
Insights into the Pharmacological Effects of Flavonoids: The Systematic Review of Computer Modeling

 ,
,
Abstract
:1. Introduction
2. Methods
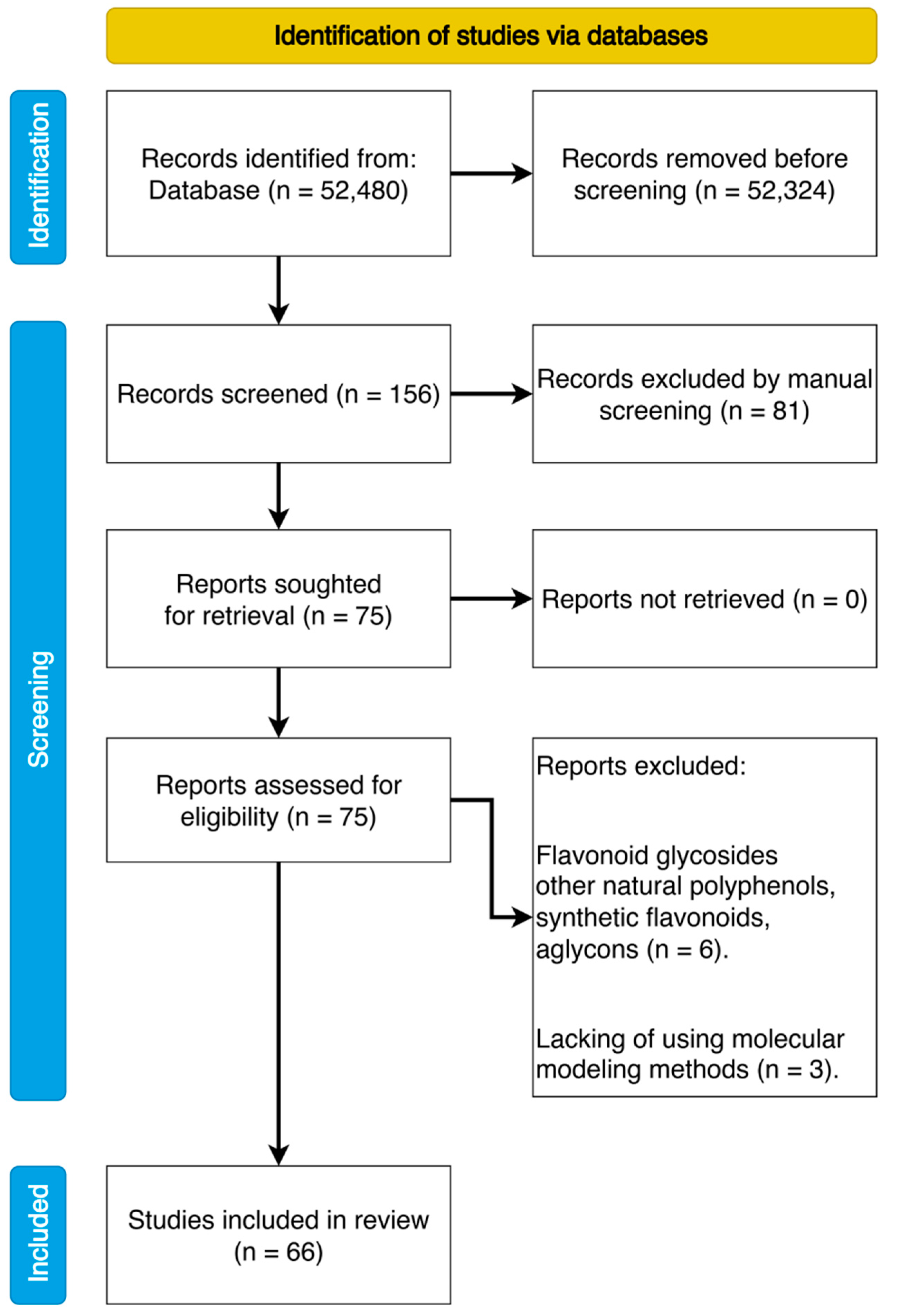
2.1. Search Strategy
2.2. Review Protocol and Data Extraction
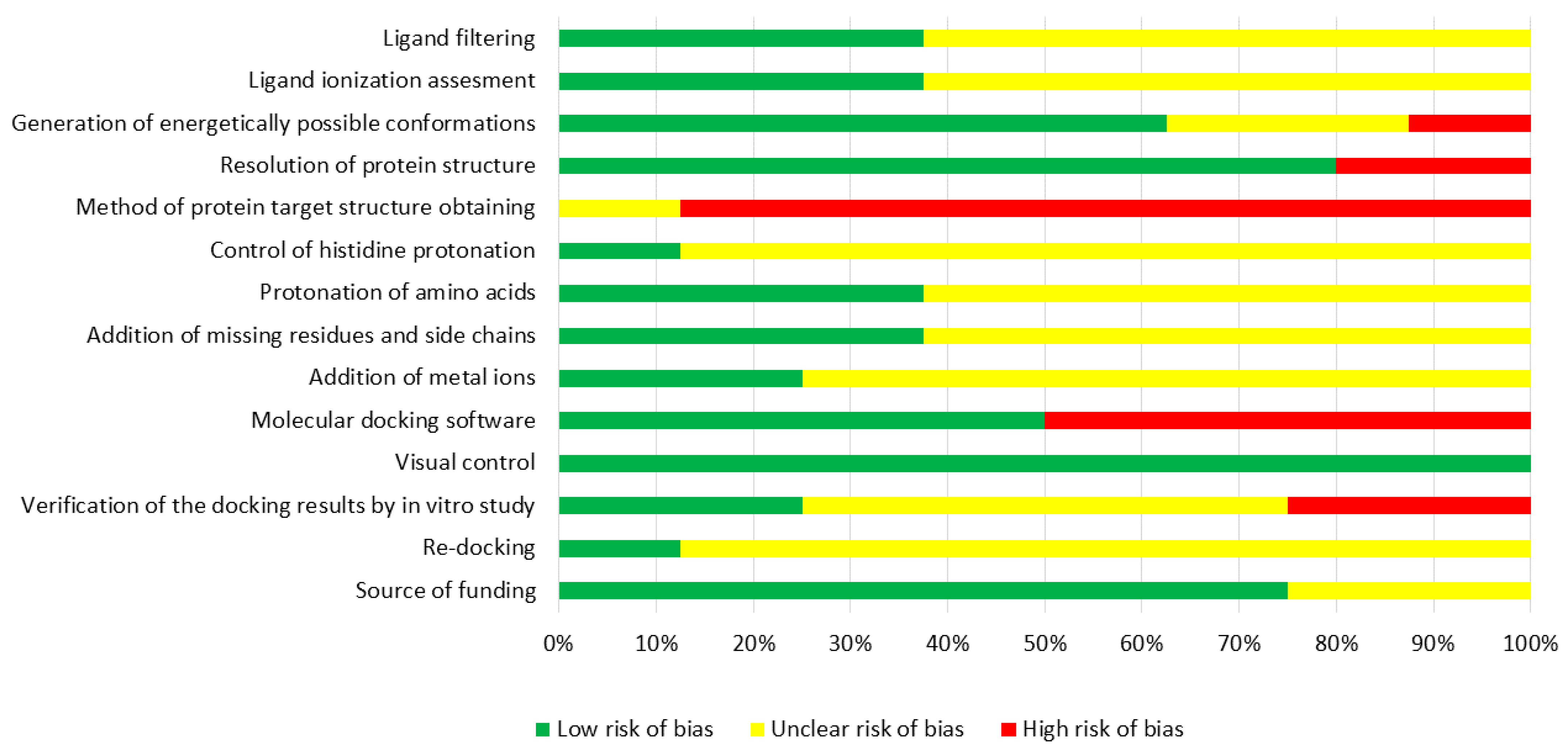
2.3. Assessment of Risk of Bias
3. Results and Discussion
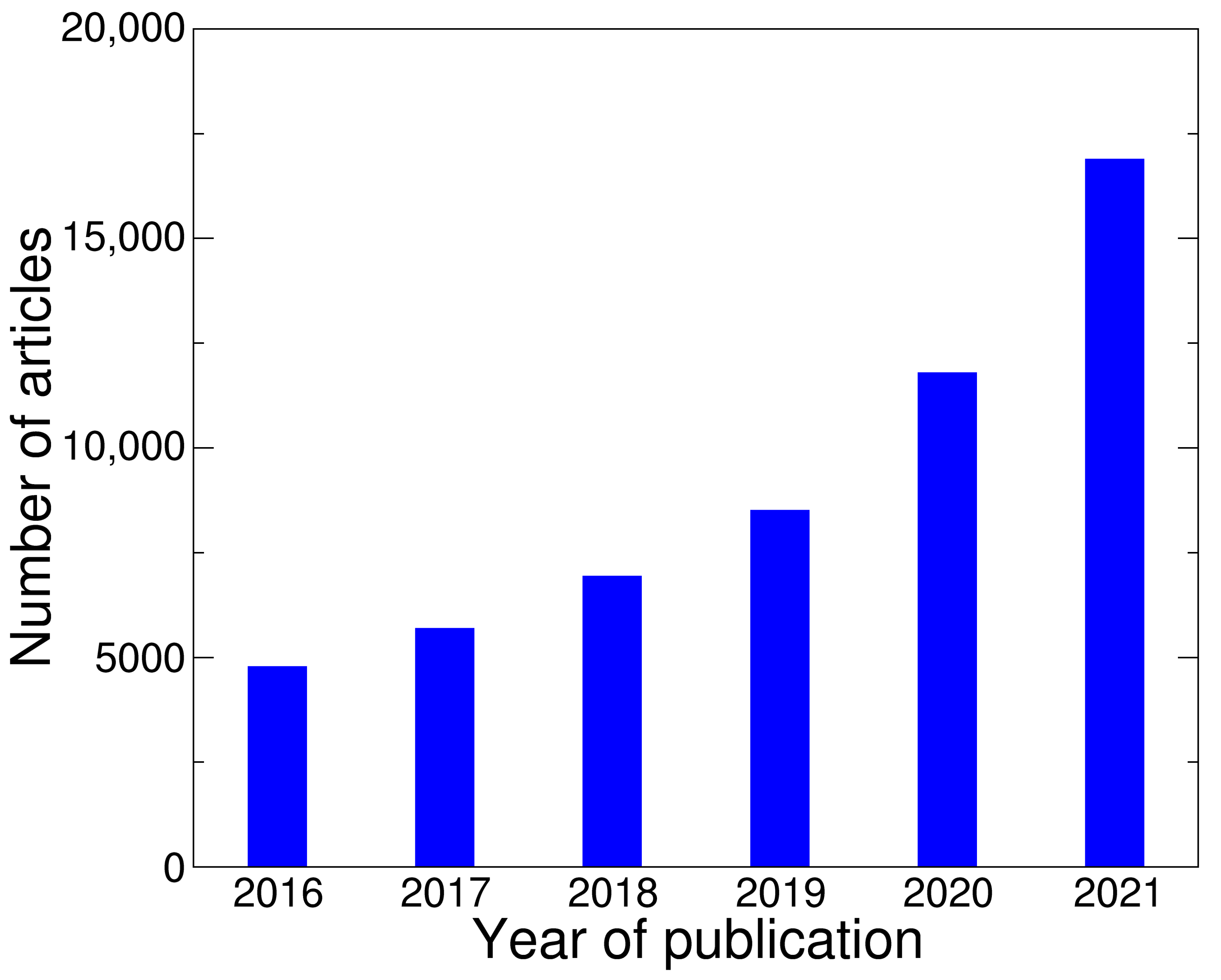
