
The Interplay of Endothelial P2Y Receptors in Cardiovascular Health: From Vascular Physiology to Pathology


Abstract
:1. Introduction: An Overview of P2Y Receptors in Endothelium
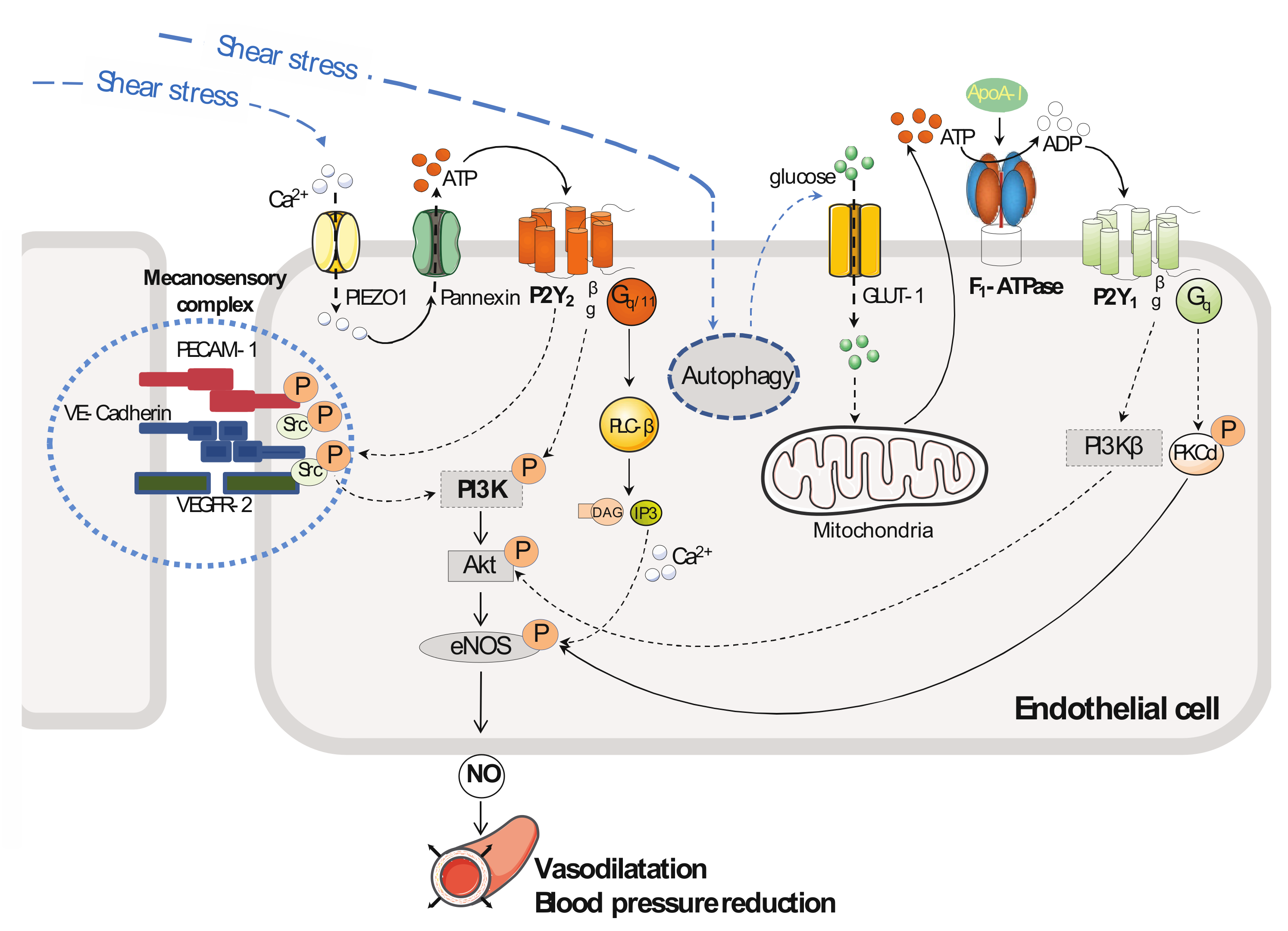
2. Vascular Tone and Blood Pressure

{kind=link}
{kind=link}
| P2Y-R Subtype (Agonist, G Protein) | Endothelial Cell Type or Tissue and Species | Function | References |
|---|---|---|---|
| P2Y1 (ADP, Gq) | BAECs | Endothelial NO release | [61] |
| HUVECs | Endothelial NO release | [8] | |
| Endothelial cell proliferation | [31] | ||
| Endothelial cell migration | [32] | ||
| Mouse lung endothelial cells | Increase in ICAM-1, VCAM-1, and P-Selectin, Leucocyte recruitment and transmigration | [7] | |
| Mouse mesenteric venules, femoral artery | Leucocyte recruitment (in vivo) * | [7] | |
| Entire aorta, aortic sinus | Development of atherosclerosis lesions * | [63] | |
| Mouse aorta | Endothelial NO release, vasorelaxation | [8,36] | |
| Mouse femoral artery | Vasodilatation NO-dependent (in vivo) | ||
| Human veins (umbilical and chorionic) | Vasorelaxation | [8] | |
| P2Y2 (ATP = UTP, Gq/11) | HUVECs | Endothelial NO release | [23,60] |
| Endothelial cell migration | [64] | ||
| Endothelial cell sprouting, vascular network formation | [65] | ||
| Cytoskeletal rearrangement, mechanical properties, cell alignment under shear stress | [66,67] | ||
| BAECs | Endothelial NO release | [23] | |
| HUAECs | Endothelial NOS activation under high laminar flow | [59] | |
| NF-κB activation, VCAM-1 expression under oscillatory flow | [59] | ||
| Mouse aorta | Vasorelaxation | [68,69] | |
| Mouse mesenteric artery | Vasorelaxation NO-dependent—blood pressure (in vivo) | [23] | |
| Mouse entire aorta, aortic sinus | Development of atherosclerosis lesions (in vivo) | [69] | |
| Mouse left common carotid artery | Intimal hyperplasia (in vivo) | [59] | |
| P2Y4 (UTP = Gq) | HUVECs | Endothelial cell migration * | [64] |
| P2Y12 (ADP = Gi) | BAECs | apoA-I and HDL transcytosis | [34] |
| HUVECs | Endothelial cell proliferation ** | [70] | |
| HMEC-1 | Reduction in the endothelial production of thrombospondin-1 ** | [71] |
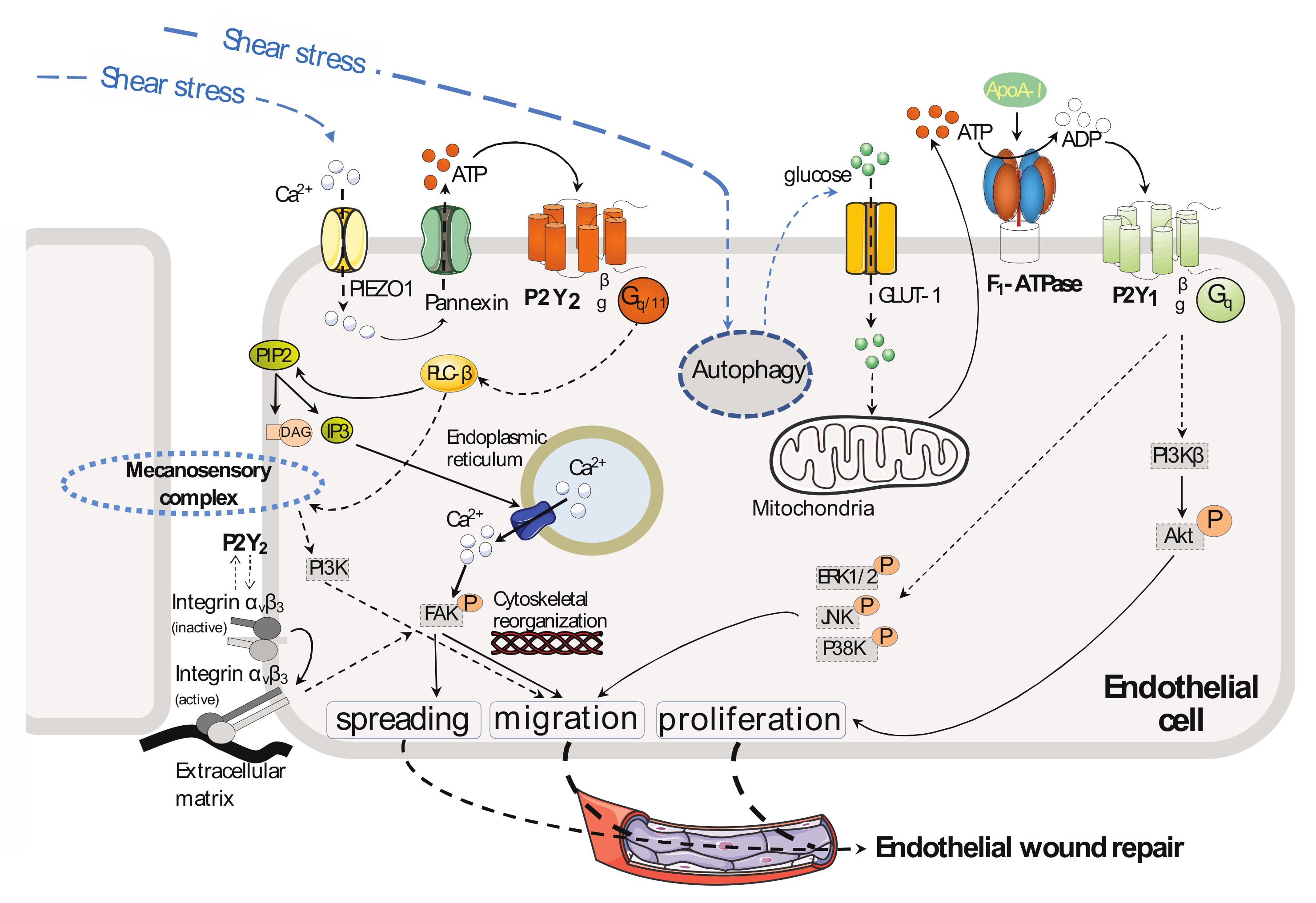
3. Endothelial Cell Migration and Proliferation
4. Vascular Inflammation and Atherogenesis
5. HDL Transcytosis in Endothelial Cells
6. Fluid Shear Stress-Induced Change in Endothelial Phenotype, Vascular Remodeling, and Atherogenesis
7. Discussion: Pharmacological Approaches Targeting Endothelial P2Y-R
8. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Heiss, C.; Rodriguez-Mateos, A.; Kelm, M. Central role of eNOS in the maintenance of endothelial homeostasis. Antioxid. Redox Signal. 2015, 22, 1230–1242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Douguet, D.; Patel, A.; Xu, A.; Vanhoutte, P.M.; Honoré, E. Piezo Ion Channels in Cardiovascular Mechanobiology. Trends Pharmacol. Sci. 2019, 40, 956–970. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koupenova, M.; Clancy, L.; Corkrey, H.A.; Freedman, J.E. Circulating Platelets as Mediators of Immunity, Inflammation, and Thrombosis. Circ. Res. 2018, 122, 337–351. [Google Scholar] [CrossRef] [PubMed]
- Tabas, I.; Lichtman, A.H. Monocyte-Macrophages and T Cells in Atherosclerosis. Immunity 2017, 47, 621–634. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Allen, S. Lipid/vascular wall interaction. Curr. Opin. Cardiol. 1998, 13, 439–446. [Google Scholar] [CrossRef] [PubMed]
- Cid, M.C.; Schnaper, H.W.; Kleinman, H.K. Estrogens and the Vascular Endothelium. Ann. N. Y. Acad. Sci. 2002, 966, 143–157. [Google Scholar] [CrossRef]
- Zerr, M.; Hechler, B.; Freund, M.; Magnenat, S.; Lanois, I.; Cazenave, J.P.; Léon, C.; Gachet, C.; Leon, C.; Gachet, C. Major contribution of the P2Y1 receptor in purinergic regulation of tnfα-induced vascular inflammation. Circulation 2011, 123, 2404–2413. [Google Scholar] [CrossRef] [Green Version]
- Cabou, C.; Honorato, P.; Briceño, L.; Ghezali, L.; Duparc, T.; León, M.; Combes, G.; Frayssinhes, L.; Fournel, A.; Abot, A.; et al. Pharmacological inhibition of the F1-ATPase/P2Y1 pathway suppresses the effect of apolipoprotein A1 on endothelial nitric oxide synthesis and vasorelaxation. Acta Physiol. 2019, 226, e13268. [Google Scholar] [CrossRef] [Green Version]
- Radojkovic, C.; Genoux, A.; Pons, V.; Combes, G.; De Jonge, H.; Champagne, E.; Rolland, C.; Perret, B.; Collet, X.; Tercé, F.; et al. Stimulation of Cell Surface F1-ATPase Activity by Apolipoprotein A-I Inhibits Endothelial Cell Apoptosis and Promotes Proliferation. Arter. Thromb Vasc Biol 2009, 29, 1125–1130. [Google Scholar] [CrossRef] [Green Version]
- Hermann, C.; Assmus, B.; Urbich, C.; Zeiher, A.M.; Dimmeler, S. Insulin-Mediated Stimulation of Protein Kinase Akt. Arterioscler. Thromb. Vasc. Biol. 2000, 20, 402–409. [Google Scholar] [CrossRef] [Green Version]
