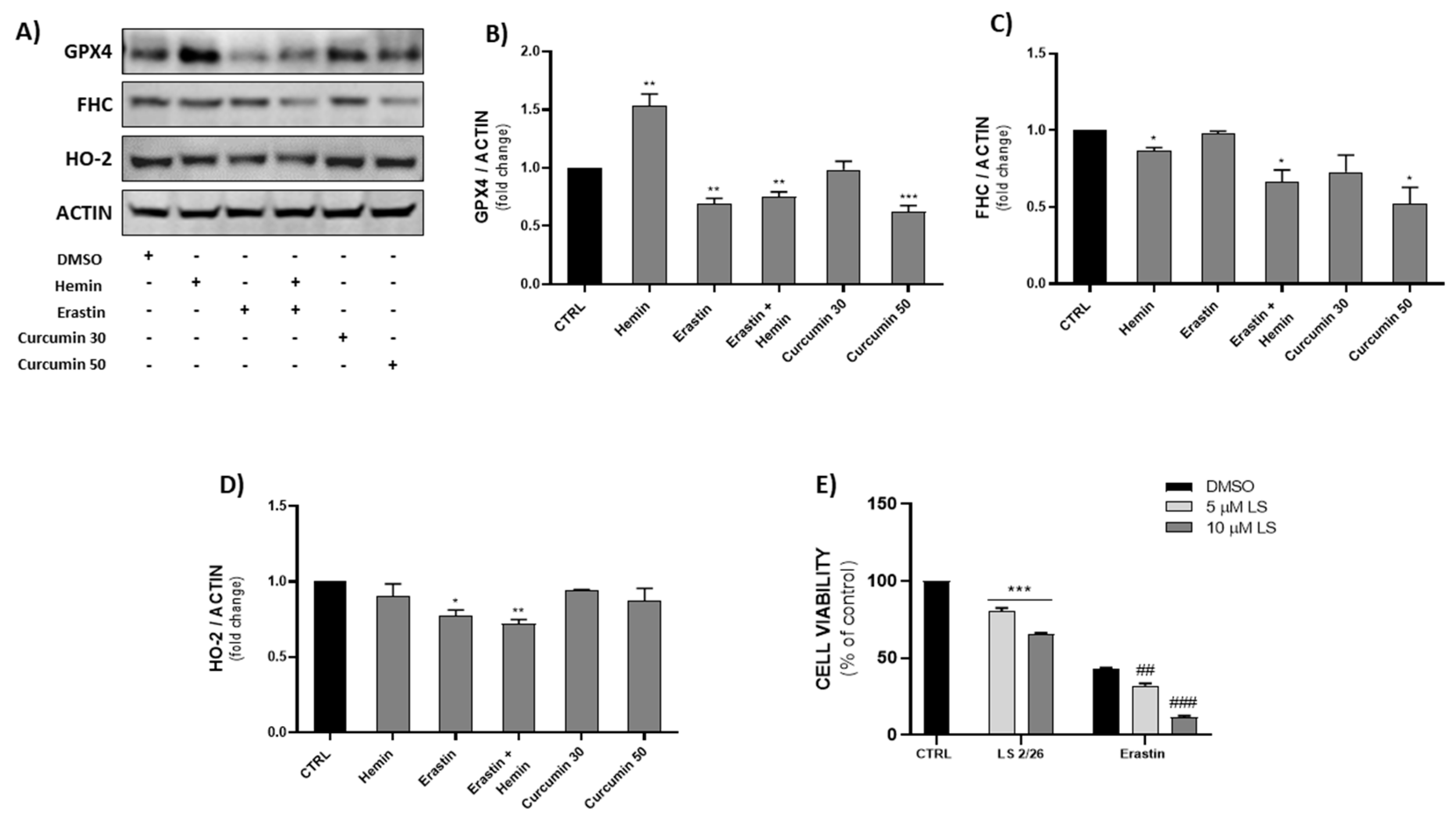
2.5. Effects of Ferroptosis Inducers on GPX4, FHC and HO-2 Protein Expression
2.6. Investigating HO-2′s Involvement in Erastin-Induced Ferroptosis
3. Discussion
4. Materials and Methods
4.1. Cell Culture and Viability Assay
4.2. Cell Transfection
4.3. Western Blot Analysis
4.4. Measurement of HO Enzymatic Activity
4.5. Determination of HO-1 Levels (ELISA)
4.6. Measurement of Intracellular Fe2+ Content
4.7. Measurement of Lipid Peroxidation
4.8. Measurement of Mitochondrial Membrane Potential
4.9. Determination of Thiol Groups
4.10. Measurement of ROS Levels
4.11. Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Dixon, S.J.; Lemberg, K.M.; Lamprecht, M.R.; Skouta, R.; Zaitsev, E.M.; Gleason, C.E.; Patel, D.N.; Bauer, A.J.; Cantley, A.M.; Yang, W.S.; et al. Ferroptosis: An iron-dependent form of nonapoptotic cell death. Cell 2012, 149, 1060–1072. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mou, Y.; Wang, J.; Wu, J.; He, D.; Zhang, C.; Duan, C.; Li, B. Ferroptosis, a new form of cell death: Opportunities and challenges in cancer. J. Hematol. Oncol. 2019, 12, 34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, Y.; Hou, W.; Song, X.; Yu, Y.; Huang, J.; Sun, X.; Kang, R.; Tang, D. Ferroptosis: Process and function. Cell Death Differ. 2016, 23, 369–379. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, Y.; Li, Y.; Zhang, R.; Wang, F.; Wang, T.; Jiao, Y. The Role of Erastin in Ferroptosis and Its Prospects in Cancer Therapy. OncoTargets Ther. 2020, 13, 5429–5441. [Google Scholar] [CrossRef] [PubMed]
- Jiang, L.; Kon, N.; Li, T.; Wang, S.J.; Su, T.; Hibshoosh, H.; Baer, R.; Gu, W. Ferroptosis as a p53-mediated activity during tumour suppression. Nature 2015, 520, 57–62. [Google Scholar] [CrossRef] [Green Version]
- Kim, M.J.; Yun, G.J.; Kim, S.E. Metabolic Regulation of Ferroptosis in Cancer. Biology 2021, 10, 83. [Google Scholar] [CrossRef]
- Liang, C.; Zhang, X.; Yang, M.; Dong, X. Recent Progress in Ferroptosis Inducers for Cancer Therapy. Adv. Mater. 2019, 31, e1904197. [Google Scholar] [CrossRef]
- Nie, J.; Lin, B.; Zhou, M.; Wu, L.; Zheng, T. Role of ferroptosis in hepatocellular carcinoma. J. Cancer Res. Clin. Onco.l. 2018, 144, 2329–2337. [Google Scholar] [CrossRef]
- Zou, J.; Wang, L.; Tang, H.; Liu, X.; Peng, F.; Peng, C. Ferroptosis in Non-Small Cell Lung Cancer: Progression and Therapeutic Potential on It. Int. J. Mol. Sci. 2021, 22, 13335. [Google Scholar] [CrossRef]
- Li, K.; Lin, C.; Li, M.; Xu, K.; He, Y.; Mao, Y.; Lu, L.; Geng, W.; Li, X.; Luo, Z.; et al. Multienzyme-like Reactivity Cooperatively Impairs Glutathione Peroxidase 4 and Ferroptosis Suppressor Protein 1 Pathways in Triple-Negative Breast Cancer for Sensitized Ferroptosis Therapy. ACS Nano 2022, 16, 2381–2398. [Google Scholar] [CrossRef]