3.1. General View on Articles Sample
3.2. Molecular Modeling Methods
3.3. Software and Databases for Molecular Docking
- User-friendly software;
- Intuitive graphical interface;
- Lack of need for high-performance computing resources;
- Good predictive ability.
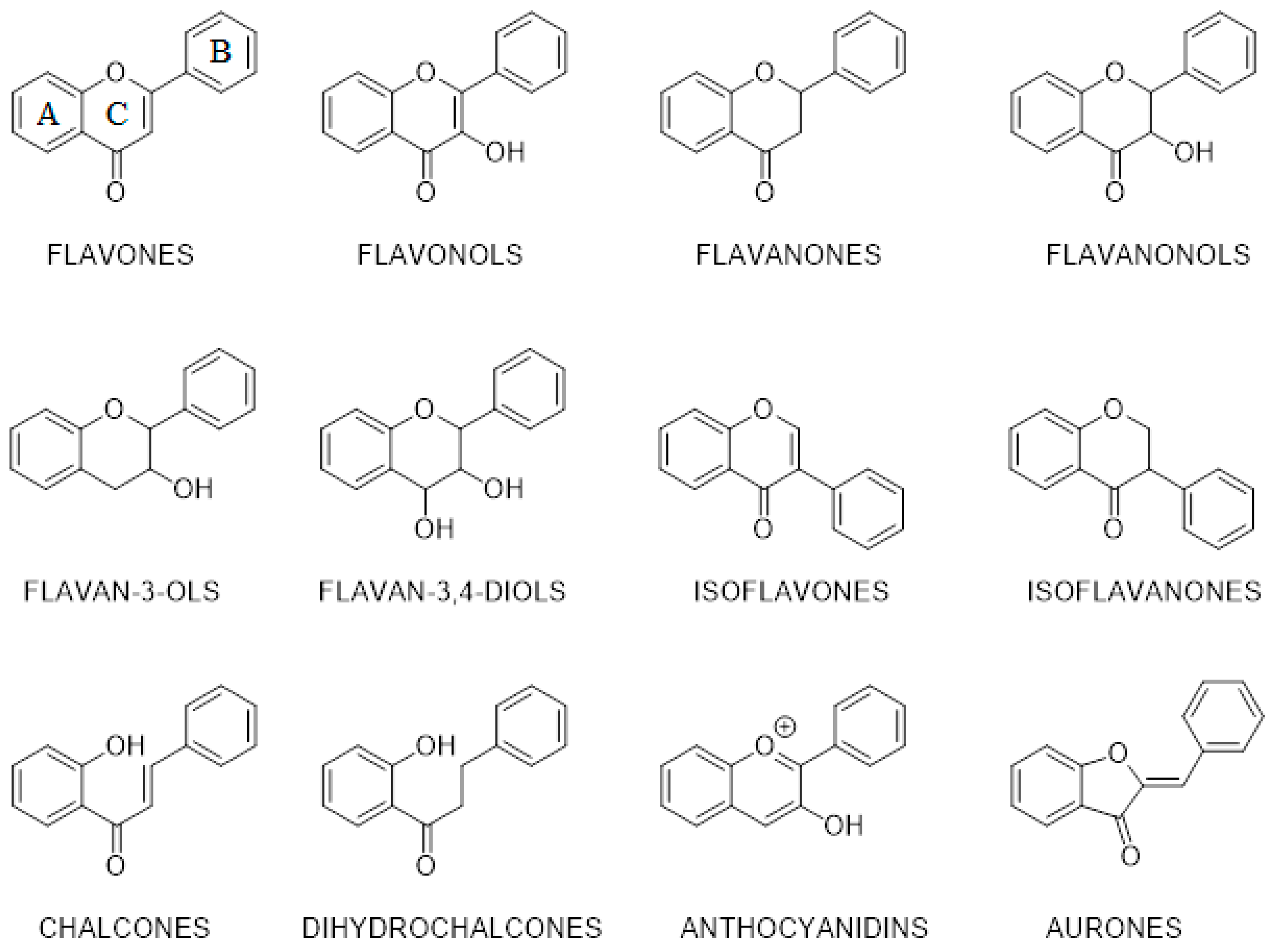
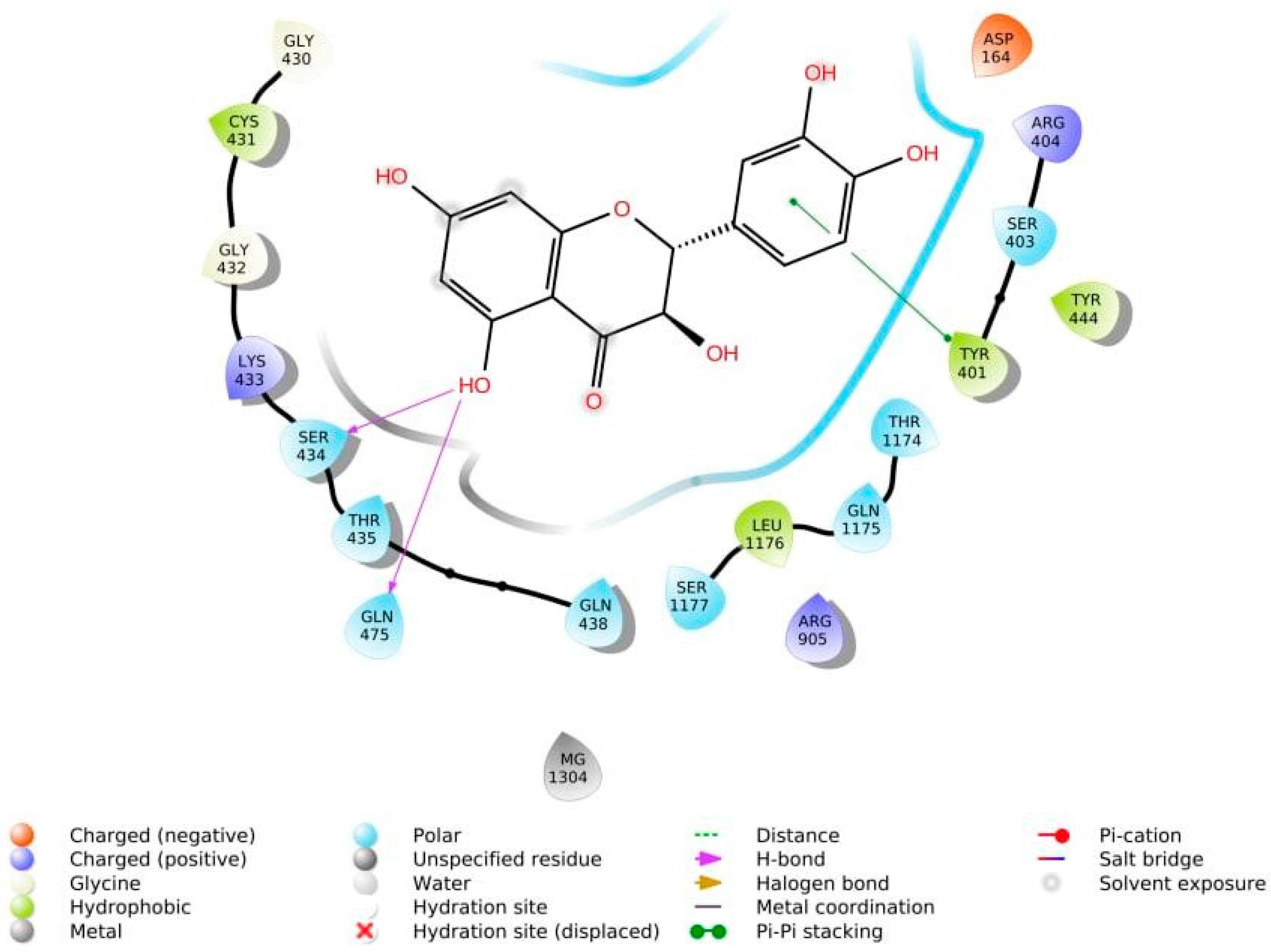
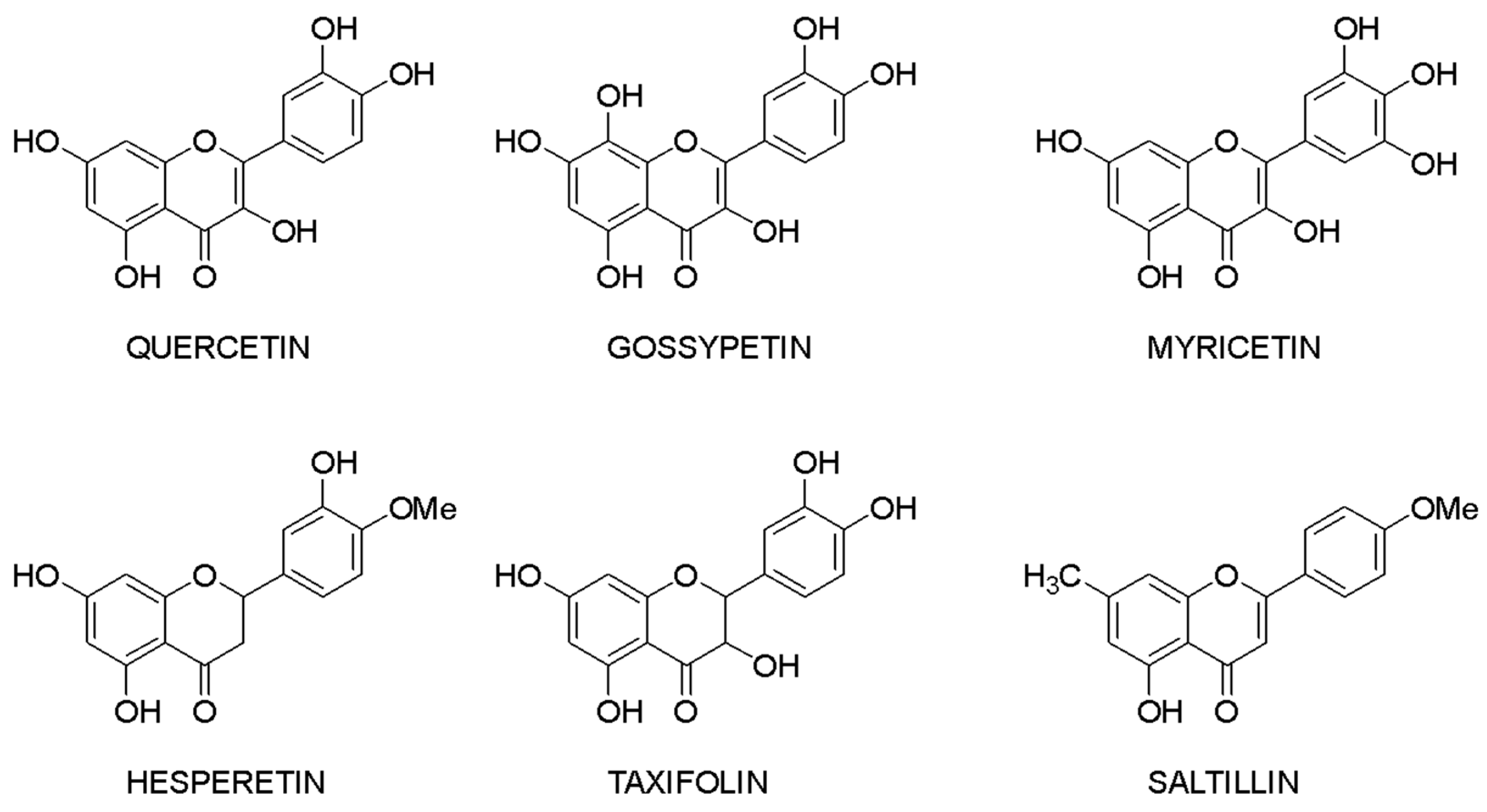
3.4. Structure—Biological Activity Relationship: Qualitative Analysis
3.5. Structure—Biological Activity Relationship: Quantitative Analysis
3.6. Limitation Section
- Bias arising from ligands selection process;
- Bias due to ligands structure optimization;
- Bias arising from protein target selection process;
- Bias due to protein target processing;
- Bias arising from molecular docking process;
- Bias in results assessment.