- Burnstock, G. Purinergic Signaling in the Cardiovascular System. Circ. Res. 2017, 120, 207–228. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Strassheim, D.; Verin, A.; Batori, R.; Nijmeh, H.; Burns, N.; Kovacs-Kasa, A.; Umapathy, N.S.; Kotamarthi, J.; Gokhale, Y.S.; Karoor, V.; et al. P2y purinergic receptors, endothelial dysfunction, and cardiovascular diseases. Int. J. Mol. Sci. 2020, 21, 6855. [Google Scholar] [CrossRef] [PubMed]
- North, R.A.; Surprenant, A. Pharmacology of Cloned P2X Receptors. Annu. Rev. Pharmacol. Toxicol. 2000, 40, 563–580. [Google Scholar] [CrossRef] [PubMed]
- von Kügelgen, I. Molecular pharmacology of P2Y receptor subtypes. Biochem. Pharmacol. 2021, 187, 114361. [Google Scholar] [CrossRef]
- Boeynaems, J.-M.; Communi, D.; Robaye, B. Overview of the pharmacology and physiological roles of P2Y receptors. Wiley Interdiscip. Rev. Membr. Transp. Signal. 2012, 1, 581–588. [Google Scholar] [CrossRef]
- Jacobson, K.A.; Delicado, E.G.; Gachet, C.; Kennedy, C.; von Kügelgen, I.; Li, B.; Miras-Portugal, M.T.; Novak, I.; Schöneberg, T.; Perez-Sen, R.; et al. Update of P2Y receptor pharmacology: IUPHAR Review 27. Br. J. Pharmacol. 2020, 177, 2413–2433. [Google Scholar] [CrossRef]
- Wang, L.; Karlsson, L.; Moses, S.; Hultgardh-Nilsson, A.; Andersson, M.; Borna, C.; Gudbjartsson, T.; Jern, S.; Erlinge, D. P2 receptor expression profiles in human vascular smooth muscle and endothelial cells. J. Cardiovasc. Pharmacol. 2002, 40, 841–853. [Google Scholar] [CrossRef]
- Yamamoto, K.; Sokabe, T.; Matsumoto, T.; Yoshimura, K.; Shibata, M.; Ohura, N.; Fukuda, T.; Sato, T.; Sekine, K.; Kato, S.; et al. Impaired flow-dependent control of vascular tone and remodeling in P2X4-deficient mice. Nat. Med. 2006, 12, 133–137. [Google Scholar] [CrossRef]
- Sathanoori, R.; Rosi, F.; Gu, B.J.; Wiley, J.S.; Müller, C.E.; Olde, B.; Erlinge, D. Shear stress modulates endothelial KLF2 through activation of P2X4. Purinergic Signal. 2015, 11, 139–153. [Google Scholar] [CrossRef] [Green Version]
- Ralevic, V. P2X Receptors in the Cardiovascular System and their Potential as Therapeutic Targets in Disease. Curr. Med. Chem. 2015, 22, 851–865. [Google Scholar] [CrossRef]
- Burnstock, G.; Knight, G.E. Cell culture: Complications due to mechanical release of ATP and activation of purinoceptors. Cell Tissue Res. 2017, 370, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bodin, P.; Bailey, D.; Burnstock, G. Increased flow-induced ATP release from isolated vascular endothelial cells but not smooth muscle cells. Br. J. Pharmacol. 1991, 103, 1203–1205. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, S.P.; Iring, A.; Strilic, B.; Juárez, J.A.; Kaur, H.; Troidl, K.; Tonack, S.; Burbiel, J.C.; Müller, C.E.; Fleming, I.; et al. P2Y2 and Gq/G11 control blood pressure by mediating endothelial mechanotransduction. J. Clin. Investig. 2015, 125, 3077–3086. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamamoto, K.; Furuya, K.; Nakamura, M.; Kobatake, E.; Sokabe, M.; Ando, J. Visualization of flow-induced ATP release and triggering of Ca2+ waves at caveolae in vascular endothelial cells. J. Cell Sci. 2011, 124, 3477–3483. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- John, K.; Barakat, A.I. Modulation of ATP/ADP Concentration at the Endothelial Surface by Shear Stress: Effect of Flow-Induced ATP Release. Ann. Biomed. Eng. 2001, 29, 740–751. [Google Scholar] [CrossRef]
- Gachet, C.; Hechler, B. The Platelet P2 Receptors in Thrombosis. Semin. Thromb. Hemost. 2005, 31, 162–167. [Google Scholar] [CrossRef]
- Goepfert, C.; Imai, M.; Brouard, S.; Csizmadia, E.; Kaczmarek, E.; Robson, S.C. CD39 modulates endothelial cell activation and apoptosis. Mol. Med. 2000, 6, 591–603. [Google Scholar] [CrossRef]
- Zimmermann, H.; Zebisch, M.; Sträter, N. Cellular function and molecular structure of ecto-nucleotidases. Purinergic Signal. 2012, 8, 437–502. [Google Scholar] [CrossRef] [Green Version]
- Cardouat, G.; Duparc, T.; Fried, S.; Perret, B.; Najib, S.; Martinez, L.O. Ectopic adenine nucleotide translocase activity controls extracellular ADP levels and regulates the F1-ATPase-mediated HDL endocytosis pathway on hepatocytes. Biochim. Biophys. Acta 2017, 1862, 832–841. [Google Scholar] [CrossRef]
- Fabre, A.C.S.; Vantourout, P.; Champagne, E.; Tercé, F.; Rolland, C.; Perret, B.; Collet, X.; Barbaras, R.; Martinez, L.O. Cell surface adenylate kinase activity regulates the F1-ATPase/P2Y13-mediated HDL endocytosis pathway on human hepatocytes. Cell. Mol. Life Sci. 2006, 63, 2829–2837. [Google Scholar] [CrossRef] [Green Version]
- Castaing-Berthou, A.; Malet, N.; Radojkovic, C.; Cabou, C.; Gayral, S.; Martinez, L.O.; Laffargue, M. PI3Kβ Plays a Key Role in Apolipoprotein A-I-Induced Endothelial Cell Proliferation Through Activation of the Ecto-F1-ATPase/P2Y1 Receptors. Cell. Physiol. Biochem. 2017, 42, 579–593. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shen, J.; DiCorleto, P.E. ADP stimulates human endothelial cell migration via P2Y1 nucleotide receptor-mediated mitogen-activated protein kinase pathways. Circ. Res. 2008, 102, 448–456. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiao, Z.; Yang, M.; Lv, Q.; Wang, W.; Deng, M.; Liu, X.; He, Q.; Chen, X.; Chen, M.; Fang, L.; et al. P2Y11 impairs cell proliferation by induction of cell cycle arrest and sensitizes endothelial cells to cisplatin-induced cell death. J. Cell. Biochem. 2011, 112, 2257–2265. [Google Scholar] [CrossRef]