- Jin, L.Y.; Gu, Y.L.; Zhu, Q.; Li, X.H.; Jiang, G.Q. The role of ferroptosis-related genes for overall survival prediction in breast cancer. J. Clin. Lab. Anal. 2021, 35, e24094. [Google Scholar] [CrossRef] [PubMed]
- Yao, X.; Xie, R.; Cao, Y.; Tang, J.; Men, Y.; Peng, H.; Yang, W. Simvastatin induced ferroptosis for triple-negative breast cancer therapy. J. Nanobiotechnology 2021, 19, 311. [Google Scholar] [CrossRef] [PubMed]
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Foulkes, W.D.; Smith, I.E.; Reis-Filho, J.S. Triple-negative breast cancer. N. Engl. J. Med. 2010, 363, 1938–1948. [Google Scholar] [CrossRef] [Green Version]
- Ovcaricek, T.; Frkovic, S.G.; Matos, E.; Mozina, B.; Borstnar, S. Triple negative breast cancer—prognostic factors and survival. Radiol. Oncol. 2011, 45, 46–52. [Google Scholar] [CrossRef] [Green Version]
- Dent, R.; Trudeau, M.; Pritchard, K.I.; Hanna, W.M.; Kahn, H.K.; Sawka, C.A.; Lickley, L.A.; Rawlinson, E.; Sun, P.; Narod, S.A. Triple-negative breast cancer: Clinical features and patterns of recurrence. Clin. Cancer Res. 2007, 13, 4429–4434. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Kong, X.; Wang, Z.; Xuan, L. Recent advances of transcriptomics and proteomics in triple-negative breast cancer prognosis assessment. J. Cell Mol. Med. 2022, 26, 1351–1362. [Google Scholar] [CrossRef]
- Gierach, G.L.; Burke, A.; Anderson, W.F. Epidemiology of triple negative breast cancers. Breast Dis. 2010, 32, 5–24. [Google Scholar] [CrossRef] [Green Version]
- Xiang, M.; Namani, A.; Wu, S.; Wang, X. Nrf2: Bane or blessing in cancer? J. Cancer Res. Clin. Oncol. 2014, 140, 1251–1259. [Google Scholar] [CrossRef]
- Consoli, V.; Sorrenti, V.; Grosso, S.; Vanella, L. Heme Oxygenase-1 Signaling and Redox Homeostasis in Physiopathological Conditions. Biomolecules 2021, 11, 589. [Google Scholar] [CrossRef]
- Trachootham, D.; Alexandre, J.; Huang, P. Targeting cancer cells by ROS-mediated mechanisms: A radical therapeutic approach? Nat. Rev. Drug Discov. 2009, 8, 579–591. [Google Scholar] [CrossRef] [PubMed]
- Na, H.K.; Surh, Y.J. Oncogenic potential of Nrf2 and its principal target protein heme oxygenase-1. Free Radic. Biol. Med. 2014, 67, 353–365. [Google Scholar] [CrossRef] [PubMed]
- Loboda, A.; Damulewicz, M.; Pyza, E.; Jozkowicz, A.; Dulak, J. Role of Nrf2/HO-1 system in development, oxidative stress response and diseases: An evolutionarily conserved mechanism. Cell. Mol. Life Sci. 2016, 73, 3221–3247. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ryter, S.W.; Tyrrell, R.M. The heme synthesis and degradation pathways: Role in oxidant sensitivity. Heme oxygenase has both pro- and antioxidant properties. Free Radic. Biol. Med. 2000, 28, 289–309. [Google Scholar] [CrossRef]
