
Redox Homeostasis in Thyroid Cancer: Implications in Na+/I− Symporter (NIS) Regulation



{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Redox Homeostasis in the Thyroid Gland
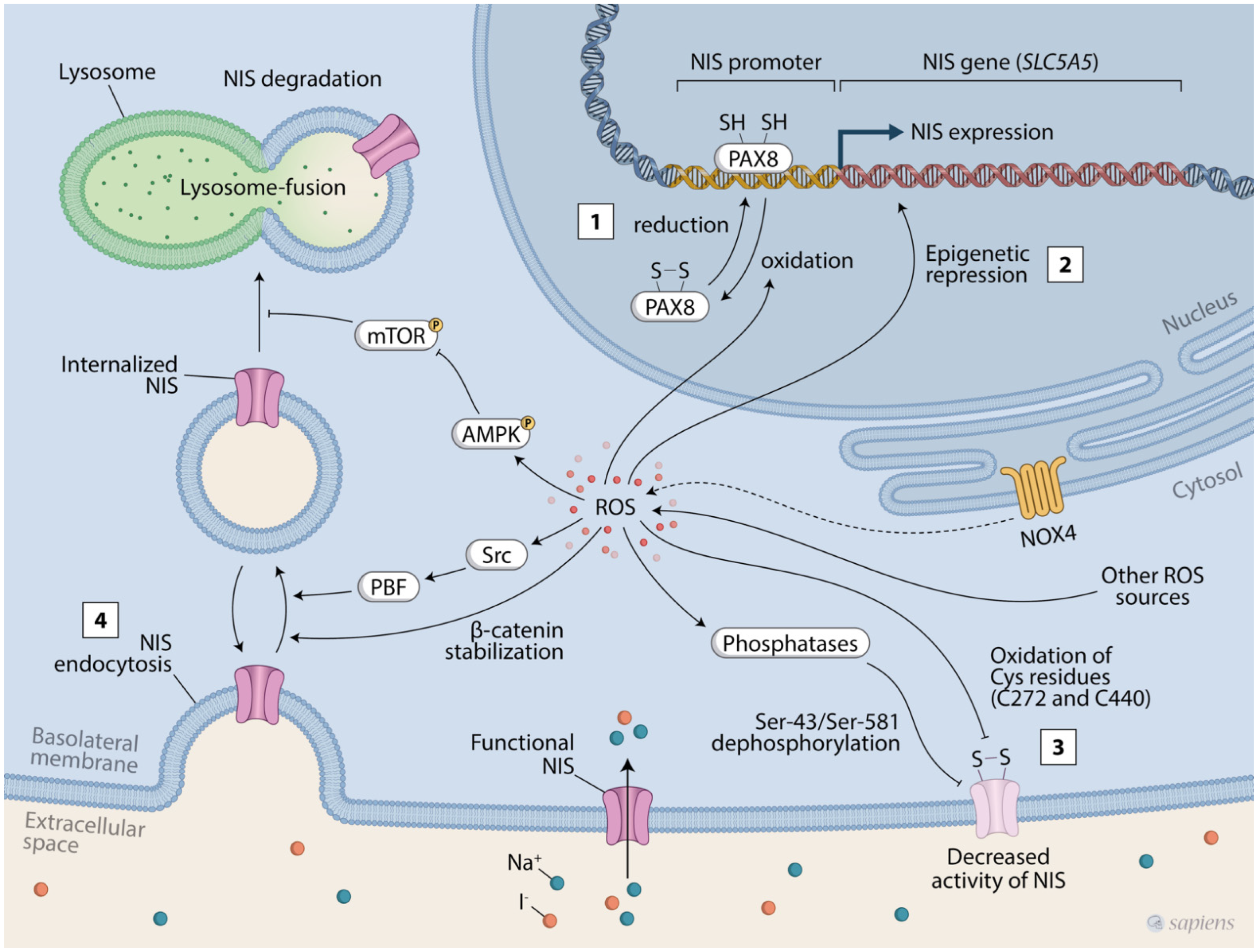
3. Evidence of NIS Regulation by ROS
3.1. Regulation of NIS Expression by ROS
3.2. Regulation of NIS Subcellular Location and Protein Stability by ROS
3.3. Regulation of NIS Activity by ROS
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Seidlin, S.L.; Marinelli, L.D.; Oshry, E. Radioactive iodine therapy; effect on functioning metastases of adenocarcinoma of the thyroid. J. Am. Med. Assoc. 1946, 132, 838. [Google Scholar] [CrossRef] [PubMed]
- De la Vieja, A.; Riesco-Eizaguirre, G. Radio-Iodide Treatment: From Molecular Aspects to the Clinical View. Cancers 2021, 13, 995. [Google Scholar] [CrossRef] [PubMed]
- Schlumberger, M. Management of refractory thyroid cancers. Ann. Endocrinol. 2011, 72, 149–157. [Google Scholar] [CrossRef] [PubMed]
- Worden, F. Treatment strategies for radioactive iodine-refractory differentiated thyroid cancer. Ther. Adv. Med. Oncol. 2014, 6, 267–279. [Google Scholar] [CrossRef] [Green Version]
- Durante, C.; Haddy, N.; Baudin, E.; Leboulleux, S.; Hartl, D.; Travagli, J.P.; Caillou, B.; Ricard, M.; Lumbroso, J.D.; De Vathaire, F.; et al. Long-Term Outcome of 444 Patients with Distant Metastases from Papillary and Follicular Thyroid Carcinoma: Benefits and Limits of Radioiodine Therapy. J. Clin. Endocrinol. Metab. 2006, 91, 2892–2899. [Google Scholar] [CrossRef]
- Schmidt, A.; Iglesias, L.; Klain, M.; Pitoia, F.; Schlumberger, M.J. Radioactive iodine-refractory differentiated thyroid cancer: An uncommon but challenging situation. Arch. Endocrinol. Metab. 2017, 61, 81–89. [Google Scholar] [CrossRef] [Green Version]
- Montero-Conde, C.; Ruiz-Llorente, S.; Dominguez, J.M.; Knauf, J.A.; Viale, A.; Sherman, E.J.; Ryder, M.; Ghossein, R.A.; Rosen, N.; Fagin, J.A. Relief of feedback inhibition of HER3 transcription by RAF and MEK inhibitors attenuates their antitumor effects in BRAF -mutant thyroid carcinomas. Cancer Discov. 2013, 3, 520–533. [Google Scholar] [CrossRef] [Green Version]
- Spitzweg, C.; Bible, K.C.; Hofbauer, L.C.; Morris, J.C. Advanced radioiodine-refractory differentiated thyroid cancer: The sodium iodide symporter and other emerging therapeutic targets. Lancet Diabetes Endocrinol. 2014, 2, 830–842. [Google Scholar] [CrossRef]
- Dohán, O.; De La Vieja, A.; Paroder, V.; Riedel, C.; Artani, M.; Reed, M.; Ginter, C.S.; Carrasco, N. The sodium/iodide symporter (NIS): Characterization, regulation, and medical significance. Endocr. Rev. 2003, 24, 48–77. [Google Scholar] [CrossRef] [Green Version]
- Dai, G.; Levy, O.; Carrasco, N. Cloning and characterization of the thyroid iodide transporter. Nature 1996, 379, 458–460. [Google Scholar] [CrossRef]
- Eskandari, S.; Loo, D.D.F.; Dai, G.; Levy, O.; Wright, E.M.; Carrasco, N. Thyroid Na+/I− Symporter. J. Biol. Chem. 1997, 272, 27230–27238. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roepke, T.K.; King, E.C.; Reyna-Neyra, A.; Paroder, M.; Purtell, K.; Koba, W.; Fine, E.; Lerner, D.J.; Carrasco, N.; Abbott, G.W. Kcne2 deletion uncovers its crucial role in thyroid hormone biosynthesis. Nat. Med. 2009, 15, 1186–1194. [Google Scholar] [CrossRef] [PubMed]
- Purtell, K.; Paroder-Belenitsky, M.; Reyna-Neyra, A.; Nicola, J.P.; Koba, W.; Fine, E.; Carrasco, N.; Abbott, G.W. The KCNQ1-KCNE2 K+ channel is required for adequate thyroid I− uptake. FASEB J. 2012, 26, 3252–3259. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Jeso, B.; Arvan, P. Thyroglobulin From Molecular and Cellular Biology to Clinical Endocrinology. Endocr. Rev. 2016, 37, 2–36. [Google Scholar] [CrossRef] [Green Version]
- Spitzweg, C.; Morris, J.C. The sodium iodide symporter: Its pathophysiological and therapeutic implications. Clin. Endocrinol. 2002, 57, 559–574. [Google Scholar] [CrossRef]
- Hingorani, M.; Spitzweg, C.; Vassaux, G.; Newbold, K.; Melcher, A.; Pandha, H.; Vile, R.; Harrington, K. The Biology of the Sodium Iodide Symporter and its Potential for Targeted Gene Delivery. Curr. Cancer Drug Targets 2010, 10, 242–267. [Google Scholar] [CrossRef] [Green Version]
- Xing, M. Molecular pathogenesis and mechanisms of thyroid cancer. Nat. Rev. Cancer 2013, 13, 184–199. [Google Scholar] [CrossRef]
- Jaber, T.; Waguespack, S.G.; Cabanillas, M.E.; Elbanan, M.; Vu, T.; Dadu, R.; Sherman, S.I.; Amit, M.; Santos, E.B.; Zafereo, M.; et al. Targeted Therapy in Advanced Thyroid Cancer to Resensitize Tumors to Radioactive Iodine. J. Clin. Endocrinol. Metab. 2018, 103, 3698–3705. [Google Scholar] [CrossRef] [Green Version]
- Dohán, O.; Baloch, Z.; Bánrévi, Z.; Livolsi, V.; Carrasco, N. RAPID COMMUNICATION: Predominant Intracellular Overexpression of the Na+/I− Symporter (NIS) in a Large Sampling of Thyroid Cancer Cases. J. Clin. Endocrinol. Metab. 2001, 86, 2697–2700. [Google Scholar] [CrossRef]