- Cavelier, C.; Ohnsorg, P.M.; Rohrer, L.; von Eckardstein, A. The beta-Chain of Cell Surface F0F1 ATPase Modulates ApoA-I and HDL Transcytosis Through Aortic Endothelial Cells. Arter. Thromb Vasc Biol 2012, 32, 131–139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lyubchenko, T.; Woodward, H.; Veo, K.D.; Burns, N.; Nijmeh, H.; Liubchenko, G.A.; Stenmark, K.R.; Gerasimovskaya, E.V.; Ga, L.; Kr, S.; et al. P2Y1 and P2Y13 purinergic receptors mediate Ca 2ϩ signaling and proliferative responses in pulmonary artery vasa vasorum endothelial cells. Am. J. Physiol.—Cell Physiol. 2011, 300, 266–275. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guns, P.-J.D.F.; Korda, A.; Crauwels, H.M.; Van Assche, T.; Robaye, B.; Boeynaems, J.-M.; Bult, H. Pharmacological characterization of nucleotide P2Y receptors on endothelial cells of the mouse aorta. Br. J. Pharmacol. 2005, 146, 288–295. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bender, S.B.; Berwick, Z.C.; Laughlin, M.H.; Tune, J.D. Functional contribution of P2Y1 receptors to the control of coronary blood flow. J. Appl. Physiol. 2011, 111, 1744–1750. [Google Scholar] [CrossRef] [Green Version]
- Uehara, K.; Uehara, A. P2Y1, P2Y6, and P2Y12 receptors in rat splenic sinus endothelial cells: An immunohistochemical and ultrastructural study. Histochem. Cell Biol. 2011, 136, 557–567. [Google Scholar] [CrossRef]
- Yelovitch, S.; Barr, H.M.; Camden, J.; Weisman, G.A.; Shai, E.; Varon, D.; Fischer, B. Identification of a Promising Drug Candidate for the Treatment of Type 2 Diabetes Based on a P2Y 1 Receptor Agonist. J. Med. Chem. 2012, 55, 7623–7635. [Google Scholar] [CrossRef] [Green Version]
- Meister, J.; Le Duc, D.; Ricken, A.; Burkhardt, R.; Thiery, J.; Pfannkuche, H.; Polte, T.; Grosse, J.; Schöneberg, T.; Schulz, A. The G Protein-coupled Receptor P2Y14 Influences Insulin Release and Smooth Muscle Function in Mice. J. Biol. Chem. 2014, 289, 23353–23366. [Google Scholar] [CrossRef] [Green Version]
- Bertrand, G.; Chapal, J.; Loubatières-Mariani, M.M.; Roye, M. Evidence for two different P2-purinoceptors on β cell and pancreatic vascular bed. Br. J. Pharmacol. 1987, 91, 783–787. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Genoux, A.; Ruidavets, J.B.; Laffargue, M.; Combes, G.; Carrié, D.; Galinier, M.; Ferrières, J.; Perret, B.; Martinez, L.O. Serum Inhibitory Factor 1 Concentration Positively Correlates with HDL-Cholesterol Level and is a New Independent Determinant of Cardiovascular Heart Disease Risk. Circulation 2011, 124, A10590. [Google Scholar]
- Lichtenstein, L.; Serhan, N.; Espinosa-Delgado, S.; Fabre, A.; Annema, W.; Tietge, U.J.F.; Robaye, B.; Boyenaems, J.-M.; Laffargue, M.; Perret, B.; et al. Increased Atherosclerosis in P2Y13/Apolipoprotein E Double-Knockout Mice: Contribution of P2Y13 to Reverse Cholesterol Transport. Cardiovasc. Res. 2015, 106, 315–323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Serhan, N.; Cabou, C.; Verdier, C.; Lichtenstein, L.; Malet, N.; Perret, B.; Laffargue, M.; Martinez, L.O.L.O. Chronic pharmacological activation of P2Y13 receptor in mice decreases HDL-cholesterol level by increasing hepatic HDL uptake and bile acid secretion. Biochim. Biophys. Acta—Mol. Cell Biol. Lipids 2013, 1831, 719–725. [Google Scholar] [CrossRef] [PubMed]
- Lichtenstein, L.; Serhan, N.; Annema, W.; Combes, G.; Robaye, B.; Boeynaems, J.-M.M.; Perret, B.; Tietge, U.J.F.; Laffargue, M.; Martinez, L.O. Lack of P2Y13 in mice fed a high cholesterol diet results in decreased hepatic cholesterol content, biliary lipid secretion and reverse cholesterol transport. Nutr. Metab. 2013, 10, 67. [Google Scholar] [CrossRef] [Green Version]
- Martinez, L.O.L.O.; Cabou, C.; Pons, V.; Malaval, C. P2Y receptors in atherosclerosis: From lipid metabolism to vascular functions. Wiley Interdiscip. Rev. Membr. Transp. Signal. 2012, 1, 743–754. [Google Scholar] [CrossRef]
- Martinez, L.O.; Najib, S.; Perret, B.; Cabou, C.; Lichtenstein, L. Ecto-F1-ATPase/P2Y pathways in metabolic and vascular functions of high density lipoproteins. Atherosclerosis 2015, 238, 89–100. [Google Scholar] [CrossRef]
- Burnstock, G.; Gentile, D. The involvement of purinergic signalling in obesity. Purinergic Signal. 2018, 14, 97–108. [Google Scholar] [CrossRef] [Green Version]
- Fleming, I.; Fisslthaler, B.; Dixit, M.; Busse, R. Role of PECAM-1 in the shear-stress-induced activation of Akt and the endothelial nitric oxide synthase (eNOS) in endothelial cells. J. Cell Sci. 2005, 118, 4103–4111. [Google Scholar] [CrossRef] [Green Version]
- Lincoln, T.M.; Dey, N.; Sellak, H. Invited Review: cGMP-dependent protein kinase signaling mechanisms in smooth muscle: From the regulation of tone to gene expression. J. Appl. Physiol. 2001, 91, 1421–1430. [Google Scholar] [CrossRef]