- Noh, S.J.; Bae, J.S.; Jamiyandorj, U.; Park, H.S.; Kwon, K.S.; Jung, S.H.; Youn, H.J.; Lee, H.; Park, B.H.; Chung, M.J.; et al. Expression of nerve growth factor and heme oxygenase-1 predict poor survival of breast carcinoma patients. BMC Cancer 2013, 13, 516. [Google Scholar] [CrossRef] [Green Version]
- Maines, M.D.; Abrahamsson, P.A. Expression of heme oxygenase-1 (HSP32) in human prostate: Normal, hyperplastic, and tumor tissue distribution. Urology 1996, 47, 727–733. [Google Scholar] [CrossRef]
- Yin, H.; Fang, J.; Liao, L.; Maeda, H.; Su, Q. Upregulation of heme oxygenase-1 in colorectal cancer patients with increased circulation carbon monoxide levels, potentially affects chemotherapeutic sensitivity. BMC Cancer 2014, 14, 436. [Google Scholar] [CrossRef] [Green Version]
- Tsuji, M.H.; Yanagawa, T.; Iwasa, S.; Tabuchi, K.; Onizawa, K.; Bannai, S.; Toyooka, H.; Yoshida, H. Heme oxygenase-1 expression in oral squamous cell carcinoma as involved in lymph node metastasis. Cancer Lett. 1999, 138, 53–59. [Google Scholar] [CrossRef]
- Fallica, A.N.; Sorrenti, V.; D’Amico, A.G.; Salerno, L.; Romeo, G.; Intagliata, S.; Consoli, V.; Floresta, G.; Rescifina, A.; D’Agata, V.; et al. Discovery of Novel Acetamide-Based Heme Oxygenase-1 Inhibitors with Potent. J. Med. Chem. 2021, 64, 13373–13393. [Google Scholar] [CrossRef]
- Salerno, L.; Vanella, L.; Sorrenti, V.; Consoli, V.; Ciaffaglione, V.; Fallica, A.N.; Canale, V.; Zajdel, P.; Pignatello, R.; Intagliata, S. Novel mutual prodrug of 5-fluorouracil and heme oxygenase-1 inhibitor (5-FU/HO-1 hybrid): Design and preliminary. J. Enzym. Inhib. Med. Chem. 2021, 36, 1378–1386. [Google Scholar] [CrossRef]
- Ciaffaglione, V.; Intagliata, S.; Pittalà, V.; Marrazzo, A.; Sorrenti, V.; Vanella, L.; Rescifina, A.; Floresta, G.; Sultan, A.; Greish, K.; et al. New Arylethanolimidazole Derivatives as HO-1 Inhibitors with Cytotoxicity against MCF-7 Breast Cancer Cells. Int. J. Mol. Sci. 2020, 21, 1923. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mucha, O.; Podkalicka, P.; Mikulski, M.; Barwacz, S.; Andrysiak, K.; Biela, A.; Mieczkowski, M.; Kachamakova-Trojanowska, N.; Ryszawy, D.; Białas, A.; et al. Development and characterization of a new inhibitor of heme oxygenase activity for cancer treatment. Arch. Biochem. Biophys. 2019, 671, 130–142. [Google Scholar] [CrossRef] [PubMed]
- Okunlola, F.O.; Soremekun, O.S.; Olotu, F.A.; Soliman, M.E.S. East to West not North-West: Structure-Based Mechanistic Resolution of 8-Hydroxyl Replacement and Resulting Effects on the Activities of Imidazole-Based Heme Oxygenase-1 Inhibitors. Protein. J. 2021, 40, 166–174. [Google Scholar] [CrossRef] [PubMed]