- Riesco-Eizaguirre, G.; Santisteban, P. A perspective view of sodium iodide symporter research and its clinical implications. Eur. J. Endocrinol. 2006, 155, 495–512. [Google Scholar] [CrossRef] [Green Version]
- Ho, A.L.; Grewal, R.K.; Leboeuf, R.; Sherman, E.J.; Pfister, D.G.; Deandreis, D.; Pentlow, K.S.; Zanzonico, P.B.; Haque, S.; Gavane, S.; et al. Selumetinib-enhanced radioiodine uptake in advanced thyroid cancer. N. Engl. J. Med. 2013, 368, 623–632. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rothenberg, S.M.; McFadden, D.G.; Palmer, E.L.; Daniels, G.H.; Wirth, L.J. Redifferentiation of iodine-refractory BRAF V600E-mutant metastatic papillary thyroid cancer with dabrafenib. Clin. Cancer Res. 2015, 21, 1028–1035. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buffet, C.; Wassermann, J.; Hecht, F.; Leenhardt, L.; Dupuy, C.; Groussin, L.; Lussey-Lepoutre, C. Redifferentiation of radioiodine-refractory thyroid cancers. Endocr. Relat. Cancer 2020, 27, R113–R132. [Google Scholar] [CrossRef] [PubMed]
- Ameziane-El-Hassani, R.; Talbot, M.; De Souza Dos Santos, M.C.; Ghuzlan, A.A.; Hartl, D.; Bidart, J.M.; De Deken, X.; Miot, F.; Diallo, I.; De Vathaire, F.; et al. NADPH oxidase DUOX1 promotes long-term persistence of oxidative stress after an exposure to irradiation. Proc. Natl. Acad. Sci. USA 2015, 112, 5051–5056. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Azouzi, N.; Cailloux, J.; Cazarin, J.M.; Knauf, J.A.; Cracchiolo, J.; Al Ghuzlan, A.; Hartl, D.; Polak, M.; Carré, A.; El Mzibri, M.; et al. NADPH Oxidase NOX4 Is a Critical Mediator of BRAF V600E -Induced Downregulation of the Sodium/Iodide Symporter in Papillary Thyroid Carcinomas. Antioxid. Redox Signal. 2017, 26, 864–877. [Google Scholar] [CrossRef] [Green Version]
- Metere, A.; Frezzotti, F.; Graves, C.E.; Vergine, M.; De Luca, A.; Pietraforte, D.; Giacomelli, L. A possible role for selenoprotein glutathione peroxidase (GPx1) and thioredoxin reductases (TrxR1) in thyroid cancer: Our experience in thyroid surgery. Cancer Cell Int. 2018, 18, 7. [Google Scholar] [CrossRef]
- Sies, H. Hydrogen peroxide as a central redox signaling molecule in physiological oxidative stress: Oxidative eustress. Redox Biol. 2017, 11, 613–619. [Google Scholar] [CrossRef]
- Liou, G.-Y.; Storz, P. Reactive oxygen species in cancer. Free Radic. Res. 2010, 44, 479–496. [Google Scholar] [CrossRef] [Green Version]
- Gorrini, C.; Harris, I.S.; Mak, T.W. Modulation of oxidative stress as an anticancer strategy. Nat. Rev. Drug Discov. 2013, 12, 931–947. [Google Scholar] [CrossRef]
- Bizhanova, A.; Kopp, P. Minireview: The sodium-iodide symporter NIS and pendrin in iodide homeostasis of the thyroid. Endocrinology 2009, 150, 1084–1090. [Google Scholar] [CrossRef] [Green Version]
- De Deken, X.; Wang, D.; Many, M.C.; Costagliola, S.; Libert, F.; Vassart, G.; Dumont, J.E.; Miot, F. Cloning of two human thyroid cDNAs encoding new members of the NADPH oxidase family. J. Biol. Chem. 2000, 275, 23227–23233. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dupuy, C.; Ohayon, R.; Valent, A.; Noël-Hudson, M.S.; Dème, D.; Virion, A. Purification of a novel flavoprotein involved in the thyroid NADPH oxidase. Cloning of the porcine and human cdnas. J. Biol. Chem. 1999, 274, 37265–37269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grasberger, H. Defects of thyroidal hydrogen peroxide generation in congenital hypothyroidism. Mol. Cell. Endocrinol. 2010, 322, 99–106. [Google Scholar] [CrossRef] [PubMed]
- Grasberger, H.; Refetoff, S. Identification of the Maturation Factor for Dual Oxidase. J. Biol. Chem. 2006, 281, 18269–18272. [Google Scholar] [CrossRef] [Green Version]
- Grasberger, H.; De Deken, X.; Miot, F.; Pohlenz, J.; Refetoff, S. Missense mutations of dual oxidase 2 (DUOX2) implicated in congenital hypothyroidism have impaired trafficking in cells reconstituted with DUOX2 maturation factor. Mol. Endocrinol. 2007, 21, 1408–1421. [Google Scholar] [CrossRef] [Green Version]
- Johnson, K.R.; Marden, C.C.; Ward-Bailey, P.; Gagnon, L.H.; Bronson, R.T.; Donahue, L.R. Congenital hypothyroidism, dwarfism, and hearing impairment caused by a missense mutation in the mouse dual oxidase 2 gene, Duox2. Mol. Endocrinol. 2007, 21, 1593–1602. [Google Scholar] [CrossRef]
- Moreno, J.C.; Bikker, H.; Kempers, M.J.E.; van Trotsenburg, A.S.P.; Baas, F.; de Vijlder, J.J.M.; Vulsma, T.; Ris-Stalpers, C. Inactivating Mutations in the Gene for Thyroid Oxidase 2 ( THOX2 ) and Congenital Hypothyroidism. N. Engl. J. Med. 2002, 347, 95–102. [Google Scholar] [CrossRef] [Green Version]
- Corvilain, B.; Collyn, L.; Van Sande, J.; Dumont, J.E. Stimulation by iodide of H2O2 generation in thyroid slices from several species. Am. J. Physiol.-Endocrinol. Metab. 2000, 278, E692–E699. [Google Scholar] [CrossRef] [Green Version]
- Song, Y.; Driessens, N.; Costa, M.; De Deken, X.; Detours, V.; Corvilain, B.; Maenhaut, C.; Miot, F.; Van Sande, J.; Many, M.C.; et al. Review: Roles of hydrogen peroxide in thyroid physiology and disease. J. Clin. Endocrinol. Metab. 2007, 92, 3764–3773. [Google Scholar] [CrossRef]