3.7. Lead Compounds
3.8. COVID-19 Treatment
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Bossel, H. Modeling and Simulation; CRC Press: Boca Raton, FL, USA, 2018; pp. 3–6. [Google Scholar]
- Goh, G.B.; Hodas, N.O.; Vishnu, A. Deep learning for computational chemistry. J. Comput. Chem. 2017, 38, 1291–1307. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Durrant, J.D.; McCammon, J.A. Molecular dynamics simulations and drug discovery. BMC Biol. 2011, 9, 71. [Google Scholar] [CrossRef] [Green Version]
- Sliwoski, G.; Kothiwale, S.; Meiler, J.; Lowe, E.W. Computational methods in drug discovery. Pharmacol. Rev. 2014, 66, 334–395. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sabe, V.T.; Ntombela, T.; Jhamba, L.A.; Maguire, G.E.M.; Govender, T.; Naicker, T.; Kruger, H.G. Current trends in computer aided drug design and a highlight of drugs discovered via computational techniques: A review. Eur. J. Med. Chem. 2021, 224, 113705. [Google Scholar] [CrossRef] [PubMed]
- Cherkasov, A.; Muratov, E.N.; Fourches, D.; Varnek, A.; Baskin, I.I.; Cronin, M.; Dearden, J.; Gramatica, P.; Martin, Y.C.; Todeschini, R.; et al. QSAR modeling: Where have you been? Where are you going to? J. Med. Chem. 2014, 57, 4977–5010. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef] [Green Version]
- Yuriev, E.; Holien, J.; Ramsland, P.A. Improvements, trends, and new ideas in molecular docking: 2012–2013 in review. J. Mol. Recognit. 2015, 28, 581–604. [Google Scholar] [CrossRef]
- Hollingsworth, S.A.; Dror, R.O. Molecular dynamics simulation for all. Neuron 2018, 99, 1129–1143. [Google Scholar] [CrossRef] [Green Version]
- Polkovnikova, Y.A.; Lenshin, A.S.; Glushko, A.A.; Khishova, O.M. Modeling and analysis of molecular dynamics of silicon-solvent-vinpocetin systems and silicon-solvent-vinpocetin dioxide. Drug Dev. Reg. 2017, 21, 44–49. (In Russian) [Google Scholar]
- Polkovnikova, Y.A. Modeling the formation of liposomes with vinpocetine from soy lecithin phospholipids by molecular dynamics. Drug Dev. Reg. 2021, 10, 83–87. (In Russian) [Google Scholar] [CrossRef]
- Il’In, M.V.; Sysoeva, A.A.; Bolotin, D.S.; Novikov, A.S.; Suslonov, V.V.; Rogacheva, E.V.; Kraeva, L.A.; Kukushkin, V.Y. Aminonitrones as highly reactive bifunctional synthons. An expedient one-pot route to 5-amino-1,2,4-triazoles and 5-amino-1,2,4-oxadiazoles—Potential antimicrobials targeting multi-drug resistant bacteria. New J. Chem. 2019, 43, 17358–17366. [Google Scholar] [CrossRef]
- Novikov, A.S.; Kuznetsov, M.L. Theoretical study of Re(IV) and Ru(II) bis-isocyanide complexes and their reactivity in cycloaddition reactions with nitrones. Inorg. Chim. Acta 2012, 380, 78–89. [Google Scholar] [CrossRef]
- Bitew, M.; Desalegn, T.; Demissie, T.B.; Belayneh, A.; Endale, M.; Eswaramoorthy, R. Pharmacokinetics and drug-likeness of antidiabetic flavonoids: Molecular docking and DFT study. PLoS ONE 2021, 16, e0260853. [Google Scholar] [CrossRef] [PubMed]
- Kirchmair, J.; Göller, A.H.; Lang, D.; Kunze, J.; Testa, B.; Wilson, I.D.; Glen, R.C.; Schneider, G. Predicting drug metabolism: Experiment and/or computation? Nat. Rev. Drug Discov. 2015, 14, 387–404. [Google Scholar] [CrossRef] [Green Version]
- Fröhlich, E.; Salar-Behzadi, S. Toxicological assessment of inhaled nanoparticles: Role of in vivo, ex vivo, in vitro, and in silico studies. Int. J. Mol. Sci. 2014, 15, 4795–4822. [Google Scholar] [CrossRef]
- PCC|Insilico Medicine. Available online: https://insilico.com/blog/pcc (accessed on 19 December 2021).
- Shaker, B.; Ahmad, S.; Lee, J.; Jung, C.; Na, D. In silico methods and tools for drug discovery. Comput. Biol. Med. 2021, 137, 104851. [Google Scholar] [CrossRef]
- Juca, M.M.; Filho, F.M.S.C.; de Almeida, J.C.; da Silva Mesquita, D.; de Moraes Barriga, J.R.; Dias, K.C.F.; Bardosa, T.M.; Vasconcelos, L.C.; Leal, L.K.A.M.; Robeiro, J.E.; et al. Flavonoids: Biological activities and therapeutic potential. Nat. Prod. Res. 2020, 34, 692–705. [Google Scholar] [CrossRef]
- Heinz, S.A.; Henson, D.A.; Austin, M.D.; Jin, F.; Nieman, D.C. Quercetin supplementation and upper respiratory tract infection: A randomized community clinical trial. Pharmacol. Res. 2010, 62, 237–242. [Google Scholar] [CrossRef]
- Terekhov, R.P.; Selivanova, I.A.; Anurova, M.N.; Zhevlakova, A.K.; Nikitin, I.D.; Cong, Z.; Ma, S.; Yang, F.; Dong, Z.; Liao, Y. Comparative study of wound-healing activity of dihydroquercetin pseudopolymorphic modifications. Bull. Exp. Biol. Med. 2021, 170, 444–447. [Google Scholar] [CrossRef]
- Lucas, C.D.; Allen, K.C.; Dorward, D.A.; Hoodless, L.J.; Melrose, L.A.; Marwick, J.A.; Tucker, C.S.; Haslett, C.; Duffin, R.; Rossi, A.G. Flavones induce neutrophil apoptosis by down-regulation of Mcl-1 via a proteasomal-dependent pathway. FASEB J. 2013, 27, 1084–1094. [Google Scholar] [CrossRef] [Green Version]
- Sheen, Y.S.; Huang, H.Y.; Liao, Y.H. The efficacy and safety of an antiaging topical serum containing hesperetin and sodium cyclic lysophosphatidic acid: A single-center clinical trial. J. Cosmet. Dermatol. 2021, 20, 3960–3967. [Google Scholar] [CrossRef] [PubMed]
- Godos, J.; Caraci, F.; Castellano, S.; Currenti, W.; Galvano, F.; Ferri, R.; Grosso, G. Association between dietary flavonoids intake and cognitive function in an italian cohort. Biomolecules 2020, 10, 1300. [Google Scholar] [CrossRef] [PubMed]
- Godos, J.; Caraci, F.; Micek, A.; Castellano, S.; D’amico, E.; Paladino, N.; Ferri, R.; Galvano, F.; Grosso, G. Dietary phenolic acids and their major food sources are associated with cognitive status in older italian adults. Antioxidants 2021, 10, 700. [Google Scholar] [CrossRef]
- Grosso, G.; Godos, J.; Currenti, W.; Micek, A.; Falzone, L.; Libra, M.; Giampieri, F.; Forbes-Hernández, T.Y.; Quiles, J.L.; Battino, M.; et al. The Effect of dietary polyphenols on vascular health and hypertension: Current evidence and mechanisms of action. Nutrients 2022, 14, 545. [Google Scholar] [CrossRef] [PubMed]
- Grosso, G.; Micek, A.; Godos, J.; Pajak, A.; Sciacca, S.; Galvano, F.; Giovannucci, E.L. Dietary flavonoid and lignan intake and mortality in prospective cohort studies: Systematic review and dose-response meta-analysis. Am. J. Epidemiol. 2017, 185, 1304–1316. [Google Scholar] [CrossRef] [PubMed]
- Marranzano, M.; Ray, S.; Godos, J.; Galvano, F. Association between dietary flavonoids intake and obesity in a cohort of adults living in the Mediterranean area. Int. J. Food Sci. Nutr. 2018, 69, 1020–1029. [Google Scholar] [CrossRef]
- Tyukavkina, N.A.; Selivanova, I.A.; Terekhov, R.P. Modern Trends in the Design of Drugs Based on Flavonoids. In Phenolic Compounds: Properties, Activity, Innovation, Fundamental and Applied Aspects; Zagoskina, N.V., Tyukavkina, N.A., Lapshin, P.V., Nechaev, T.L., Eds.; IFR RAN: Moscow, Russia, 2018; pp. 526–532. (In Russian) [Google Scholar]
- Ilyasov, I.R.; Beloborodov, V.L.; Selivanova, I.A. Three ABTS•+ radical cation-based approaches for the evaluation of antioxidant activity: Fast- and slow-reacting antioxidant behavior. Chem. Pap. 2018, 72, 1917–1925. [Google Scholar] [CrossRef]