- Rahman, M.N.; Vukomanovic, D.; Vlahakis, J.Z.; Szarek, W.A.; Nakatsu, K.; Jia, Z. Structural insights into human heme oxygenase-1 inhibition by potent and selective azole-based compounds. J. R. Soc. Interface 2013, 10, 20120697. [Google Scholar] [CrossRef] [PubMed]
- Salerno, L.; Floresta, G.; Ciaffaglione, V.; Gentile, D.; Margani, F.; Turnaturi, R.; Rescifina, A.; Pittalà, V. Progress in the development of selective heme oxygenase-1 inhibitors and their potential therapeutic application. Eur. J. Med. Chem. 2019, 167, 439–453. [Google Scholar] [CrossRef]
- Tertil, M.; Golda, S.; Skrzypek, K.; Florczyk, U.; Weglarczyk, K.; Kotlinowski, J.; Maleszewska, M.; Czauderna, S.; Pichon, C.; Kieda, C.; et al. Nrf2-heme oxygenase-1 axis in mucoepidermoid carcinoma of the lung: Antitumoral effects associated with down-regulation of matrix metalloproteinases. Free Radic. Biol. Med. 2015, 89, 147–157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sorrenti, V.; D’Amico, A.G.; Barbagallo, I.; Consoli, V.; Grosso, S.; Vanella, L. Tin Mesoporphyrin Selectively Reduces Non-Small-Cell Lung Cancer Cell Line A549 Proliferation by Interfering with Heme Oxygenase and Glutathione Systems. Biomolecules 2021, 11, 917. [Google Scholar] [CrossRef]
- Kwon, M.Y.; Park, E.; Lee, S.J.; Chung, S.W. Heme oxygenase-1 accelerates erastin-induced ferroptotic cell death. Oncotarget 2015, 6, 24393–24403. [Google Scholar] [CrossRef] [Green Version]
- Lee, N.; Carlisle, A.E.; Peppers, A.; Park, S.J.; Doshi, M.B.; Spears, M.E.; Kim, D. xCT-Driven Expression of GPX4 Determines Sensitivity of Breast Cancer Cells to Ferroptosis Inducers. Antioxidants 2021, 10, 317. [Google Scholar] [CrossRef]
- Shibata, Y.; Yasui, H.; Higashikawa, K.; Miyamoto, N.; Kuge, Y. Erastin, a ferroptosis-inducing agent, sensitized cancer cells to X-ray irradiation via glutathione starvation in vitro and in vivo. PLoS ONE 2019, 14, e0225931. [Google Scholar] [CrossRef] [Green Version]
- Pan, X.; Lin, Z.; Jiang, D.; Yu, Y.; Yang, D.; Zhou, H.; Zhan, D.; Liu, S.; Peng, G.; Chen, Z.; et al. Erastin decreases radioresistance of NSCLC cells partially by inducing GPX4-mediated ferroptosis. Oncol. Lett. 2019, 17, 3001–3008. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jin, Z.L.; Gao, W.Y.; Liao, S.J.; Yu, T.; Shi, Q.; Yu, S.Z.; Cai, Y.F. Paeonol inhibits the progression of intracerebral haemorrhage by mediating the HOTAIR/UPF1/ACSL4 axis. ASN Neuro 2021, 13, 17590914211010647. [Google Scholar] [CrossRef] [PubMed]
- Salerno, L.; Pittalà, V.; Romeo, G.; Modica, M.N.; Marrazzo, A.; Siracusa, M.A.; Sorrenti, V.; Di Giacomo, C.; Vanella, L.; Parayath, N.N.; et al. Novel imidazole derivatives as heme oxygenase-1 (HO-1) and heme oxygenase-2 (HO-2) inhibitors and their cytotoxic activity in human-derived cancer cell lines. Eur. J. Med. Chem. 2015, 96, 162–172. [Google Scholar] [CrossRef] [PubMed]