- Tarbell, J.M.; Simon, S.I.; Curry, F.-R.E. Mechanosensing at the vascular interface. Annu. Rev. Biomed. Eng. 2014, 16, 505–532. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dimmeler, S.; Fleming, I.; Fisslthaler, B.; Hermann, C.; Busse, R.; Zeiher, A.M. Activation of nitric oxide synthase in endothelial cells by Akt-dependent phosphorylation. Nature 1999, 399, 601–605. [Google Scholar] [CrossRef] [PubMed]
- Fulton, D.; Gratton, J.-P.; McCabe, T.J.; Fontana, J.; Fujio, Y.; Walsh, K.; Franke, T.F.; Papapetropoulos, A.; Sessa, W.C. Regulation of endothelium-derived nitric oxide production by the protein kinase Akt. Nature 1999, 399, 597–601. [Google Scholar] [CrossRef] [PubMed]
- Fleming, I.; Busse, R. Molecular mechanisms involved in the regulation of the endothelial nitric oxide synthase. Am. J. Physiol. Integr. Comp. Physiol. 2003, 284, R1–R12. [Google Scholar] [CrossRef] [Green Version]
- Egorova, A.D.; van der Heiden, K.; Poelmann, R.E.; Hierck, B.P. Primary cilia as biomechanical sensors in regulating endothelial function. Differentiation 2012, 83, S56–S61. [Google Scholar] [CrossRef]
- Tzima, E.; Irani-Tehrani, M.; Kiosses, W.B.; Dejana, E.; Schultz, D.A.; Engelhardt, B.; Cao, G.; DeLisser, H.; Schwartz, M.A. A mechanosensory complex that mediates the endothelial cell response to fluid shear stress. Nature 2005, 437, 426–431. [Google Scholar] [CrossRef]
- Yamashiro, Y.; Yanagisawa, H. The molecular mechanism of mechanotransduction in vascular homeostasis and disease. Clin. Sci. 2020, 134, 2399–2418. [Google Scholar] [CrossRef]
- Wang, S.P.; Chennupati, R.; Kaur, H.; Iring, A.; Wettschureck, N.; Offermanns, S. Endothelial cation channel PIEZO1 controls blood pressure by mediating flow-induced ATP release. J. Clin. Investig. 2016, 126, 4527–4536. [Google Scholar] [CrossRef]
- Albarrán-Juárez, J.; Iring, A.; Wang, S.P.; Joseph, S.; Grimm, M.; Strilic, B.; Wettschureck, N.; Althoff, T.F.; Offermanns, S. Piezo1 and Gq/G11 promote endothelial inflammation depending on flow pattern and integrin activation. J. Exp. Med. 2018, 215, 2655–2672. [Google Scholar] [CrossRef] [Green Version]
- Raqeeb, A.; Sheng, J.; Ao, N.; Braun, A.P. Purinergic P2Y2 receptors mediate rapid Ca2+ mobilization, membrane hyperpolarization and nitric oxide production in human vascular endothelial cells. Cell Calcium 2011, 49, 240–248. [Google Scholar] [CrossRef]
- Bharath, L.P.; Cho, J.M.; Park, S.-K.; Ruan, T.; Li, Y.; Mueller, R.; Bean, T.; Reese, V.; Richardson, R.S.; Cai, J.; et al. Endothelial Cell Autophagy Maintains Shear Stress–Induced Nitric Oxide Generation via Glycolysis-Dependent Purinergic Signaling to Endothelial Nitric Oxide Synthase. Arterioscler. Thromb. Vasc. Biol. 2017, 37, 1646–1656. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- da Silva, C.G.; Specht, A.; Wegiel, B.; Ferran, C.; Kaczmarek, E. Mechanism of Purinergic Activation of Endothelial Nitric Oxide Synthase in Endothelial Cells. Circulation 2009, 119, 871–879. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hechler, B.; Freund, M.; Ravanat, C.; Magnenat, S.; Cazenave, J.P.; Gachet, C. Reduced atherosclerotic lesions in P2Y1/Apolipoprotein E double-knockout mice: The contribution of non-hematopoietic-derived P2Y 1 receptors. Circulation 2008, 118, 754–763. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaczmarek, E.; Erb, L.; Koziak, K.; Jarzyna, R.; Wink, M.; Guckelberger, O.; Blusztajn, K.; Trinkaus-Randall, V.; Weisman, G.; Robson, S. Modulation of endothelial cell migration by extracellular nucleotides. Thromb. Haemost. 2005, 93, 735–742. [Google Scholar]
- Mühleder, S.; Fuchs, C.; Basílio, J.; Szwarc, D.; Pill, K.; Labuda, K.; Slezak, P.; Siehs, C.; Pröll, J.; Priglinger, E.; et al. Purinergic P2Y2 receptors modulate endothelial sprouting. Cell. Mol. Life Sci. 2020, 77, 885–901. [Google Scholar] [CrossRef]
- Sathanoori, R.; Bryl-Gorecka, P.; Müller, C.E.; Erb, L.; Weisman, G.A.; Olde, B.; Erlinge, D. P2Y2 receptor modulates shear stress-induced cell alignment and actin stress fibers in human umbilical vein endothelial cells. Cell. Mol. Life Sci. 2017, 74, 731–746. [Google Scholar] [CrossRef] [Green Version]
- Gil-Redondo, J.C.; Iturri, J.; Ortega, F.; Pérez-Sen, R.; Weber, A.; Miras-Portugal, M.T.; Toca-Herrera, J.L.; Delicado, E.G. Nucleotides-induced changes in the mechanical properties of living endothelial cells and astrocytes, analyzed by atomic force microscopy. Int. J. Mol. Sci. 2021, 22, 624. [Google Scholar] [CrossRef]
- Guns, P.-J.J.D.F.; Van Assche, T.; Fransen, P.; Robaye, B.; Boeynaems, J.M.; Bult, H. Endothelium-dependent relaxation evoked by ATP and UTP in the aorta of P2Y2-deficient mice. Br. J. Pharmacol. 2006, 147, 569–574. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.; Qian, S.; Hoggatt, A.; Tang, H.; Hacker, T.A.; Obukhov, A.G.; Herring, P.B.; Seye, C.I. Endothelial Cell-Specific Deletion of P2Y2 Receptor Promotes Plaque Stability in Atherosclerosis-Susceptible ApoE-Null Mice. Arterioscler. Thromb. Vasc. Biol. 2017, 37, 75–83. [Google Scholar] [CrossRef] [Green Version]