- Coclet, J.; Foureau, F.; Ketelbant, P.; Galand, P.; Dumont, J.E. Cell population kinetics in dog and human adult thyroid. Clin. Endocrinol. 1989, 31, 655–665. [Google Scholar] [CrossRef]
- Ekholm, R. Iodination of thyroglobulin. Mol. Cell. Endocrinol. 1981, 24, 141–163. [Google Scholar] [CrossRef]
- Versteyhe, S.; Driessens, N.; Ghaddhab, C.; Tarabichi, M.; Hoste, C.; Dumont, J.-E.; Miot, F.; Corvilain, B.; Detours, V. Comparative analysis of the thyrocytes and T cells: Responses to H2O2 and radiation reveals an H2O2-induced antioxidant transcriptional program in thyrocytes. J. Clin. Endocrinol. Metab. 2013, 98, E1645–E1654. [Google Scholar] [CrossRef] [PubMed]
- Howie, A.F.; Arthur, J.R.; Nicol, F.; Walker, S.W.; Beech, S.G.; Beckett, G.J. Identification of a 57-kilodalton selenoprotein in human thyrocytes as thioredoxin reductase and evidence that its expression is regulated through the calcium-phosphoinositol signaling pathway. J. Clin. Endocrinol. Metab. 1998, 83, 2052–2058. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; Lee, T.H.; Park, E.S.; Suh, J.M.; Park, S.J.; Chung, H.K.; Kwon, O.Y.; Kim, Y.K.; Ro, H.K.; Shong, M. Role of peroxiredoxins in regulating intracellular hydrogen peroxide and hydrogen peroxide-induced apoptosis in thyroid cells. J. Biol. Chem. 2000, 275, 18266–18270. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ziros, P.G.; Habeos, I.G.; Chartoumpekis, D.V.; Ntalampyra, E.; Somm, E.; Renaud, C.O.; Bongiovanni, M.; Trougakos, I.P.; Yamamoto, M.; Kensler, T.W.; et al. NFE2-Related transcription factor 2 coordinates antioxidant defense with thyroglobulin production and iodination in the thyroid gland. Thyroid 2018, 28, 780–798. [Google Scholar] [CrossRef] [PubMed]
- Hybertson, B.M.; Gao, B.; Bose, S.K.; McCord, J.M. Oxidative stress in health and disease: The therapeutic potential of Nrf2 activation. Mol. Asp. Med. 2011, 32, 234–246. [Google Scholar] [CrossRef]
- Hornsveld, M.; Dansen, T.B. The Hallmarks of Cancer from a Redox Perspective. Antioxid. Redox Signal. 2016, 25, 300–325. [Google Scholar] [CrossRef]
- Muzza, M.; Colombo, C.; Cirello, V.; Perrino, M.; Vicentini, L.; Fugazzola, L. Oxidative stress and the subcellular localization of the telomerase reverse transcriptase (TERT) in papillary thyroid cancer. Mol. Cell. Endocrinol. 2016, 431, 54–61. [Google Scholar] [CrossRef]
- Weyemi, U.; Caillou, B.; Talbot, M.; Ameziane-El-Hassani, R.; Lacroix, L.; Lagent-Chevallier, O.; Al Ghuzlan, A.; Roos, D.; Bidart, J.-M.; Virion, A.; et al. Intracellular expression of reactive oxygen species-generating NADPH oxidase NOX4 in normal and cancer thyroid tissues. Endocr. Relat. Cancer 2010, 17, 27–37. [Google Scholar] [CrossRef]
- Caillou, B.; Dupuy, C.; Lacroix, L.; Nocera, M.; Talbot, M.; Ohayon, R.; Dème, D.; Bidart, J.M.; Schlumberger, M.; Virion, A. Expression of reduced nicotinamide adenine dinucleotide phosphate oxidase (Thox, LNOX, Duox) genes and proteins in human thyroid tissues. J. Clin. Endocrinol. Metab. 2001, 86, 3351–3358. [Google Scholar] [CrossRef]
- Weyemi, U.; Lagente-Chevallier, O.; Boufraqech, M.; Prenois, F.; Courtin, F.; Caillou, B.; Talbot, M.; Dardalhon, M.; Al Ghuzlan, A.; Bidart, J.-M.; et al. ROS-generating NADPH oxidase NOX4 is a critical mediator in oncogenic H-Ras-induced DNA damage and subsequent senescence. Oncogene 2012, 31, 1117–1129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Williams, D. Radiation carcinogenesis: Lessons from Chernobyl. Oncogene 2008, 27, S9–S18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ameziane-El-Hassani, R.; Boufraqech, M.; Lagente-Chevallier, O.; Weyemi, U.; Talbot, M.; Métivier, D.; Courtin, F.; Bidart, J.M.; El Mzibri, M.; Schlumberger, M.; et al. Role of H2O2 in RET/PTC1 chromosomal rearrangement produced by ionizing radiation in human thyroid cells. Cancer Res. 2010, 70, 4123–4132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bedard, K.; Krause, K.-H. The NOX family of ROS-generating NADPH oxidases: Physiology and pathophysiology. Physiol. Rev. 2007, 87, 245–313. [Google Scholar] [CrossRef]
- Chen, K.; Kirber, M.T.; Xiao, H.; Yang, Y.; Keaney, J.F. Regulation of ROS signal transduction by NADPH oxidase 4 localization. J. Cell Biol. 2008, 181, 1129–1139. [Google Scholar] [CrossRef] [Green Version]
- Graham, K.A.; Kulawiec, M.; Owens, K.M.; Li, X.; Desouki, M.M.; Chandra, D.; Singh, K.K. NADPH oxidase 4 is an oncoprotein localized to mitochondria. Cancer Biol. Ther. 2010, 10, 223–231. [Google Scholar] [CrossRef] [Green Version]
- Lee, C.F.; Ullevig, S.; Kim, H.S.; Asmis, R. Regulation of Monocyte Adhesion and Migration by Nox4. PLoS ONE 2013, 8, e66964. [Google Scholar] [CrossRef] [Green Version]
- von Löhneysen, K.; Noack, D.; Wood, M.R.; Friedman, J.S.; Knaus, U.G. Structural insights into Nox4 and Nox2: Motifs involved in function and cellular localization. Mol. Cell. Biol. 2010, 30, 961–975. [Google Scholar] [CrossRef] [Green Version]