- Yanagawa, T.; Omura, K.; Harada, H.; Nakaso, K.; Iwasa, S.; Koyama, Y.; Onizawa, K.; Yusa, H.; Yoshida, H. Heme oxygenase-1 expression predicts cervical lymph node metastasis of tongue squamous cell carcinomas. Oral. Oncol. 2004, 40, 21–27. [Google Scholar] [CrossRef] [Green Version]
- Han, S.; Lin, F.; Qi, Y.; Liu, C.; Zhou, L.; Xia, Y.; Chen, K.; Xing, J.; Liu, Z.; Yu, W.; et al. HO-1 Contributes to Luteolin-Triggered Ferroptosis in Clear Cell Renal Cell Carcinoma via Increasing the Labile Iron Pool and Promoting Lipid Peroxidation. Oxidative Med. Cell. Longev. 2022, 2022, 3846217. [Google Scholar] [CrossRef]
- Gueron, G.; De Siervi, A.; Ferrando, M.; Salierno, M.; De Luca, P.; Elguero, B.; Meiss, R.; Navone, N.; Vazquez, E.S. Critical role of endogenous heme oxygenase 1 as a tuner of the invasive potential of prostate cancer cells. Mol. Cancer Res. 2009, 7, 1745–1755. [Google Scholar] [CrossRef] [Green Version]
- Wagener, F.A.; da Silva, J.L.; Farley, T.; de Witte, T.; Kappas, A.; Abraham, N.G. Differential effects of heme oxygenase isoforms on heme mediation of endothelial intracellular adhesion molecule 1 expression. J. Pharmacol. Exp. Ther. 1999, 291, 416–423. [Google Scholar]
- Li, Z.; Wang, Y.; Man, R.Y.; Vanhoutte, P.M. Upregulation of heme oxygenase-1 potentiates EDH-type relaxations in the mesenteric artery of the spontaneously hypertensive rat. Am. J. Physiol. Heart Circ. Physiol. 2013, 305, H1471–H1483. [Google Scholar] [CrossRef] [Green Version]
- Tang, D.; Chen, X.; Kang, R.; Kroemer, G. Ferroptosis: Molecular mechanisms and health implications. Cell Res. 2021, 31, 107–125. [Google Scholar] [CrossRef]
- NaveenKumar, S.K.; Hemshekhar, M.; Kemparaju, K.; Girish, K.S. Hemin-induced platelet activation and ferroptosis is mediated through ROS-driven proteasomal activity and inflammasome activation: Protection by Melatonin. Biochim. Biophys. Acta Mol. Basis Dis. 2019, 1865, 2303–2316. [Google Scholar] [CrossRef]
- Fiorito, V.; Chiabrando, D.; Petrillo, S.; Bertino, F.; Tolosano, E. The Multifaceted Role of Heme in Cancer. Front. Oncol. 2019, 9, 1540. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seiwert, N.; Wecklein, S.; Demuth, P.; Hasselwander, S.; Kemper, T.A.; Schwerdtle, T.; Brunner, T.; Fahrer, J. Heme oxygenase 1 protects human colonocytes against ROS formation, oxidative DNA damage and cytotoxicity induced by heme iron, but not inorganic iron. Cell Death Dis. 2020, 11, 787. [Google Scholar] [CrossRef] [PubMed]