- Markova, E.Y.; Matveev, A.V.; Cox, Y.D.; Kovalevskaya, M.A.; Dergacheva, L.I. Modern antioxidant therapy in pediatric ophthalmology. Eff. Pharmacother. 2011, 29, 46–51. (In Russian) [Google Scholar]
- Shikov, A.N.; Pozharitskaya, O.N.; Miroshnyk, I.; Mirza, S.; Urakova, I.N.; Hirsjärvi, S.; Makarova, V.G.; Heinämäki, J.; Yliruusi, J.; Hiltunen, R. Nanodispersions of taxifolin: Impact of solid-state properties on dissolution behavior. Int. J. Pharm. 2009, 377, 148–152. [Google Scholar] [CrossRef] [PubMed]
- Moher, D.; Liberati, A.; Tetzlaff, J.; Altman, D.G.; Altman, D.; Antes, G.; Atkins, D.; Barbour, V.; Barrowman, N.; Berlin, J.A.; et al. Preferred reporting items for systematic reviews and meta-analyses: The PRISMA statement. PLoS Med. 2009, 6, e1000097. [Google Scholar] [CrossRef] [Green Version]
- Chen, H.J.; Chung, Y.L.; Li, C.Y.; Chang, Y.T.; Wang, C.C.N.; Lee, H.Y.; Lin, H.Y.; Hung, C.C. Taxifolin resensitizes multidrug resistance cancer cells via uncompetitive inhibition of P-glycoprotein. Funct. Mol. 2018, 23, 3055. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, J.; Wang, X.; Geng, S.; Liu, B.; Liang, G. Inhibitory mechanism of taxifolin against α-glucosidase based on spectrofluorimetry and molecular docking. Nat. Prod. Commun. 2017, 12, 1725–1728. [Google Scholar] [CrossRef] [PubMed]
- Mahdavimehr, M.; Meratan, A.A.; Ghobeh, M.; Ghasemi, A.; Saboury, A.A.; Nemat-Gorgani, M. Inhibition of HEWL fibril formation by taxifolin: Mechanism of action. PLoS ONE 2017, 12, e0187841. [Google Scholar] [CrossRef] [PubMed]
- Haque, M.; Mohd Siddique, M.; Bose, P.; Pattanayak, S. Taxifolin possesses anti-cancer activity on the 7,12-Dimethylbenz(a)anthracene-Induced breast cancer in the sprague dawley rats by remodeling nuclear factor Erythroid 2- Kelch-like ECH-associated protein 1-heme oxygenase 1 and anti-oxidant pathways. Pharmacogn. Mag. 2018, 14, 110. [Google Scholar] [CrossRef]
- George, T.K.; Joy, A.; Divya, K.; Jisha, M.S. In vitro and in silico docking studies of antibacterial compounds derived from endophytic Penicillium setosum. Microb. Pathog. 2019, 131, 87–97. [Google Scholar] [CrossRef]
- Terekhov, R.P.; Selivanova, I.A. Molecular modeling of the interaction of the dihydroquercetin and its metabolites with cyclooxygenase-2. Bull. Exp. Biol. Med. 2019, 18, 101–106. [Google Scholar] [CrossRef]
- Mohseni-Shahri, F.S.; Housaindokht, M.R.; Bozorgmehr, M.R.; Moosavi-Movahedi, A.A. Comparative study of the effects of the structurally similar flavonoids quercetin and taxifolin on the therapeutic behavior of alprazolam. Can. J. Chem. 2016, 94, 458–469. [Google Scholar] [CrossRef]
- Chilingaryan, G.V. Comparative analysis of quercetin and taxifolin interaction with human telomeric G-quadruplex DNA hybrid form based on molecular dynamic simulations. Biol. J. Armenia 2018, 7, 57–61. [Google Scholar]
- Verma, S.; Grover, S.; Tyagi, C.; Goyal, S.; Jamal, S.; Singh, A.; Grover, A. Hydrophobic interactions are a key to MDM2 inhibition by polyphenols as revealed by molecular dynamics simulations and MM/PBSA free energy calculations. PLoS ONE 2016, 11, e0149014. [Google Scholar] [CrossRef]
- Mary, V.; Haris, P.; Varghese, M.K.; Aparna, P.; Sudarsanakumar, C. Experimental probing and molecular dynamics simulation of the molecular recognition of DNA duplexes by the flavonoid luteolin. J. Chem. Inf. Model. 2017, 57, 2237–2249. [Google Scholar] [CrossRef]
- Ginex, T.; Trius, M.; Luque, F.J. Computational study of the aza-michael addition of the flavonoid (+)-taxifolin in the inhibition of β-amyloid fibril aggregation. Chem. A Eur. J. 2018, 24, 5813–5824. [Google Scholar] [CrossRef] [PubMed]
- Proença, C.; Freitas, M.; Ribeiro, D.; Oliveira, E.F.T.; Sousa, J.L.C.; Tomé, S.M.; Ramos, M.J.; Silva, A.M.S.; Fernandes, P.A.; Fernandes, E. α-glucosidase inhibition by flavonoids: An in vitro and in silico structure–activity relationship study. J. Enzyme Inhib. Med. Chem. 2017, 32, 1216–1228. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meshram, R.J.; Bagul, K.T.; Pawnikar, S.P.; Barage, S.H.; Kolte, B.S.; Gacche, R.N. Known compounds and new lessons: Structural and electronic basis of flavonoid-based bioactivities. J. Biomol. Struct. Dyn. 2019, 38, 1168–1184. [Google Scholar] [CrossRef] [PubMed]
- Manjula, S.; Kumaradhas, P. Evaluating the suitability of RNA intervention mechanism exerted by some flavonoid molecules against dengue virus MTase RNA capping site: A molecular docking, molecular dynamics simulation, and binding free energy study. J. Biomol. Struct. Dyn. 2019, 38, 3533–3543. [Google Scholar] [CrossRef] [PubMed]
- Hashemi, S.; Sharifi, A.; Zareei, S.; Mohamedi, G.; Biglar, M.; Amanlou, M. Discovery of direct inhibitor of KRAS oncogenic protein by natural products: A combination of pharmacophore search, molecular docking, and molecular dynamic studies. Res. Pharm. Sci. 2020, 15, 226. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Ge, F.; Wang, Y.; Dong, Y.; Shan, Y.; Zhu, Q.; Wu, X.; Wu, C.; Ge, R.S. Taxifolin is a rat and human 11β-hydroxysteroid dehydrogenase 1 inhibitor as a possible drug to treat the metabolic syndrome. J. Funct. Foods 2018, 49, 181–187. [Google Scholar] [CrossRef]
- Haque, M.W.; Pattanayak, S.P. Taxifolin inhibits 7,12-dimethylbenz(a)anthracene-induced breast carcinogenesis by regulating AhR/CYP1A1 signaling pathway. Pharmacogn. Mag. 2017, 13, S749. [Google Scholar] [CrossRef]
- Thao, N.P.; Luyen, B.T.T.; Kim, J.H.; Jo, A.R.; Dat, N.T.; van Kiem, P.; van Minh, C.; Kim, Y.H. Identification, characterization, kinetics, and molecular docking of flavonoid constituents from Archidendron clypearia (Jack.) Nielsen leaves and twigs. Bioorg. Med. Chem. 2016, 24, 3125–3132. [Google Scholar] [CrossRef]
- Rehman, K.; Chohan, T.A.; Waheed, I.; Gilani, Z.; Akash, M.S.H. Taxifolin prevents postprandial hyperglycemia by regulating the activity of α-amylase: Evidence from an in vivo and in silico studies. J. Cell. Biochem. 2019, 120, 425–438. [Google Scholar] [CrossRef] [Green Version]
- Al-Khayyat, M.Z. In silico homology modeling and docking studies of RecA from Campylobacter jejuni. Int. J. Boautoimmun. 2019, 23, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Ismail, N.A.; Jusoh, S.A. Molecular docking and molecular dynamics simulation studies to predict flavonoid binding on the surface of DENV2 E protein. Interdiscip. Sci. Comput. Life Sci. 2016, 9, 499–511. [Google Scholar] [CrossRef] [PubMed]
- Fang, Y.; Cao, W.; Liang, F.; Xia, M.; Pan, S.; Xu, X. Structure affinity relationship and docking studies of flavonoids as substrates of multidrug-resistant associated protein 2 (MRP2) in MDCK/MRP2 cells. Food Chem. 2019, 291, 101–109. [Google Scholar] [CrossRef] [PubMed]