- Ambasta, R.K.; Kumar, P.; Griendling, K.K.; Schmidt, H.H.H.W.; Busse, R.; Brandes, R.P. Direct interaction of the novel Nox proteins with p22phox is required for the formation of a functionally active NADPH oxidase. J. Biol. Chem. 2004, 279, 45935–45941. [Google Scholar] [CrossRef] [Green Version]
- Fortunato, R.S.; Braga, W.M.O.; Ortenzi, V.H.; Rodrigues, D.C.; Andrade, B.M.; Miranda-Alves, L.; Rondinelli, E.; Dupuy, C.; Ferreira, A.C.F.; Carvalho, D.P. Sexual Dimorphism of Thyroid Reactive Oxygen Species Production Due to Higher NADPH Oxidase 4 Expression in Female Thyroid Glands. Thyroid 2013, 23, 111–119. [Google Scholar] [CrossRef]
- Boudreau, H.E.; Casterline, B.W.; Rada, B.; Korzeniowska, A.; Leto, T.L. Nox4 involvement in TGF-beta and SMAD3-driven induction of the epithelial-to-mesenchymal transition and migration of breast epithelial cells. Free. Radic. Biol. Med. 2013, 53, 1489–1499. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jafari, N.; Kim, H.; Park, R.; Li, L.; Jang, M.; Morris, A.J.; Park, J.; Huang, C. CRISPR-Cas9 mediated nox4 knockout inhibits cell proliferation and invasion in hela cells. PLoS ONE 2017, 12, e0170327. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ju, H.-Q.; Ying, H.; Tian, T.; Ling, J.; Fu, J.; Lu, Y.; Wu, M.; Yang, L.; Achreja, A.; Chen, G.; et al. Mutant Kras- and p16-regulated NOX4 activation overcomes metabolic checkpoints in development of pancreatic ductal adenocarcinoma. Nat. Commun. 2017, 8, 14437. [Google Scholar] [CrossRef] [Green Version]
- Lin, X.; Yang, L.; Fu, S.; Lin, W.; Gao, Y. Overexpression of NOX4 predicts poor prognosis and promotes tumor progression in human colorectal cancer. Oncotarget 2017, 8, 33586–33600. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meitzler, J.L.; Makhlouf, H.R.; Antony, S.; Wu, Y.; Butcher, D.; Jiang, G.; Juhasz, A.; Lu, J.; Dahan, I.; Jansen-Dürr, P.; et al. Decoding NADPH oxidase 4 expression in human tumors. Redox Biol. 2017, 13, 182–195. [Google Scholar] [CrossRef] [PubMed]
- Zeng, C.; Wu, Q.; Wang, J.; Yao, B.; Ma, L.; Yang, Z.; Li, J.; Liu, B. NOX4 supports glycolysis and promotes glutamine metabolism in non-small cell lung cancer cells. Free Radic. Biol. Med. 2016, 101, 236–248. [Google Scholar] [CrossRef]
- Tang, P.; Dang, H.; Huang, J.; Xu, T.; Yuan, P.; Hu, J.; Sheng, J. NADPH oxidase NOX4 is a glycolytic regulator through mROS-HIF1α axis in thyroid carcinomas. Sci. Rep. 2018, 8, 15897. [Google Scholar] [CrossRef]
- Oglio, R.; Salvarredi, L.; Rossich, L.; Copelli, S.; Pisarev, M.; Juvenal, G.; Thomasz, L. Participation of NADPH 4 oxidase in thyroid regulation. Mol. Cell. Endocrinol. 2019, 480, 65–73. [Google Scholar] [CrossRef]
- Lenaz, G. The Mitochondrial Production of Reactive Oxygen Species: Mechanisms and Implications in Human Pathology. IUBMB Life (Int. Union Biochem. Mol. Biol. Life) 2001, 52, 159–164. [Google Scholar] [CrossRef]
- Máximo, V.; Lima, J.; Prazeres, H.; Soares, P.; Sobrinho-Simões, M. The biology and the genetics of Hürthle cell tumors of the thyroid. Endocr. Relat. Cancer 2012, 19, R131–R147. [Google Scholar] [CrossRef] [Green Version]
- Bonora, E.; Porcelli, A.M.; Gasparre, G.; Biondi, A.; Ghelli, A.; Carelli, V.; Baracca, A.; Tallini, G.; Martinuzzi, A.; Lenaz, G.; et al. Defective Oxidative Phosphorylation in Thyroid Oncocytic Carcinoma Is Associated with Pathogenic Mitochondrial DNA Mutations Affecting Complexes I and III. Cancer Res. 2006, 66, 6087–6096. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Máximo, V.; Sobrinho-Simões, M. Hürthle cell tumours of the thyroid. A review with emphasis on mitochondrial abnormalities with clinical relevance. Virchows Arch. 2000, 437, 107–115. [Google Scholar] [CrossRef] [PubMed]
- Cavadas, B.; Pereira, J.B.; Correia, M.; Fernandes, V.; Eloy, C.; Sobrinho-Simões, M.; Soares, P.; Samuels, D.C.; Máximo, V.; Pereira, L. Genomic and transcriptomic characterization of the mitochondrial-rich oncocytic phenotype on a thyroid carcinoma background. Mitochondrion 2019, 46, 123–133. [Google Scholar] [CrossRef]
- Shanmugasundaram, K.; Nayak, B.K.; Friedrichs, W.E.; Kaushik, D.; Rodriguez, R.; Block, K. NOX4 functions as a mitochondrial energetic sensor coupling cancer metabolic reprogramming to drug resistance. Nat. Commun. 2017, 8, 997. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wen, Q.L.; Yi, H.Q.; Yang, K.; Yin, C.T.; Yin, W.J.; Xiang, F.Y.; Bao, M.; Shuai, J.; Song, Y.W.; Ge, M.H.; et al. Role of oncogene PIM-1 in the development and progression of papillary thyroid carcinoma: Involvement of oxidative stress. Mol. Cell. Endocrinol. 2021, 523, 111144. [Google Scholar] [CrossRef] [PubMed]
- Cammarota, F.; Fiscardi, F.; Esposito, T.; Vita, G.; Salvatore, M.; Laukkanen, M.O. Clinical relevance of thyroid cell models in redox research. Cancer Cell Int. 2015, 15, 113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stanley, J.A.; Neelamohan, R.; Suthagar, E.; Vengatesh, G.; Jayakumar, J.; Chandrasekaran, M.; Banu, S.K.; Aruldhas, M.M. Lipid peroxidation and antioxidants status in human malignant and non-malignant thyroid tumours. Hum. Exp. Toxicol. 2016, 35, 585–597. [Google Scholar] [CrossRef]