- Sorrenti, V.; Raffaele, M.; Vanella, L.; Acquaviva, R.; Salerno, L.; Pittalà, V.; Intagliata, S.; Di Giacomo, C. Protective Effects of Caffeic Acid Phenethyl Ester (CAPE) and Novel Cape Analogue as Inducers of Heme Oxygenase-1 in Streptozotocin-Induced Type 1 Diabetic Rats. Int. J. Mol. Sci. 2019, 20, 2441. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mohammed, F.; Rashid-Doubell, F.; Taha, S.; Cassidy, S.; Fredericks, S. Effects of curcumin complexes on MDA-MB-231 breast cancer cell proliferation. Int. J. Oncol. 2020, 57, 445–455. [Google Scholar] [CrossRef]
- Lima, C.F.; Pereira-Wilson, C.; Rattan, S.I. Curcumin induces heme oxygenase-1 in normal human skin fibroblasts through redox signaling: Relevance for anti-aging intervention. Mol. Nutr. Food Res. 2011, 55, 430–442. [Google Scholar] [CrossRef] [Green Version]
- Balogun, E.; Hoque, M.; Gong, P.; Killeen, E.; Green, C.J.; Foresti, R.; Alam, J.; Motterlini, R. Curcumin activates the haem oxygenase-1 gene via regulation of Nrf2 and the antioxidant-responsive element. Biochem. J. 2003, 371, 887–895. [Google Scholar] [CrossRef] [Green Version]
- Scapagnini, G.; Foresti, R.; Calabrese, V.; Giuffrida Stella, A.M.; Green, C.J.; Motterlini, R. Caffeic acid phenethyl ester and curcumin: A novel class of heme oxygenase-1 inducers. Mol. Pharmacol. 2002, 61, 554–561. [Google Scholar] [CrossRef]
- Cao, X.; Li, Y.; Wang, Y.; Yu, T.; Zhu, C.; Zhang, X.; Guan, J. Curcumin suppresses tumorigenesis by ferroptosis in breast cancer. PLoS ONE 2022, 17, e0261370. [Google Scholar] [CrossRef]
- Jang, H.Y.; Hong, O.Y.; Chung, E.Y.; Park, K.H.; Kim, J.S. Roles of JNK/Nrf2 Pathway on Hemin-Induced Heme Oxygenase-1 Activation in MCF-7 Human Breast Cancer Cells. Medicina 2020, 56, 268. [Google Scholar] [CrossRef]
- Georgiou-Siafis, S.K.; Tsiftsoglou, A.S. Activation of KEAP1/NRF2 stress signaling involved in the molecular basis of hemin-induced cytotoxicity in human pro-erythroid K562 cells. Biochem. Pharmacol. 2020, 175, 113900. [Google Scholar] [CrossRef]
- Song, X.; Long, D. Nrf2 and Ferroptosis: A New Research Direction for Neurodegenerative Diseases. Front. Neurosci. 2020, 14, 267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Imoto, S.; Sawamura, T.; Shibuya, Y.; Kono, M.; Ohbuchi, A.; Suzuki, T.; Mizokoshi, Y.; Saigo, K. Labile iron, ROS, and cell death are prominently induced by haemin, but not by non-transferrin-bound iron. Transfus. Apher. Sci. 2021, 61, 103319. [Google Scholar] [CrossRef]
- Liang, D.; Minikes, A.M.; Jiang, X. Ferroptosis at the intersection of lipid metabolism and cellular signaling. Mol. Cell 2022. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Yu, C.; Kang, R.; Tang, D. Iron Metabolism in Ferroptosis. Front. Cell Dev. Biol. 2020, 8, 590226. [Google Scholar] [CrossRef] [PubMed]