- Trezza, A.; Cicaloni, V.; Porciatti, P.; Langella, A.; Fusi, F.; Saponara, S.; Spiga, O. From in silico to in vitro: A trip to reveal flavonoid binding on the Rattus norvegicus Kir6.1 ATP-sensitive inward rectifier potassium channel. PeerJ 2018, 6, e4680. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, H.; He, S.; Wang, T.; Orang-Ojong, B.; Lu, Q.; Zhang, Z.; Pan, L.; Chai, X.; Wu, H.; Fan, G.; et al. Selected phytoestrogens distinguish roles of ERα transactivation and ligand binding for anti-inflammatory activity. Endocrinology 2018, 159, 3351–3364. [Google Scholar] [CrossRef] [PubMed]
- Vijayasri, S.; Hopper, W. Towards the identification of novel phytochemical leads as macrodomain inhibitors of chikungunya virus using molecular docking approach. J. Appl. Pharm. Sci. 2017, 7, 74–082. [Google Scholar] [CrossRef] [Green Version]
- Xiong, Z.; Liu, W.; Zhou, L.; Zou, L.; Chen, J. Mushroom (Agaricus bisporus) polyphenoloxidase inhibited by apigenin: Multi-spectroscopic analyses and computational docking simulation. Food Chem. 2016, 203, 430–439. [Google Scholar] [CrossRef] [PubMed]
- Maharani, M.G.; Lestari, S.R.; Lukiati, B. Molecular docking studies flavonoid (quercetin, isoquercetin, and kaempferol) of single bulb garlic (Allium sativum) to inhibit lanosterol synthase as anti-hypercholesterol therapeutic strategies. AIP Conf. Proc. 2020, 2231, 040021. [Google Scholar] [CrossRef]
- Kurakin, G.F.; Lopina, N.P.; Bordina, G.E. Computational modelling of the interaction between flavonoids and adenosine receptors. Probl. Biol. Med. Pharm. Chem. 2019, 22, 42–47. (In Russian) [Google Scholar] [CrossRef]
- Zeng, L.; Zhang, G.; Lin, S.; Gong, D. Inhibitory mechanism of apigenin on α-glucosidase and synergy analysis of flavonoids. J. Agric. Food Chem. 2016, 64, 6939–6949. [Google Scholar] [CrossRef]
- Bhuiya, S.; Haque, L.; Dutta, T.; Chowdhury, S.; Das, S. Binding aspects of dietary flavone, luteolin, with polymorphic forms of natural DNA: A spectroscopic and molecular docking approach. New J. Chem. 2018, 43, 249–260. [Google Scholar] [CrossRef]
- Geethalakshmi, R.; Sundaramurthi, J.C.; Sarada, D.V.L. Antibacterial activity of flavonoid isolated from Trianthema decandra against Pseudomonas aeruginosa and molecular docking study of FabZ. Microb. Pathog. 2018, 121, 87–92. [Google Scholar] [CrossRef] [PubMed]
- Bhuiya, S.; Haque, L.; Pradhan, A.B.; Das, S. Inhibitory effects of the dietary flavonoid quercetin on the enzyme activity of zinc(II)-dependent yeast alcohol dehydrogenase: Spectroscopic and molecular docking studies. Int. J. Biol. Macromol. 2017, 95, 177–184. [Google Scholar] [CrossRef] [PubMed]
- Pérez-Abril, M.; Lucas-Abellán, C.; Castillo-Sánchez, J.; Pérez-Sánchez, H.; Cerón-Carrasco, J.P.; Fortea, I.; Gabaldón, J.A.; Núñez-Delicado, E. Systematic investigation and molecular modelling of complexation between several groups of flavonoids and HP-β-cyclodextrins. J. Funct. Foods 2017, 36, 122–131. [Google Scholar] [CrossRef]
- Kurkin, V.A. Phenylpropanoids from medicinal plants: Distribution, classification, structural analysis, and biological activity. Chem. Nat. Compd 2003, 39, 123–153. [Google Scholar] [CrossRef]
- Shanmugasundaram, J.; Subramanian, V.; Nadipelly, J.; Kathirvelu, P.; Sayeli, V.; Cheriyan, B.V. Anxiolytic–like activity of 5–methoxyflavone in mice with involvement of GABAergic and serotonergic systems—In vivo and in silico evidences. Eur. Neuropsychopharmacol. 2020, 36, 100–110. [Google Scholar] [CrossRef] [PubMed]
- Taldaev, A.K.; Terekhov, R.P.; Selivanova, I.A. Interaction of Taxifolin and P-Glycoprotein in silico. In Book of Abstracts XI International Conference on Chemistry for Young Scientists; Izdatelstvo VVM: Moscow, Russia, 2019; p. 436. [Google Scholar]
- Gacche, R.N.; Shegokar, H.D.; Gond, D.S.; Yang, Z.; Jadhav, A.D. Evaluation of selected flavonoids as antiangiogenic, anticancer, and radical scavenging agents: An experimental and in silico analysis. Cell Biochem. Biophys. 2011, 61, 651–663. [Google Scholar] [CrossRef]
- Yugandhar, P.; Kumar, K.K.; Neeraja, P.; Savithramma, N. Isolation, characterization and in silico docking studies of synergistic estrogen receptor a anticancer polyphenols from Syzygium alternifolium (Wt.) Walp. J. Intercult. Ethnopharmacol. 2017, 6, 296. [Google Scholar] [CrossRef]
- Guerrero, L.; Castillo, J.; Quiñones, M.; Garcia-Vallvé, S.; Arola, L.; Pujadas, G.; Muguerza, B. Inhibition of angiotensin-converting enzyme activity by flavonoids: Structure-activity relationship studies. PLoS ONE 2012, 7, e49493. [Google Scholar] [CrossRef] [Green Version]
- Ye, H.; Shaw, I.C. Food flavonoid ligand structure/estrogen receptor-α affinity relationships—Toxicity or food functionality? Food Chem. Toxicol. 2019, 129, 328–336. [Google Scholar] [CrossRef]
- Katzenellenbogen, B.S.; Choi, I.; Delage-Mourroux, R.; Ediger, T.R.; Martini, P.G.V.; Montano, M.; Sun, J.; Weis, K.; Katzenellenbogen, J.A. Molecular mechanisms of estrogen action: Selective ligands and receptor pharmacology. J. Steroid Biochem. Mol. Biol. 2000, 74, 279–285. [Google Scholar] [CrossRef]
- Anasir, M.I.; Ramanathan, B.; Poh, C.L. Structure-based design of antivirals against envelope glycoprotein of dengue virus. Viruses 2020, 12, 367. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jouimyi, M.R.; Bounder, G.; Essaidi, I.; Boura, H.; Zerouali, K.; Lebrazi, H.; Kettani, A.; Maachi, F. Molecular docking of a set of flavonoid compounds with Helicobacter pylori virulence factors CagA and VacA. J. Herbmed. Pharmacol. 2020, 9, 412–419. [Google Scholar] [CrossRef]
- Da Silva, E.R.; Brogi, S.; Lucon-Júnior, J.F.; Campiani, G.; Gemma, S.; Maquiaveli, C.D.C. Dietary polyphenols rutin, taxifolin and quercetin related compounds target Leishmania amazonensis arginase. Food Funct. 2019, 10, 3172–3180. [Google Scholar] [CrossRef] [PubMed]
- Validandi, V.; Khandare, A.L. Reduction of fluoride toxicity by tamarind components: An in silico study. Fluoride 2018, 51, 122–136. [Google Scholar]
- Mishra, S.; Singh, S.; Misra, K. Restraining pathogenicity in Candida albicans by taxifolin as an inhibitor of Ras1-pka pathway. Mycopathologia 2017, 182, 953–965. [Google Scholar] [CrossRef]
- Garg, A.X.; Hackam, D.; Tonelli, M. Systematic review and meta-analysis: When one study is just not enough. Clin. J. Am. Soc. Nephrol. 2008, 3, 253–260. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Zhang, K.Y.J. Hierarchical virtual screening approaches in small molecule drug discovery. Methods 2015, 71, 26–37. [Google Scholar] [CrossRef]
- Lyne, P.D. Structure-based virtual screening: An overview. Drug Discov. Today 2002, 7, 1047–1055. [Google Scholar] [CrossRef]
- Onufriev, A.V.; Alexov, E. Protonation and pK changes in protein–ligand binding. Q. Rev. Biophys. 2013, 46, 181–209. [Google Scholar] [CrossRef] [Green Version]
- Kalliokoski, T.; Salo, H.S.; Lahtela-Kakkonen, M.; Poso, A. The effect of ligand-based tautomer and protomer prediction on structure-based virtual screening. J. Chem. Inf. Model. 2009, 49, 2742–2748. [Google Scholar] [CrossRef]
- Vieth, M.; Hirst, J.D.; Brooks, C.L. Do active site conformations of small ligands correspond to low free-energy solution structures? J. Comput. Mol. Des. 1998, 12, 563–572. [Google Scholar] [CrossRef] [PubMed]