- Eng, P.H.K.; Cardona, G.R.; Fang, S.; Previti, M.; Alex, S.; Carrasco, N.; Chin, W.W.; Braverman, L.E. Escape from the Acute Wolff-Chaikoff Effect Is Associated with a Decrease in Thyroid Sodium/Iodide Symporter Messenger Ribonucleic Acid and Protein 1. Endocrinology 1999, 140, 3404–3410. [Google Scholar] [CrossRef]
- Calil-Silveira, J.; Serrano-Nascimento, C.; Kopp, P.A.; Nunes, M.T. Iodide excess regulates its own efflux: A possible involvement of pendrin. Am. J. Physiol. Cell Physiol. 2016, 310, C576–C582. [Google Scholar] [CrossRef] [Green Version]
- Leoni, S.G.; Kimura, E.T.; Santisteban, P.; De la Vieja, A. Regulation of thyroid oxidative state by thioredoxin reductase has a crucial role in thyroid responses to iodide excess. Mol. Endocrinol. 2011, 25, 1924–1935. [Google Scholar] [CrossRef] [Green Version]
- Vitale, M.; Di Matola, T.; D’Ascoli, F.; Salzano, S.; Bogazzi, F.; Fenzi, G.; Martino, E.; Rossi, G. Iodide Excess Induces Apoptosis in Thyroid Cells through a p53-Independent Mechanism Involving Oxidative Stress1. Endocrinology 2000, 141, 598–605. [Google Scholar] [CrossRef] [PubMed]
- Yao, X.; Li, M.; He, J.; Zhang, G.; Wang, M.; Ma, J.; Sun, Y.; Zhang, W.; Li, L. Effect of early acute high concentrations of iodide exposure on mitochondrial superoxide production in FRTL cells. Free Radic. Biol. Med. 2012, 52, 1343–1352. [Google Scholar] [CrossRef] [PubMed]
- Serrano-Nascimento, C.; da Silva Teixeira, S.; Nicola, J.P.; Nachbar, R.T.; Masini-Repiso, A.M.; Nunes, M.T. The acute inhibitory effect of iodide excess on sodium/iodide symporter expression and activity involves the PI3K/Akt signaling pathway. Endocrinology 2014, 155, 1145–1156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arriagada, A.A.; Albornoz, E.; Opazo, M.C.; Becerra, A.; Vidal, G.; Fardella, C.; Michea, L.; Carrasco, N.; Simon, F.; Elorza, A.A.; et al. Excess iodide induces an acute inhibition of the sodium/iodide symporter in thyroid male rat cells by increasing reactive oxygen species. Endocrinology 2015, 156, 1540–1551. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- da Silva, M.M.; Xavier, L.L.F.; Gonçalves, C.F.L.; Santos-Silva, A.P.; Paiva-Melo, F.D.; de Freitas, M.L.; Fortunato, R.S.; Miranda-Alves, L.; Ferreira, A.C.F. Bisphenol A increases hydrogen peroxide generation by thyrocytes both in vivo and in vitro. Endocr. Connect. 2018, 7, 1196–1207. [Google Scholar] [CrossRef] [Green Version]
- Agrawal, N.; Akbani, R.; Aksoy, B.A.; Ally, A.; Arachchi, H.; Asa, S.L.; Auman, J.T.; Balasundaram, M.; Balu, S.; Baylin, S.B.; et al. Integrated Genomic Characterization of Papillary Thyroid Carcinoma. Cell 2014, 159, 676–690. [Google Scholar] [CrossRef] [Green Version]
- Eloy, C.; Santos, J.; Soares, P.; Sobrinho-Simões, M. The preeminence of growth pattern and invasiveness and the limited influence of BRAF and RAS mutations in the occurrence of papillary thyroid carcinoma lymph node metastases. Virchows Arch. 2011, 459, 265–276. [Google Scholar] [CrossRef]
- Kim, T.H.; Park, Y.J.; Lim, J.A.; Ahn, H.Y.; Lee, E.K.; Lee, Y.J.; Kim, K.W.; Hahn, S.K.; Youn, Y.K.; Kim, K.H.; et al. The association of the BRAFV600E mutation with prognostic factors and poor clinical outcome in papillary thyroid cancer. Cancer 2012, 118, 1764–1773. [Google Scholar] [CrossRef]
- Trovisco, V.; Soares, P.; Preto, A.; De Castro, I.V.; Lima, J.; Castro, P.; Máximo, V.; Botelho, T.; Moreira, S.; Meireles, A.M.; et al. Type and prevalence of BRAF mutations are closely associated with papillary thyroid carcinoma histotype and patients’ age but not with tumour aggressiveness. Virchows Arch. 2005, 446, 589–595. [Google Scholar] [CrossRef]
- Xing, M.; Westra, W.H.; Tufano, R.P.; Cohen, Y.; Rosenbaum, E.; Rhoden, K.J.; Carson, K.A.; Vasko, V.; Larin, A.; Tallini, G.; et al. BRAF Mutation Predicts a Poorer Clinical Prognosis for Papillary Thyroid Cancer. J. Clin. Endocrinol. Metab. 2005, 90, 6373–6379. [Google Scholar] [CrossRef] [Green Version]
- Riesco-Eizaguirre, G.; Rodriguez, I.; De la Vieja, a.; Costamagna, E.; Carrasco, N.; Nistal, M.; Santisteban, P. The BRAFV600E Oncogene Induces Transforming Growth Factor Secretion Leading to Sodium Iodide Symporter Repression and Increased Malignancy in Thyroid Cancer. Cancer Res. 2009, 69, 8317–8325. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chakravarty, D.; Santos, E.; Ryder, M.; Knauf, J.A.; Liao, X.-H.; West, B.L.; Bollag, G.; Kolesnick, R.; Thin, T.H.; Rosen, N.; et al. Small-molecule MAPK inhibitors restore radioiodine incorporation in mouse thyroid cancers with conditional BRAF activation. J. Clin. Investig. 2011, 121, 4700–4711. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Riesco-Eizaguirre, G.; Gutiérrez-Martínez, P.; García-Cabezas, M.A.; Nistal, M.; Santisteban, P. The oncogene BRAFV600E is associated with a high risk of recurrence and less differentiated papillary thyroid carcinoma due to the impairment of Na+/I− targeting to the membrane. Endocr. Relat. Cancer 2006, 13, 257–269. [Google Scholar] [CrossRef] [PubMed]