- Korybalska, K.; Rutkowski, R.; Luczak, J.; Czepulis, N.; Karpinski, K.; Witowski, J. The role of purinergic P2Y12 receptor blockers on the angiogenic properties of endothelial cells: An in vitro study. J. Physiol. Pharmacol. 2018, 69. [Google Scholar] [CrossRef]
- Gdula, A.M.; Swiatkowska, M. A2A receptor agonists and P2Y12 receptor antagonists modulate expression of thrombospondin-1 (TSP-1) and its secretion from Human Microvascular Endothelial Cells (HMEC-1). Microvasc. Res. 2021, 138, 104218. [Google Scholar] [CrossRef] [PubMed]
- Vantourout, P.; Radojkovic, C.; Lichtenstein, L.; Pons, V.; Champagne, E.; Martinez, L.O. Ecto-F1-ATPase: A moonlighting protein complex and an unexpected apoA-I receptor. World J. Gastroenterol. 2010, 16, 5925–5935. [Google Scholar] [PubMed]
- Martinez, L.O.; Jacquet, S.; Esteve, J.P.; Rolland, C.; Cabezon, E.; Champagne, E.; Pineau, T.; Georgeaud, V.; Walker, J.E.; Terce, F.; et al. Ectopic beta-chain of ATP synthase is an apolipoprotein A-I receptor in hepatic HDL endocytosis. Nature 2003, 421, 75–79. [Google Scholar] [CrossRef] [PubMed]
- Dol-Gleizes, F.; Marés, A.M.; Savi, P.; Herbert, J.M. Relaxant effect of 2-methyl-thio-adenosine diphosphate on rat thoracic aorta: Effect of clopidogrel. Eur. J. Pharmacol. 1999, 367, 247–253. [Google Scholar] [CrossRef]
- Kauffenstein, G.; Fürstenau, C.R.; D’Orléans-Juste, P.; Sévigny, J. The ecto-nucleotidase NTPDase1 differentially regulates P2Y1 and P2Y2 receptor-dependent vasorelaxation. Br. J. Pharmacol. 2010, 159, 576–585. [Google Scholar] [CrossRef] [Green Version]
- Mercier, N.; Kiviniemi, T.O.; Saraste, A.; Miiluniemi, M.; Silvola, J.; Jalkanen, S.; Yegutkin, G.G. Impaired ATP-Induced Coronary Blood Flow and Diminished Aortic NTPDase Activity Precede Lesion Formation in Apolipoprotein E–Deficient Mice. Am. J. Pathol. 2012, 180, 419–428. [Google Scholar] [CrossRef]
- Robson, S.C.; Kaczmarek, E.; Siegel, J.B.; Candinas, D.; Koziak, K.; Millan, M.; Hancock, W.W.; Bach, F.H. Loss of ATP Diphosphohydrolase Activity with Endothelial Cell Activation. J. Exp. Med. 1997, 185, 153–164. [Google Scholar] [CrossRef]
- Eltzschig, H.K.; Ibla, J.C.; Furuta, G.T.; Leonard, M.O.; Jacobson, K.A.; Enjyoji, K.; Robson, S.C.; Colgan, S.P. Coordinated Adenine Nucleotide Phosphohydrolysis and Nucleoside Signaling in Posthypoxic Endothelium. J. Exp. Med. 2003, 198, 783–796. [Google Scholar] [CrossRef] [Green Version]
- Kauffenstein, G.; Drouin, A.; Thorin-Trescases, N.; Bachelard, H.; Robaye, B.; D’Orleans-Juste, P.; Marceau, F.; Thorin, E.; Sevigny, J. NTPDase1 (CD39) controls nucleotide-dependent vasoconstriction in mouse. Cardiovasc. Res. 2010, 85, 204–213. [Google Scholar] [CrossRef] [Green Version]
- Roy, C.; Tabiasco, J.; Caillon, A.; Delneste, Y.; Merot, J.; Favre, J.; Guihot, A.L.; Martin, L.; Nascimento, D.C.; Ryffel, B.; et al. Loss of vascular expression of nucleoside triphosphate diphosphohydrolase-1/CD39 in hypertension. Purinergic Signal. 2018, 14, 73–82. [Google Scholar] [CrossRef]
- Van Daele, P.; Van Coevorden, A.; Roger, P.P.; Boeynaems, J.M. Effects of adenine nucleotides on the proliferation of aortic endothelial cells. Circ. Res. 1992, 70, 82–90. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burnstock, G. Purinergic signaling and vascular cell proliferation and death. Arter. Thromb. Vasc. Biol. 2002, 22, 364–373. [Google Scholar] [CrossRef] [PubMed]
- Tran-Dinh, A.; Diallo, D.; Delbosc, S.; Varela-Perez, L.M.; Dang, Q.B.; Lapergue, B.; Burillo, E.; Michel, J.B.; Levoye, A.; Martin-Ventura, J.L.; et al. HDL and endothelial protection. Br. J. Pharmacol. 2013, 169, 493–511. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robert, J.; Osto, E.; von Eckardstein, A. The endothelium is both a target and a barrier of hdl’s protective functions. Cells 2021, 10, 1041. [Google Scholar] [CrossRef]
- Albuquerque, M.L.; Waters, C.M.; Savla, U.; Schnaper, H.W.; Flozak, A.S. Shear stress enhances human endothelial cell wound closure in vitro. Am. J. Physiol. Heart Circ. Physiol. 2000, 279, H293–H302. [Google Scholar] [CrossRef] [Green Version]
- Vyalov, S.; Langille, B.L.; Gotlieb, A.I. Decreased blood flow rate disrupts endothelial repair in vivo. Am. J. Pathol. 1996, 149, 2107–2118. [Google Scholar]
- Seetharam, D.; Mineo, C.; Gormley, A.K.; Gibson, L.L.; Vongpatanasin, W.; Chambliss, K.L.; Hahner, L.D.; Cummings, M.L.; Kitchens, R.L.; Marcel, Y.L.; et al. High-density lipoprotein promotes endothelial cell migration and reendothelialization via scavenger receptor-B type I. Circ. Res. 2006, 98, 63–72. [Google Scholar] [CrossRef] [Green Version]
- Kimura, T.; Sato, K.; Malchinkhuu, E.; Tomura, H.; Tamama, K.; Kuwabara, A.; Murakami, M.; Okajima, F. High-density lipoprotein stimulates endothelial cell migration and survival through sphingosine 1-phosphate and its receptors. Arter. Thromb. Vasc. Biol. 2003, 23, 1283–1288. [Google Scholar] [CrossRef] [Green Version]
- Suc, I.; Escargueil-Blanc, I.; Troly, M.; Salvayre, R.; Negre-Salvayre, A. HDL and ApoA prevent cell death of endothelial cells induced by oxidized LDL. Arter. Thromb. Vasc. Biol. 1997, 17, 2158–2166. [Google Scholar] [CrossRef]