- Patrick Walters, W.; Stahl, M.T.; Murcko, M.A. Virtual screening—An overview. Drug Discov. Today 1998, 3, 160–178. [Google Scholar] [CrossRef]
- Mannhold, R.; Kubinyi, H.; Timmerman, H. Molecular Modeling Basic Principles and Applications; John Wiley & Sons: New York, NY, USA, 2008. [Google Scholar]
- Billeter, M. Comparison of protein structures determined by NMR in solution and by X-ray diffraction in single crystals. Q. Rev. Biophys. 1992, 25, 325–377. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Forli, S.; Huey, R.; Pique, M.E.; Sanner, M.F.; Goodsell, D.S.; Olson, A.J. Computational protein–ligand docking and virtual drug screening with the AutoDock suite. Nat. Protoc. 2016, 11, 905–919. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lohning, A.E.; Levonis, S.M.; Williams-Noonan, B.; Schweiker, S.S. A practical guide to molecular docking and homology modelling for medicinal chemists. Curr. Top. Med. Chem. 2017, 17, 2023–2040. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, M.O.; Nichols, S.E.; Wang, Y.; McCammon, J.A. Effects of histidine protonation and rotameric states on virtual screening of M. tuberculosis RmlC. J. Comput. Aided. Mol. Des. 2013, 27, 235–246. [Google Scholar] [CrossRef] [Green Version]
- Riccardi, L.; Genna, V.; de Vivo, M. Metal–ligand interactions in drug design. Nat. Rev. Chem. 2018, 2, 100–112. [Google Scholar] [CrossRef]
- Kellenberger, E.; Rodrigo, J.; Muller, P.; Rognan, D. Comparative evaluation of eight docking tools for docking and virtual screening accuracy. Proteins Struct. Funct. Bioinforma. 2004, 57, 225–242. [Google Scholar] [CrossRef]
- Chen, Y.C. Beware of docking! Trends Pharmacol. Sci. 2015, 36, 78–95. [Google Scholar] [CrossRef]
- Bender, B.J.; Gahbauer, S.; Luttens, A.; Lyu, J.; Webb, C.M.; Stein, R.M.; Fink, E.A.; Balius, T.E.; Carlsson, J.; Irwin, J.J.; et al. A practical guide to large-scale docking. Nat. Protoc. 2021, 16, 4799–4832. [Google Scholar] [CrossRef]
- Macip, G.; Garcia-Segura, P.; Mestres-Truyol, J.; Saldivar-Espinoza, B.; Ojeda-Montes, M.J.; Gimeno, A.; Cereto-Massagué, A.; Garcia-Vallvé, S.; Pujadas, G. Haste makes waste: A critical review of docking-based virtual screening in drug repurposing for SARS-CoV-2 main protease (M-pro) inhibition. Med. Res. Rev. 2022, 42, 744–769. [Google Scholar] [CrossRef] [PubMed]
- Davis, C.K.; Nasla, K.; Anjana, A.K.; Rajanikant, G.K. Taxifolin as dual inhibitor of Mtb DNA gyrase and isoleucyl-tRNA synthetase: In silico molecular docking, dynamics simulation and in vitro assays. Silico Pharmacol. 2018, 6, 8. [Google Scholar] [CrossRef] [PubMed]
- Raj, U.; Varadwaj, P.K. Flavonoids as multi-target inhibitors for proteins associated with ebola virus: In silico discovery using virtual screening and molecular docking studies. Interdiscip. Sci. Comput. Life Sci. 2015, 8, 132–141. [Google Scholar] [CrossRef] [PubMed]
- Meenambiga, S.S.; Rajagopal, K. Antibiofilm activity and molecular docking studies of bioactive secondary metabolites from endophytic fungus Aspergillus nidulans on oral Candida albicans. Artic. J. Appl. Pharm. Sci. 2018, 8, 37–45. [Google Scholar] [CrossRef]
- Rajendran, P.; Maheshwari, U.; Muthukrishnan, A.; Muthuswamy, R.; Anand, K.; Ravindran, B.; Dhanaraj, P.; Balamuralikrishnan, B.; Chang, S.W.; Chung, W.J. Myricetin: Versatile plant based flavonoid for cancer treatment by inducing cell cycle arrest and ROS–reliant mitochondria-facilitated apoptosis in A549 lung cancer cells and in silico prediction. Mol. Cell. Biochem. 2020, 476, 57–68. [Google Scholar] [CrossRef]
- Xu, Z.; Li, K.; Pan, T.; Liu, J.; Li, B.; Li, C.; Wang, S.; Diao, Y.; Liu, X. Lonicerin, an anti-algE flavonoid against Pseudomonas aeruginosa virulence screened from Shuanghuanglian formula by molecule docking based strategy. J. Ethnopharmacol. 2019, 239, 111909. [Google Scholar] [CrossRef]
- Lu, J.J.; Zhou, F.M.; Hu, X.J.; Fang, J.J.; Liu, C.X.; Zhu, B.Q.; Ding, Z.S. Molecular docking simulation and in vitro studies on estrogenic activities of flavonoids from leaves of Carya cathayensis Sarg. Steroids 2020, 163, 108726. [Google Scholar] [CrossRef]
- Esfahani, A.N.; Mirzaei, M. Flavonoid derivatives for monoamine oxidase–A inhibition. Adv. J. Chem. Sect. B 2019, 1, 17–22. [Google Scholar] [CrossRef]
- Orlova, S.V.; Tatarinov, V.V.; Nikitina, E.A.; Sheremeta, A.V.; Ivlev, V.A.; Vasil’ev, V.G.; Paliy, K.V.; Goryainov, S.V. Molecular-biological problems of drug design and mechanism of drug action bioavailability and safety of dihydroquercetin (Review). Pharm. Chem. J. 2021, 55, 3–8. [Google Scholar] [CrossRef]
- Weidmann, A.E. Dihydroquercetin: More than just an impurity? Eur. J. Pharmacol. 2012, 684, 19–26. [Google Scholar] [CrossRef]
- Das, A.; Baidya, R.; Chakraborty, T.; Samanta, A.K.; Roy, S. Pharmacological basis and new insights of taxifolin: A comprehensive review. Biomed. Pharmacother. 2021, 142, 112004. [Google Scholar] [CrossRef] [PubMed]
- Turck, D.; Bresson, J.; Burlingame, B.; Dean, T.; Fairweather-Tait, S.; Heinonen, M.; Hirsch-Ernst, K.I.; Mangelsdorf, I.; McArdle, H.J.; Naska, A.; et al. Statement on the safety of taxifolin-rich extract from Dahurian Larch (Larix gmelinii). EFSA J. 2017, 15, e05059. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gogoi, N.; Chowdhury, P.; Goswami, A.K.; Das, A.; Chetia, D.; Gogoi, B. Computational guided identification of a citrus flavonoid as potential inhibitor of SARS-CoV-2 main protease. Mol. Divers. 2021, 25, 1745–1759. [Google Scholar] [CrossRef] [PubMed]
- Fischer, A.; Sellner, M.; Neranjan, S.; Smieško, M.; Lill, M.A. Potential inhibitors for novel coronavirus protease identified by virtual screening of 606 million compounds. Int. J. Mol. Sci. 2020, 21, 3626. [Google Scholar] [CrossRef]
- Yang, C.; Wang, Z.; Mi, Y.; Gao, M.; Lv, J.; Meng, Y.; Yang, B.; Kuang, H. UHPLC-MS/MS determination, pharmacokinetic and bioavailability study of taxifolin in rat plasma after oral administration of its nanodispersion. Molecules 2016, 21, 494. [Google Scholar] [CrossRef]
- Zu, Y.; Wu, W.; Zhao, X.; Li, Y.; Wang, W.; Zhong, C. Enhancement of solubility, antioxidant ability and bioavailability of taxifolin nanoparticles by liquid antisolvent precipitation technique. Int. J. Pharm. 2014, 471, 366–376. [Google Scholar] [CrossRef]
- Yang, P.; Xu, F.; Li, H.F.; Wang, Y.; Li, F.C.; Shang, M.Y.; Liu, G.X.; Wang, X.; Cai, S.Q. Detection of 191 taxifolin metabolites and their distribution in rats using HPLC-ESI-IT-TOF-MSn. Molecules 2016, 21, 1209. [Google Scholar] [CrossRef] [Green Version]
- Terekhov, R.P.; Selivanova, I.A.; Tyukavkina, N.A.; Shylov, G.V.; Utenishev, A.N.; Porozov, Y.B. Taxifolin tubes: Crystal engineering and characteristics. Acta Crystallogr. Sect. B Struct. Sci. Cryst. Eng. Mater. 2019, 75, 175–182. [Google Scholar] [CrossRef]
- WHO Coronavirus (COVID-19) Dashboard|WHO Coronavirus (COVID-19) Dashboard with Vaccination Data. Available online: https://covid19.who.int/ (accessed on 9 March 2022).