- Romei, C.; Ciampi, R.; Faviana, P.; Agate, L.; Molinaro, E.; Bottici, V.; Basolo, F.; Miccoli, P.; Pacini, F.; Pinchera, A.; et al. BRAFV600E mutation, but not RET/PTC rearrangements, is correlated with a lower expression of both thyroperoxidase and sodium iodide symporter genes in papillary thyroid cancer. Endocr. Relat. Cancer 2008, 15, 511–520. [Google Scholar] [CrossRef]
- Sabra, M.M.; Dominguez, J.M.; Grewal, R.K.; Larson, S.M.; Ghossein, R.A.; Tuttle, R.M.; Fagin, J.A. Clinical outcomes and molecular profile of differentiated thyroid cancers with radioiodine-avid distant metastases. J. Clin. Endocrinol. Metab. 2013, 98, 829–836. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pratilas, C.A.; Taylor, B.S.; Ye, Q.; Viale, A.; Sander, C.; Solit, D.B.; Rosen, N. V600EBRAF is associated with disabled feedback inhibition of RAF-MEK signaling and elevated transcriptional output of the pathway. Proc. Natl. Acad. Sci. USA 2009, 106, 4519–4524. [Google Scholar] [CrossRef] [Green Version]
- Nagarajah, J.; Le, M.; Knauf, J.A.; Ferrandino, G.; Montero-Conde, C.; Pillarsetty, N.; Bolaender, A.; Irwin, C.; Krishnamoorthy, G.P.; Saqcena, M.; et al. Sustained ERK inhibition maximizes responses of BrafV600E thyroid cancers to radioiodine. J. Clin. Investig. 2016, 126, 4119–4124. [Google Scholar] [CrossRef] [Green Version]
- Brown, S.R.; Hall, A.; Buckley, H.L.; Flanagan, L.; Gonzalez De Castro, D.; Farnell, K.; Moss, L.; Gregory, R.; Newbold, K.; Du, Y.; et al. Investigating the potential clinical benefit of Selumetinib in resensitising advanced iodine refractory differentiated thyroid cancer to radioiodine therapy (SEL-I-METRY): Protocol for a multicentre UK single arm phase II trial. BMC Cancer 2019, 19, 582. [Google Scholar] [CrossRef]
- Dunn, L.A.; Sherman, E.J.; Baxi, S.S.; Tchekmedyian, V.; Grewal, R.K.; Larson, S.M.; Pentlow, K.S.; Haque, S.; Tuttle, R.M.; Sabra, M.M.; et al. Vemurafenib Redifferentiation of BRAF Mutant, RAI-Refractory Thyroid Cancers. J. Clin. Endocrinol. Metab. 2019, 104, 1417–1428. [Google Scholar] [CrossRef]
- Huillard, O.; Tenenbaum, F.; Clerc, J.; Goldwasser, F. Restoring Radioiodine Uptake in BRAF V600E–Mutated Papillary Thyroid Cancer. J. Endocr. Soc. 2017, 1, 285–287. [Google Scholar] [CrossRef] [Green Version]
- Iravani, A.; Solomon, B.; Pattison, D.A.; Jackson, P.; Ravi Kumar, A.; Kong, G.; Hofman, M.S.; Akhurst, T.; Hicks, R.J. Mitogen-Activated Protein Kinase Pathway Inhibition for Redifferentiation of Radioiodine Refractory Differentiated Thyroid Cancer: An Evolving Protocol. Thyroid 2019, 29, 1634–1645. [Google Scholar] [CrossRef] [PubMed]
- Leboulleux, S.; Dupuy, C.; Lacroix, L.; Attard, M.; Grimaldi, S.; Corre, R.; Ricard, M.; Nasr, S.; Berdelou, A.; Hadoux, J.; et al. Redifferentiation of a BRAF K601E -Mutated Poorly Differentiated Thyroid Cancer Patient with Dabrafenib and Trametinib Treatment. Thyroid 2019, 29, 735–742. [Google Scholar] [CrossRef]
- Taki, K.; Kogai, T.; Kanamoto, Y.; Hershman, J.M.; Brent, G.A. A Thyroid-Specific Far-Upstream Enhancer in the Human Sodium/Iodide Symporter Gene Requires Pax-8 Binding and Cyclic Adenosine 3′,5′-Monophosphate Response Element-Like Sequence Binding Proteins for Full Activity and Is Differentially Regulated in Normal and Thyroid Cancer Cells. Mol. Endocrinol. 2002, 16, 2266–2282. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ohno, M.; Zannini, M.; Levy, O.; Carrasco, N.; di Lauro, R. The paired-domain transcription factor Pax8 binds to the upstream enhancer of the rat sodium/iodide symporter gene and participates in both thyroid-specific and cyclic-AMP-dependent transcription. Mol. Cell. Biol. 1999, 19, 2051–2060. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leoni, S.G.; Sastre-Perona, A.; De la Vieja, A.; Santisteban, P. Selenium Increases Thyroid-Stimulating Hormone-Induced Sodium/Iodide Symporter Expression Through Thioredoxin/Apurinic/Apyrimidinic Endonuclease 1-Dependent Regulation of Paired Box 8 Binding Activity. Antioxid. Redox Signal. 2016, 24, 855–866. [Google Scholar] [CrossRef]
- Puppin, C.; Arturi, F.; Ferretti, E.; Russo, D.; Sacco, R.; Tell, G.; Damante, G.; Filetti, S. Transcriptional regulation of human sodium/iodide symporter gene: A role for redox factor-1. Endocrinology 2004, 145, 1290–1293. [Google Scholar] [CrossRef]
- Galrão, A.L.; Camargo, R.Y.; Friguglietti, C.U.; Moraes, L.; Cerutti, J.M.; Serrano-Nascimento, C.; Suzuki, M.F.; Medeiros-Neto, G.; Rubio, I.G.S. Hypermethylation of a new distal sodium/iodide symporter (NIS) enhancer (NDE) Is associated with reduced nis expression in thyroid tumors. J. Clin. Endocrinol. Metab. 2014, 99, 944–952. [Google Scholar] [CrossRef] [Green Version]
- Venkataraman, G.M.; Yatin, M.; Marcinek, R.; Ain, K.B. Restoration of iodide uptake in dedifferentiated thyroid carcinoma: Relationship to human Na+/I-symporter gene methylation status. J. Clin. Endocrinol. Metab. 1999, 84, 2449–2457. [Google Scholar] [CrossRef]