- Gao, M.; Monian, P.; Pan, Q.; Zhang, W.; Xiang, J.; Jiang, X. Ferroptosis is an autophagic cell death process. Cell Res. 2016, 26, 1021–1032. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gryzik, M.; Asperti, M.; Denardo, A.; Arosio, P.; Poli, M. NCOA4-mediated ferritinophagy promotes ferroptosis induced by erastin, but not by RSL3 in HeLa cells. Biochim. Biophys. Acta Mol. Cell Res. 2021, 1868, 118913. [Google Scholar] [CrossRef]
- Sodhi, K.; Inoue, K.; Gotlinger, K.H.; Canestraro, M.; Vanella, L.; Kim, D.H.; Manthati, V.L.; Koduru, S.R.; Falck, J.R.; Schwartzman, M.L.; et al. Epoxyeicosatrienoic acid agonist rescues the metabolic syndrome phenotype of HO-2-null mice. J. Pharmacol. Exp. Ther. 2009, 331, 906–916. [Google Scholar] [CrossRef] [Green Version]
- Intagliata, S.; Salerno, L.; Ciaffaglione, V.; Leonardi, C.; Fallica, A.N.; Carota, G.; Amata, E.; Marrazzo, A.; Pittalà, V.; Romeo, G. Heme Oxygenase-2 (HO-2) as a therapeutic target: Activators and inhibitors. Eur. J. Med. Chem. 2019, 183, 111703. [Google Scholar] [CrossRef]
- Vanella, L.; Di Giacomo, C.; Acquaviva, R.; Barbagallo, I.; Li Volti, G.; Cardile, V.; Abraham, N.G.; Sorrenti, V. Effects of ellagic Acid on angiogenic factors in prostate cancer cells. Cancers 2013, 5, 726–738. [Google Scholar] [CrossRef] [Green Version]
- Ge, C.; Zhang, S.; Mu, H.; Zheng, S.; Tan, Z.; Huang, X.; Xu, C.; Zou, J.; Zhu, Y.; Feng, D.; et al. Emerging Mechanisms and Disease Implications of Ferroptosis: Potential Applications of Natural Products. Front. Cell Dev. Biol. 2021, 9, 774957. [Google Scholar] [CrossRef]
- Xie, Y.; Song, X.; Sun, X.; Huang, J.; Zhong, M.; Lotze, M.T.; Zeh, H.J.; Kang, R.; Tang, D. Identification of baicalein as a ferroptosis inhibitor by natural product library screening. Biochem. Biophys. Res. Commun. 2016, 473, 775–780. [Google Scholar] [CrossRef] [PubMed]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Consoli, V.; Sorrenti, V.; Pittalà, V.; Greish, K.; D’Amico, A.G.; Romeo, G.; Intagliata, S.; Salerno, L.; Vanella, L. Heme Oxygenase Modulation Drives Ferroptosis in TNBC Cells. Int. J. Mol. Sci. 2022, 23, 5709. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23105709
Consoli V, Sorrenti V, Pittalà V, Greish K, D’Amico AG, Romeo G, Intagliata S, Salerno L, Vanella L. Heme Oxygenase Modulation Drives Ferroptosis in TNBC Cells. International Journal of Molecular Sciences. 2022; 23(10):5709. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23105709
Chicago/Turabian StyleConsoli, Valeria, Valeria Sorrenti, Valeria Pittalà, Khaled Greish, Agata Grazia D’Amico, Giuseppe Romeo, Sebastiano Intagliata, Loredana Salerno, and Luca Vanella. 2022. "Heme Oxygenase Modulation Drives Ferroptosis in TNBC Cells" International Journal of Molecular Sciences 23, no. 10: 5709. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23105709