- Franchi, F.; Angiolillo, D.J. Novel antiplatelet agents in acute coronary syndrome. Nat. Rev. Cardiol. 2015, 12, 30–47. [Google Scholar] [CrossRef]
- Sible, A.M.; Nawarskas, J.J. Cangrelor: A New Route for P2Y12 Inhibition. Cardiol. Rev. 2017, 25, 133–139. [Google Scholar] [CrossRef] [PubMed]
- Campo, G.; Dalla Sega, F.V.; Pavasini, R.; Aquila, G.; Gallo, F.; Fortini, F.; Tonet, E.; Cimaglia, P.; Del Franco, A.; Pestelli, G.; et al. Biological effects of ticagrelor over clopidogrel in patients with stable coronary artery disease and chronic obstructive pulmonary disease. Thromb. Haemost. 2017, 117, 1208–1216. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mangiacapra, F.; Panaioli, E.; Colaiori, I.; Ricottini, E.; Lauria Pantano, A.; Pozzilli, P.; Barbato, E.; Di Sciascio, G. Clopidogrel Versus Ticagrelor for Antiplatelet Maintenance in Diabetic Patients Treated With Percutaneous Coronary Intervention: Results of the CLOTILDIA Study (Clopidogrel High Dose Versus Ticagrelor for Antiplatelet Maintenance in Diabetic Patients). Circulation 2016, 134, 835–837. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Warnholtz, A.; Ostad, M.A.; Velich, N.; Trautmann, C.; Schinzel, R.; Walter, U.; Munzel, T. A single loading dose of clopidogrel causes dose-dependent improvement of endothelial dysfunction in patients with stable coronary artery disease: Results of a double-blind, randomized study. Atherosclerosis 2008, 196, 689–695. [Google Scholar] [CrossRef] [PubMed]
- Fromonot, J.; Dignat-Georges, F.; Rossi, P.; Mottola, G.; Kipson, N.; Ruf, J.; Bonello, L.; Guieu, R.; Paganelli, F. Ticagrelor Improves Peripheral Arterial Function in Acute Coronary Syndrome Patients Relationship with Adenosine Plasma Level. J. Am. Coll. Cardiol. 2016, 67, 1967–1968. [Google Scholar] [CrossRef]
- Jakubowski, A.; Chlopicki, S.; Olszanecki, R.; Jawien, J.; Lomnicka, M.; Dupin, J.P.; Gryglewski, R.J. Endothelial action of thienopyridines and thienopyrimidinones in the isolated guinea pig heart. Prostaglandins Leukot. Essent. Fat. Acids 2005, 72, 139–145. [Google Scholar] [CrossRef]
- Zhou, Z.; Chrifi, I.; Xu, Y.; Pernow, J.; Duncker, D.J.; Merkus, D.; Cheng, C. Uridine adenosine tetraphosphate acts as a proangiogenic factor in vitro through purinergic P2Y receptors. Am. J. Physiol. Heart Circ. Physiol. 2016, 311, H299–H309. [Google Scholar] [CrossRef]
- Heil, M.; Eitenmüller, I.; Schmitz-Rixen, T.; Schaper, W. Arteriogenesis versus angiogenesis: Similarities and differences. J. Cell. Mol. Med. 2006, 10, 45–55. [Google Scholar] [CrossRef] [Green Version]
- McEnaney, R.M.; Shukla, A.; Madigan, M.C.; Sachdev, U.; Tzeng, E. P2Y 2 nucleotide receptor mediates arteriogenesis in a murine model of hind limb ischemia. J. Vasc. Surg. 2016, 63, 216–225. [Google Scholar] [CrossRef] [Green Version]
- Le Duc, D.; Schulz, A.; Lede, V.; Schulze, A.; Thor, D.; Brüser, A.; Schöneberg, T. P2Y Receptors in Immune Response and Inflammation. Adv. Immunol. 2017, 136, 85–121. [Google Scholar]
- Ouimet, M.; Barrett, T.J.; Fisher, E.A. HDL and Reverse Cholesterol Transport. Circ. Res. 2019, 124, 1505–1518. [Google Scholar] [CrossRef]
- Swirski, F.K.; Nahrendorf, M. Leukocyte behavior in atherosclerosis, myocardial infarction, and heart failure. Science 2013, 339, 161–166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rohrer, L.; Ohnsorg, P.M.; Lehner, M.; Landolt, F.; Rinninger, F.; Von Eckardstein, A. High-density lipoprotein transport through aortic endothelial cells involves scavenger receptor BI and ATP-binding cassette transporter G1. Circ. Res. 2009, 104, 1142–1150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Virgilio, F.; Chiozzi, P.; Ferrari, D.; Falzoni, S.; Sanz, J.M.; Morelli, A.; Torboli, M.; Bolognesi, G.; Baricordi, O.R. Nucleotide receptors: An emerging family of regulatory molecules in blood cells. Blood 2001, 97, 587–600. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tian, G.; Zhou, J.; Quan, Y.; Kong, Q.; Wu, W.; Liu, X. P2Y1 Receptor Agonist Attenuates Cardiac Fibroblasts Activation Triggered by TGF-β1. Front. Pharmacol. 2021, 12, 627773. [Google Scholar] [CrossRef] [PubMed]
- Huo, Y.; Schober, A.; Forlow, S.B.; Smith, D.F.; Hyman, M.C.; Jung, S.; Littman, D.R.; Weber, C.; Ley, K. Circulating activated platelets exacerbate atherosclerosis in mice deficient in apolipoprotein E. Nat. Med. 2003, 9, 61–67. [Google Scholar] [CrossRef]
- Jang, E.; Robert, J.; Rohrer, L.; von Eckardstein, A.; Lee, W.L. Transendothelial transport of lipoproteins. Atherosclerosis 2020, 315, 111–125. [Google Scholar] [CrossRef]
- Cavelier, C.; Rohrer, L.; von Eckardstein, A. ATP-Binding cassette transporter A1 modulates apolipoprotein A-I transcytosis through aortic endothelial cells. Circ. Res. 2006, 99, 1060–1066. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martinez, L.O.; Genoux, A.; Ferrières, J.; Duparc, T.; Perret, B. Serum inhibitory factor 1, high-density lipoprotein and cardiovascular diseases. Curr. Opin. Lipidol. 2017, 28, 1. [Google Scholar] [CrossRef]