- Choi, Y.W.; Kim, H.-J.; Kim, Y.H.; Park, S.H.; Chwae, Y.J.; Lee, J.; Soh, E.Y.; Kim, J.-H.; Park, T.J. B-RafV600E inhibits sodium iodide symporter expression via regulation of DNA methyltransferase 1. Exp. Mol. Med. 2014, 46, e120. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.; Liu, D.; Murugan, A.K.; Liu, Z.; Xing, M. Histone deacetylation of NIS promoter underlies BRAF V600E-promoted NIS silencing in thyroid cancer. Endocr. Relat. Cancer 2014, 21, 161–173. [Google Scholar] [CrossRef]
- Cheng, W.; Liu, R.; Zhu, G.; Wang, H.; Xing, M. Robust thyroid gene expression and radioiodine uptake induced by simultaneous suppression of BRAF V600E and histone deacetylase in thyroid cancer cells. J. Clin. Endocrinol. Metab. 2016, 101, 962–971. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fortunati, N.; Catalano, M.G.; Arena, K.; Brignardello, E.; Piovesan, A.; Boccuzzi, G. Valproic Acid Induces the Expression of the Na+/I− Symporter and Iodine Uptake in Poorly Differentiated Thyroid Cancer Cells. J. Clin. Endocrinol. Metab. 2004, 89, 1006–1009. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Furuya, F.; Shimura, H.; Suzuki, H.; Taki, K.; Ohta, K.; Haraguchi, K.; Onaya, T.; Endo, T.; Kobayashi, T. Histone deacetylase inhibitors restore radioiodide uptake and retention in poorly differentiated and anaplastic thyroid cancer cells by expression of the sodium/iodide symporter thyroperoxidase and thyroglobulin. Endocrinology 2004, 145, 2865–2875. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Provenzano, M.J.; Fitzgerald, M.P.; Krager, K.; Domann, F.E. Increased iodine uptake in thyroid carcinoma after treatment with sodium butyrate and decitabine (5-Aza-dC). Otolaryngol. Head Neck Surg. 2007, 137, 722–728. [Google Scholar] [CrossRef] [PubMed]
- Zarnegar, R.; Brunaud, L.; Kanauchi, H.; Wong, M.; Fung, M.; Ginzinger, D.; Duh, Q.Y.; Clark, O.H.; Kinder, B.K.; Zeiger, M.A.; et al. Increasing the effectiveness of radioactive iodine therapy in the treatment of thyroid cancer using Trichostatin A, a histone deacetylase inhibitor. Surgery 2002, 132, 984–990. [Google Scholar] [CrossRef] [PubMed]
- Riesco-Eizaguirre, G.; Wert-Lamas, L.; Perales-Paton, J.; Sastre-Perona, A.; Fernandez, L.P.; Santisteban, P. The miR-146b-3p/PAX8/NIS regulatory circuit modulates the differentiation phenotype and function of thyroid cells during carcinogenesis. Cancer Res. 2015, 75, 4119–4130. [Google Scholar] [CrossRef] [Green Version]
- Haghpanah, V.; Fallah, P.; Tavakoli, R.; Naderi, M.; Samimi, H.; Soleimani, M.; Larijani, B. Antisense-miR-21 enhances differentiation/apoptosis and reduces cancer stemness state on anaplastic thyroid cancer. Tumor Biol. 2016, 37, 1299–1308. [Google Scholar] [CrossRef]
- Bhat, A.V.; Hora, S.; Pal, A.; Jha, S.; Taneja, R. Stressing the (Epi)Genome: Dealing with Reactive Oxygen Species in Cancer. Antioxid. Redox Signal. 2018, 29, 1273–1292. [Google Scholar] [CrossRef]
- Kang, K.A.; Zhang, R.; Kim, G.Y.; Bae, S.C.; Hyun, J.W. Epigenetic changes induced by oxidative stress in colorectalcancer cells: Methylation of tumor suppressor RUNX3. Tumor Biol. 2012, 33, 403–412. [Google Scholar] [CrossRef]
- O’Hagan, H.M.; Wang, W.; Sen, S.; DeStefano Shields, C.; Lee, S.S.; Zhang, Y.W.; Clements, E.G.; Cai, Y.; Van Neste, L.; Easwaran, H.; et al. Oxidative Damage Targets Complexes Containing DNA Methyltransferases, SIRT1, and Polycomb Members to Promoter CpG Islands. Cancer Cell 2011, 20, 606–619. [Google Scholar] [CrossRef] [Green Version]
- Jajoo, S.; Mukherjea, D.; Kaur, T.; Sheehan, K.E.; Sheth, S.; Borse, V.; Rybak, L.P.; Ramkumar, V. Essential role of NADPH oxidase-dependent reactive oxygen species generation in regulating MicroRNA-21 expression and function in prostate cancer. Antioxid. Redox Signal. 2013, 19, 1863–1876. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ameziane El Hassani, R.; Buffet, C.; Leboulleux, S.; Dupuy, C. Oxidative stress in thyroid carcinomas: Biological and clinical significance. Endocr. Relat. Cancer 2019, 26, R131–R143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H.; Peng, C.; Zhu, C.; Nie, S.; Qian, X.; Shi, Z.; Shi, M.; Liang, Y.; Ding, X.; Zhang, S.; et al. Hypoxia promotes the metastasis of pancreatic cancer through regulating NOX4/KDM5A-mediated histone methylation modification changes in a HIF1A-independent manner. Clin. Epigenetics 2021, 13, 18. [Google Scholar] [CrossRef] [PubMed]
- Boelaert, K.; Smith, V.E.; Stratford, A.L.; Kogai, T.; Tannahill, L.A.; Watkinson, J.C.; Eggo, M.C.; Franklyn, J.A.; McCabe, C.J. PTTG and PBF repress the human sodium iodide symporter. Oncogene 2007, 26, 4344–4356. [Google Scholar] [CrossRef] [Green Version]
- Read, M.L.; Lewy, G.D.; Fong, J.C.W.; Sharma, N.; Seed, R.I.; Smith, V.E.; Gentilin, E.; Warfield, A.; Eggo, M.C.; Knauf, J.A.; et al. Proto-oncogene PBF/PTTG1IP regulates thyroid cell growth and represses radioiodide treatment. Cancer Res. 2011, 71, 6153–6164. [Google Scholar] [CrossRef] [Green Version]