- Jain, A.S.; Sushma, P.; Dharmashekar, C.; Beelagi, M.S.; Prasad, S.K.; Shivamallu, C.; Prasad, A.; Syed, A.; Marraiki, N.; Prasad, K.S. In silico evaluation of flavonoids as effective antiviral agents on the spike glycoprotein of SARS-CoV-2. Saudi J. Biol. Sci. 2021, 28, 1040–1051. [Google Scholar] [CrossRef]
- Rudrapal, M.; Issahaku, A.R.; Agoni, C.; Bendale, A.R.; Nagar, A.; Soliman, M.E.S.; Lokwani, D. In silico screening of phytopolyphenolics for the identification of bioactive compounds as novel protease inhibitors effective against SARS-CoV-2. J. Biomol. Struct. Dynam. 2021, 1–17. [Google Scholar] [CrossRef]
- Guler, H.I.; Tatar, G.; Yildiz, O.; Belduz, A.O.; Kolayli, S. Investigation of potential inhibitor properties of ethanolic propolis extracts against ACE-II receptors for COVID-19 treatment by molecular docking study. Arch. Microbiol. 2021, 203, 3557–3564. [Google Scholar] [CrossRef] [PubMed]
- Taldaev, A.K.; Terekhov, R.P.; Selivanova, I.A. Flavonoids as potential inhibitors of SARS-CoV-2 infection: In silico study. Bull. Sib. Med. 2022, 21, 103–108. [Google Scholar] [CrossRef]
- Shohan, M.; Nashibi, R.; Mahmoudian-Sani, M.R.; Abolnezhadian, F.; Ghafourian, M.; Alavi, S.M.; Sharhani, A.; Khodadadi, A. The therapeutic efficacy of quercetin in combination with antiviral drugs in hospitalized COVID-19 patients: A randomized controlled trial. Eur. J. Pharmacol. 2022, 914, 174615. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Section | Criteria | Include If: |
|---|---|---|
| Language | English | Yes |
| Russian | Yes | |
| Design | In silico studies, complex translational studies with the molecular modeling stage | Yes |
| In vitro and in vivo studies, reviews, editorials, letter to the editor | No | |
| Content | Studies examining the affinity of natural flavonoids aglycons to different biological targets | Yes |
| Studies examining the affinity of synthetic flavonoids aglycons to different biological targets | No | |
| Studies examining the affinity of flavonoids glycosides or other natural polyphenols to different biological targets | No | |
| Access | Full-text article accessible | Yes |
| Bias Domain | Issue | Low Risk of Bias | High Risk of Bias | Unclear Risk of Bias |
|---|---|---|---|---|
| Ligand selection | Ligand filtering | Should be performed | Did not applied | No data |
| Ligands optimization | Ionization assessment | The ligands were ionized according to pKa and pH values of media | The research was performed without reference to pKa values of ligands and pH values of media | No data |
| Generation of energetically possible conformations | Should be performed | Generation was performed without reference to potential energy calculation | No data | |
| Target selection | Resolution of protein structure | Not more than 2.5 Å | More than 2.5 Å | No data |
| Method of protein target structure obtaining | NMR spectroscopy | X-ray crystallography or cryogenic electron microscopy | No data | |
| Target optimization | Control of histidine protonation | Should be performed | The structure of target did not reference biological conditions | No data |
| Protonation of amino acids after X-ray crystallography or cryogenic electron microscopy | Should be performed | The structure of target did not reference biological conditions | No data | |
| Addition of missing residues and side chains after X-ray crystallography or cryogenic electron microscopy | Should be performed | Was performed without special tools | No data | |
| Addition of metals | Should be performed | The structure of target did not reference biological conditions | No data | |
| Docking | Molecular docking software | Glide, GOLD | AutoDock, DOCK, FlexX | No data |
| Results assessment | Visual control | Should be performed | Structure defects were observed | No data |
| Re-docking | Should be performed | The RMSD value is too high compared with the initial structure | No data | |
| Verification of docking results by in vitro study | Binding constant should be determined | The quantitative calculations were not performed | No data |
| Flavonoid Group | Affinity to the Biological Target, kcal/mol * | ||||
|---|---|---|---|---|---|
| ERα | E Protein DENV2-Thai | E Protein DENV2-My | Potassium Channel Kir6.1 | Protein VacA | |
| Flavones | −8.3 ± 0.6 | −7.8 ± 1.3 | −7.5 ± 0.9 | −6.7 ± n/a | −8.5 ± 0.3 |
| Flavonols | −7.9 ± 0.5 | −8.4 ± n/a | −8.6 ± n/a | −8.1 ± n/a | - |
| Flavonol glycosides | - | −8.1 ± n/a | −7.7 ± n/a | - | - |
| Flavanones | −8.5 ± n/a | - | - | - | - |
| Flavanonols | −9.0 ± n/a | - | - | - | - |
| Flavonoid Group | Affinity to the Biological Target, kcal/mol * | ||||
|---|---|---|---|---|---|
| ERα | Complex CA II-F | Arginase | Tec1 | Rfg1 | |
| Flavones | −8.5 ± 0.3 | −3.3 ± 0.0 | - | - | - |
| Flavonols | −8.8 ± n/a | - | −8.1 ± n/a | - | - |
| Flavonol glycosides | - | - | −8.2 ± 0.3 | - | - |
| Flavanones | −10.2 ± n/a | −2.7 ± 0.2 | - | −7.7 ± n/a | −6.7 ± n/a |
| Flavanonols | - | −2.9 ± n/a | −8.2 ± n/a | −7.7 ± n/a | −4.9 ± n/a |
| Flavan-3-ols | - | −4.7 ± 0.6 | - | - | - |
| Isoflavones | −9.0 ± 0.20 | - | - | - | - |
| Dihydrochalcones | −8.3 ± n/a | - | - | - | - |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Taldaev, A.; Terekhov, R.; Nikitin, I.; Zhevlakova, A.; Selivanova, I. Insights into the Pharmacological Effects of Flavonoids: The Systematic Review of Computer Modeling. Int. J. Mol. Sci. 2022, 23, 6023. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23116023
Taldaev A, Terekhov R, Nikitin I, Zhevlakova A, Selivanova I. Insights into the Pharmacological Effects of Flavonoids: The Systematic Review of Computer Modeling. International Journal of Molecular Sciences. 2022; 23(11):6023. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23116023
Chicago/Turabian StyleTaldaev, Amir, Roman Terekhov, Ilya Nikitin, Anastasiya Zhevlakova, and Irina Selivanova. 2022. "Insights into the Pharmacological Effects of Flavonoids: The Systematic Review of Computer Modeling" International Journal of Molecular Sciences 23, no. 11: 6023. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23116023