- Fabre, A.C.A.; Malaval, C.; Ben Addi, A.; Verdier, C.; Pons, V.; Serhan, N.; Lichtenstein, L.; Combes, G.; Huby, T.; Briand, F.; et al. P2Y13 receptor is critical for reverse cholesterol transport. Hepatology 2010, 52, 1477–1483. [Google Scholar] [CrossRef]
- Pons, V.; Serhan, N.; Gayral, S.; Malaval, C.; Nauze, M.; Malet, N.; Laffargue, M.; Galés, C.; Martinez, L.O. Role of the ubiquitin-proteasome system in the regulation of P2Y13 receptor expression: Impact on hepatic HDL uptake. Cell. Mol. Life Sci. 2014, 71, 1775–1788. [Google Scholar] [CrossRef] [PubMed]
- Malaval, C.; Laffargue, M.; Barbaras, R.; Rolland, C.; Peres, C.; Champagne, E.; Perret, B.; Tercé, F.; Collet, X.; Martinez, L.O. RhoA/ROCK I signalling downstream of the P2Y13 ADP-receptor controls HDL endocytosis in human hepatocytes. Cell. Signal. 2009, 21, 120–127. [Google Scholar] [CrossRef] [PubMed]
- Jacquet, S.; Malaval, C.; Martinez, L.O.; Sak, K.; Rolland, C.; Perez, C.; Nauze, M.; Champagne, E.; Terce, F.; Gachet, C.; et al. The nucleotide receptor P2Y13 is a key regulator of hepatic high-density lipoprotein (HDL) endocytosis. Cell. Mol. Life Sci. 2005, 62, 2508–2515. [Google Scholar] [CrossRef] [PubMed]
- Martinez, L.O.; Georgeaud, V.; Rolland, C.; Collet, X.; Tercé, F.; Perret, B.; Barbaras, R. Characterization of Two High-Density Lipoprotein Binding Sites on Porcine Hepatocyte Plasma Membranes: Contribution of Scavenger Receptor Class B Type I (SR-BI) to the Low-Affinity Component. Biochemistry 2000, 39, 1076–1082. [Google Scholar] [CrossRef] [PubMed]
- Sprague, E.A.; Steinbach, B.L.; Nerem, R.M.; Schwartz, C.J. Influence of a laminar steady-state fluid-imposed wall shear stress on the binding, internalization, and degradation of low-density lipoproteins by cultured arterial endothelium. Circulation 1987, 76, 648–656. [Google Scholar] [CrossRef] [Green Version]
- Gimbrone, M.A.; Topper, J.N.; Nagel, T.; Anderson, K.R.; Garcia-Cardeña, G. Endothelial Dysfunction, Hemodynamic Forces, and Atherogenesisa. Ann. N. Y. Acad. Sci. 2006, 902, 230–240. [Google Scholar] [CrossRef]
- Chiu, J.-J.; Chien, S. Effects of disturbed flow on vascular endothelium: Pathophysiological basis and clinical perspectives. Physiol. Rev. 2011, 91, 327–387. [Google Scholar] [CrossRef] [Green Version]
- Ross, R. The pathogenesis of atherosclerosis. Mech. Ageing Dev. 1979, 9, 435–440. [Google Scholar] [CrossRef]
- Kader, K.N.; Akella, R.; Ziats, N.P.; Lakey, L.A.; Harasaki, H.; Ranieri, J.P.; Bellamkonda, R. V eNOS-overexpressing endothelial cells inhibit platelet aggregation and smooth muscle cell proliferation in vitro. Tissue Eng. 2000, 6, 241–251. [Google Scholar] [CrossRef]
- Jeremy, J.Y.; Rowe, D.; Emsley, A.M.; Newby, A.C. Nitric oxide and the proliferation of vascular smooth muscle cells. Cardiovasc. Res. 1999, 43, 580–594. [Google Scholar] [CrossRef] [Green Version]
- Janssens, S.; Flaherty, D.; Nong, Z.; Varenne, O.; van Pelt, N.; Haustermans, C.; Zoldhelyi, P.; Gerard, R.; Collen, D. Human endothelial nitric oxide synthase gene transfer inhibits vascular smooth muscle cell proliferation and neointima formation after balloon injury in rats. Circulation 1998, 97, 1274–1281. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vermeersch, P.; Nong, Z.; Stabile, E.; Varenne, O.; Gillijns, H.; Pellens, M.; Van Pelt, N.; Hoylaerts, M.; De Scheerder, I.; Collen, D.; et al. L-arginine administration reduces neointima formation after stent injury in rats by a nitric oxide-mediated mechanism. Arterioscler. Thromb. Vasc. Biol. 2001, 21, 1604–1609. [Google Scholar] [CrossRef] [PubMed]
- Allagnat, F.; Haefliger, J.-A.; Lambelet, M.; Longchamp, A.; Bérard, X.; Mazzolai, L.; Corpataux, J.-M.; Déglise, S. Nitric Oxide Deficit Drives Intimal Hyperplasia in Mouse Models of Hypertension. Eur. J. Vasc. Endovasc. Surg. 2016, 51, 733–742. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holt, A.W.; Tulis, D.A. Experimental Rat and Mouse Carotid Artery Surgery: Injury & Remodeling Studies. ISRN Minim. Invasive Surg. 2013, 2013, 167407. [Google Scholar] [PubMed]
- Langille, B.L.; O’Donnell, F. Reductions in arterial diameter produced by chronic decreases in blood flow are endothelium-dependent. Science 1986, 231, 405–407. [Google Scholar] [CrossRef]
- Rudic, R.D.; Shesely, E.G.; Maeda, N.; Smithies, O.; Segal, S.S.; Sessa, W.C. Direct evidence for the importance of endothelium-derived nitric oxide in vascular remodeling. J. Clin. Investig. 1998, 101, 731–736. [Google Scholar] [CrossRef]
- Kumar, A.; Lindner, V. Remodeling with neointima formation in the mouse carotid artery after cessation of blood flow. Arterioscler. Thromb. Vasc. Biol. 1997, 17, 2238–2244. [Google Scholar] [CrossRef]
- Nam, D.; Ni, C.-W.; Rezvan, A.; Suo, J.; Budzyn, K.; Llanos, A.; Harrison, D.; Giddens, D.; Jo, H. Partial carotid ligation is a model of acutely induced disturbed flow, leading to rapid endothelial dysfunction and atherosclerosis. Am. J. Physiol. Heart Circ. Physiol. 2009, 297, H1535–H1543. [Google Scholar] [CrossRef]