- Smith, V.E.; Read, M.L.; Turnell, A.S.; Watkins, R.J.; Watkinson, J.C.; Lewy, G.D.; Fong, J.C.W.; James, S.R.; Eggo, M.C.; Boelaert, K.; et al. A novel mechanism of sodium iodide symporter repression in differentiated thyroid cancer. J. Cell Sci. 2009, 122, 3393–3402. [Google Scholar] [CrossRef] [Green Version]
- Smith, V.E.; Sharma, N.; Watkins, R.J.; Read, M.L.; Ryan, G.A.; Kwan, P.P.; Martin, A.; Watkinson, J.C.; Boelaert, K.; Franklyn, J.A.; et al. Manipulation of PBF/PTTG1IP phosphorylation status; a potential new therapeutic strategy for improving radioiodine uptake in thyroid and other tumors. J. Clin. Endocrinol. Metab. 2013, 98, 2876–2886. [Google Scholar] [CrossRef] [Green Version]
- Giannoni, E.; Chiarugi, P. Redox circuitries driving Src regulation. Antioxid. Redox Signal. 2014, 20, 2011–2025. [Google Scholar] [CrossRef]
- Kim, H.; Sung, J.Y.; Park, E.K.; Kho, S.; Koo, K.H.; Park, S.Y.; Goh, S.H.; Jeon, Y.K.; Oh, S.; Park, B.K.; et al. Regulation of anoikis resistance by NADPH oxidase 4 and epidermal growth factor receptor. Br. J. Cancer 2017, 116, 370–381. [Google Scholar] [CrossRef] [Green Version]
- Xi, G.; Shen, X.; Maile, L.A.; Wai, C.; Gollahon, K.; Clemmons, D.R. Hyperglycemia enhances IGF-I-stimulated Src activation via increasing Nox4-derived reactive oxygen species in a PKCζ-dependent manner in vascular smooth muscle cells. Diabetes 2012, 61, 104–113. [Google Scholar] [CrossRef] [Green Version]
- Lan, L.; Basourakos, S.; Cui, D.; Zuo, X.; Deng, W.; Huo, L.; Chen, L.; Zhang, G.; Deng, L.; Shi, B.; et al. Inhibiting β-catenin expression promotes efficiency of radioiodine treatment in aggressive follicular thyroid cancer cells probably through mediating NIS localization. Oncol. Rep. 2017, 37, 426–434. [Google Scholar] [CrossRef] [PubMed]
- Burrows, N.; Resch, J.; Cowen, R.L.; Von Wasielewski, R.; Hoang-Vu, C.; West, C.M.; Williams, K.J.; Brabant, G. Expression of hypoxia-inducible factor 1α in thyroid carcinomas. Endocr. Relat. Cancer 2010, 17, 61–72. [Google Scholar] [CrossRef] [PubMed]
- Rossi, E.D.; Straccia, P.; Palumbo, M.; Stigliano, E.; Revelli, L.; Lombardi, C.P.; Santeusanio, G.; Pontecorvi, A.; Fadda, G. Diagnostic and prognostic role of HBME-1, galectin-3, and β-catenin in poorly differentiated and anaplastic thyroid carcinomas. Appl. Immunohistochem. Mol. Morphol. 2013, 21, 237–241. [Google Scholar] [CrossRef]
- Funato, Y.; Michiue, T.; Asashima, M.; Miki, H. The thioredoxin-related redox-regulating protein nucleoredoxin inhibits wnt–β-catenin signalling through dishevelled. Nat. Cell Biol. 2006, 8, 501–508. [Google Scholar] [CrossRef] [PubMed]
- Jun, J.C.; Rathore, A.; Younas, H.; Gilkes, D.; Polotsky, V.Y. Hypoxia-Inducible Factors and Cancer. Curr. Sleep Med. Rep. 2017, 3, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Cazarin, J.; Andrade, B.; Carvalho, D. AMP-Activated Protein Kinase Activation Leads to Lysome-Mediated NA+/I−-Symporter Protein Degradation in Rat Thyroid Cells. Horm. Metab. Res. 2014, 46, 313–317. [Google Scholar] [CrossRef]
- Chai, W.; Ye, F.; Zeng, L.; Li, Y.; Yang, L. HMGB1-mediated autophagy regulates sodium/iodide symporter protein degradation in thyroid cancer cells. J. Exp. Clin. Cancer Res. 2019, 38, 325. [Google Scholar] [CrossRef] [Green Version]
- Choi, S.L.; Kim, S.J.; Lee, K.T.; Kim, J.; Mu, J.; Birnbaum, M.J.; Soo Kim, S.; Ha, J. The regulation of AMP-activated protein kinase by H2O2. Biochem. Biophys. Res. Commun. 2001, 287, 92–97. [Google Scholar] [CrossRef]
- Vidal, A.P.; Andrade, B.M.; Vaisman, F.; Cazarin, J.; Pinto, L.F.R.; Breitenbach, M.M.D.; Corbo, R.; Caroli-Bottino, A.; Soares, F.; Vaisman, M.; et al. AMP-activated protein kinase signaling is upregulated in papillary thyroid cancer. Eur. J. Endocrinol. 2013, 169, 521–528. [Google Scholar] [CrossRef] [Green Version]
- Vadysirisack, D.D.; Chen, E.S.W.; Zhang, Z.; Tsai, M.D.; Chang, G.D.; Jhiang, S.M. Identification of in vivo phosphorylation sites and their functional significance in the sodium iodide symporter. J. Biol. Chem. 2007, 282, 36820–36828. [Google Scholar] [CrossRef] [Green Version]
- Corcoran, A.; Cotter, T.G. Redox regulation of protein kinases. FEBS J. 2013, 280, 1944–1965. [Google Scholar] [CrossRef] [PubMed]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cazarin, J.; Dupuy, C.; Pires de Carvalho, D. Redox Homeostasis in Thyroid Cancer: Implications in Na+/I− Symporter (NIS) Regulation. Int. J. Mol. Sci. 2022, 23, 6129. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23116129
Cazarin J, Dupuy C, Pires de Carvalho D. Redox Homeostasis in Thyroid Cancer: Implications in Na+/I− Symporter (NIS) Regulation. International Journal of Molecular Sciences. 2022; 23(11):6129. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23116129
Chicago/Turabian StyleCazarin, Juliana, Corinne Dupuy, and Denise Pires de Carvalho. 2022. "Redox Homeostasis in Thyroid Cancer: Implications in Na+/I− Symporter (NIS) Regulation" International Journal of Molecular Sciences 23, no. 11: 6129. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23116129