- Seye, C.I.; Gadeau, A.P.; Daret, D.; Dupuch, F.; Alzieu, P.; Capron, L.; Desgranges, C. Overexpression of P2Y2 purinoceptor in intimal lesions of the rat aorta. Arterioscler. Thromb. Vasc. Biol. 1997, 17, 3602–3610. [Google Scholar] [CrossRef]
- Seye, C.I.; Kong, Q.; Erb, L.; Garrad, R.C.; Krugh, B.; Wang, M.; Turner, J.T.; Sturek, M.; Gonzalez, F.A.; Weisman, G.A. Functional P2Y2 nucleotide receptors mediate uridine 5′-triphosphate-induced intimal hyperplasia in collared rabbit carotid arteries. Circulation 2002, 106, 2720–2726. [Google Scholar] [CrossRef] [Green Version]
- Behdad, A.; Sun, X.; Khalpey, Z.; Enjyoji, K.; Wink, M.; Wu, Y.; Usheva, A.; Robson, S.C. Vascular smooth muscle cell expression of ectonucleotidase CD39 (ENTPD1) is required for neointimal formation in mice. Purinergic Signal. 2009, 5, 335–342. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Papanikolaou, A.; Papafotika, A.; Murphy, C.; Papamarcaki, T.; Tsolas, O.; Drab, M.; Kurzchalia, T.V.; Kasper, M.; Christoforidis, S. Cholesterol-dependent lipid assemblies regulate the activity of the ecto-nucleotidase CD39. J. Biol. Chem. 2005, 280, 26406–26414. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Agca, Y.; Qian, S.; Agca, C.; Seye, C.I. Direct Evidence for P2Y2 Receptor Involvement in Vascular Response to Injury. J. Vasc. Res. 2016, 53, 163–171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seye, C.I.; Kong, Q.; Yu, N.; Gonzalez, F.A.; Erb, L.; Weisman, G.A. P2 receptors in atherosclerosis and postangioplasty restenosis. Purinergic Signal. 2007, 3, 153–162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, X.; Li, Q.; Pi, S.; Xia, Y.; Mao, L. G protein-coupled purinergic P2Y receptor oligomerization: Pharmacological changes and dynamic regulation. Biochem. Pharmacol. 2021, 192, 114689. [Google Scholar] [CrossRef]
- Garcia, C.; Maurel-Ribes, A.; Nauze, M.; N’Guyen, D.; Martinez, L.O.; Payrastre, B.; Sénard, J.M.; Galés, C.; Pons, V. Deciphering biased inverse agonism of cangrelor and ticagrelor at P2Y 12 receptor. Cell. Mol. Life Sci. 2019, 76, 561–576. [Google Scholar] [CrossRef] [PubMed]
- Nagel, T.; Resnick, N.; Dewey, C.F.; Gimbrone, M.A. Vascular endothelial cells respond to spatial gradients in fluid shear stress by enhanced activation of transcription factors. Arterioscler. Thromb. Vasc. Biol. 1999, 19, 1825–1834. [Google Scholar] [CrossRef] [Green Version]
- Lau, O.C.F.; Samarawickrama, C.; Skalicky, S.E. P2Y2 receptor agonists for the treatment of dry eye disease: A review. Clin. Ophthalmol. 2014, 8, 327–334. [Google Scholar] [PubMed] [Green Version]
- Kellerman, D.; Rossi Mospan, A.; Engels, J.; Schaberg, A.; Gorden, J.A.; Smiley, L. Denufosol: A review of studies with inhaled P2Y2 agonists that led to Phase 3. Pulm. Pharmacol. Ther. 2008, 21, 600–607. [Google Scholar] [CrossRef]
- Gremmel, T.; Yanachkov, I.B.; Yanachkova, M.I.; Wright, G.E.; Wider, J.; Undyala, V.V.R.; Michelson, A.D.; Frelinger, A.L.; Przyklenk, K. Synergistic inhibition of both P2Y1 and P2Y12 adenosine diphosphate receptors as novel approach to rapidly attenuate platelet-mediated thrombosis. Arterioscler. Thromb. Vasc. Biol. 2016, 36, 501–509. [Google Scholar] [CrossRef] [Green Version]
- Koganov, E.S.; Michelson, A.D.; Yanachkov, I.B.; Yanachkova, M.I.; Wright, G.E.; Przyklenk, K.; Frelinger, A.L. GLS-409, an Antagonist of Both P2Y1 and P2Y12, Potently Inhibits Canine Coronary Artery Thrombosis and Reversibly Inhibits Human Platelet Activation. Sci. Rep. 2018, 8, 14529. [Google Scholar] [CrossRef] [PubMed]
- Neumann, A.; Attah, I.; Al-Hroub, H.; Namasivayam, V.; Müller, C.E. Discovery of P2Y 2 Receptor Antagonist Scaffolds through Virtual High-Throughput Screening. J. Chem. Inf. Model. 2022, 62, 1538–1549. [Google Scholar] [CrossRef] [PubMed]
- Xu, P.; Feng, X.; Luan, H.; Wang, J.; Ge, R.; Li, Z.; Bian, J. Current knowledge on the nucleotide agonists for the P2Y2 receptor. Bioorg. Med. Chem. 2018, 26, 366–375. [Google Scholar] [CrossRef] [PubMed]
- Botta, J.; Appelhans, J.; McCormick, P.J. Continuing challenges in targeting oligomeric GPCR-based drugs. In Progress in Molecular Biology and Translational Science; Elsevier: Amsterdam, The Netheralnds, 2020; Volume 169, pp. 213–245. ISBN 9780128179291. [Google Scholar]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cabou, C.; Martinez, L.O. The Interplay of Endothelial P2Y Receptors in Cardiovascular Health: From Vascular Physiology to Pathology. Int. J. Mol. Sci. 2022, 23, 5883. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23115883
Cabou C, Martinez LO. The Interplay of Endothelial P2Y Receptors in Cardiovascular Health: From Vascular Physiology to Pathology. International Journal of Molecular Sciences. 2022; 23(11):5883. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23115883
Chicago/Turabian StyleCabou, Cendrine, and Laurent O. Martinez. 2022. "The Interplay of Endothelial P2Y Receptors in Cardiovascular Health: From Vascular Physiology to Pathology" International Journal of Molecular Sciences 23, no. 11: 5883. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23115883