
Synthesis and Anti-Cancer Activity of New Pyrazolinyl-Indole Derivatives: Pharmacophoric Interactions and Docking Studies for Identifying New EGFR Inhibitors

 ,
,  ,
,  , and
, and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Chemical Synthesis
2.2. General Procedure for the Synthesis of 1-(2-Aryl)-3-(1-H-indole-3-yl)prop-2-en-1-on (2a–d)
2.3. General Procedure for the Synthesis of (Substituted Aryl)-Pyrazolines (HD01–HD12)
2.3.1. 1-(3-(4-Hydroxyphenyl)-5-(1H-indol-3-yl)-4,5-dihydro-1H-pyrazol-1-yl)-2-phenylethanone, [HD01]
2.3.2. 1-(5-(1-H-Indol-3-yl)-3-(p-tolyl)-4,5-dihydro-1H-pyrazol-1-yl)-2-phenylethanone, [HD02]
2.3.3. 1-(3-(2-Hydroxyphenyl)-5-(1H-indol-3-yl)-4,5-dihydro-1H-pyrazol-1-yl)-2-phenylethanone, [HD03]
2.3.4. 1-(3-(4-Chlorophenyl)-5-(1H-indol-3-yl)-4,5-dihydro-1H-pyrazol-1-yl)-2-phenylethanone, [HD04]
2.3.5. 1-(3-(4-Chlorophenyl)-5-(1H-indol-3-yl)-4,5-dihydro-1H-pyrazol-1-yl)-2-phenoxyethanone, [HD05]
2.3.6. 1-(5-(1-H-Indol-3-yl)-3-(p-tolyl)-4,5-dihydro-1H-pyrazol-1-yl)-2-phenoxyethanone, [HD06]
2.3.7. 1-(3-(2-Hydroxyphenyl)-5-(1H-indol-3-yl)-4,5-dihydro-1H-pyrazol-1-yl)-2-phenoxyethanone, [HD07]
2.3.8. Phenyl-3-(4-hydroxyphenyl)-5-(1H-indol-3-yl)-4,5-dihydro-1H-pyrazole-1-carboxylate, [HD08]
2.3.9. (3-(4-Hydroxyphenyl)-5-(1H-indol-3-yl)-4,5-dihydro-1H-pyrazol-1-yl)(pyridin-4-yl)methanone, [HD09]
2.3.10. (5-(1-H-Indol-3-yl)-3-(p-tolyl)-4,5-dihydro-1H-pyrazol-1-yl)(pyridin-4-yl)methanone, [HD10]
2.3.11. (3-(2-Hydroxyphenyl)-5-(1H-indol-3-yl)-4,5-dihydro-1H-pyrazol-1-yl)(pyridin-4-yl)methanone, [HD11]
2.3.12. (3-(4-Chlorophenyl)-5-(1H-indol-3-yl)-4,5-dihydro-1H-pyrazol-1-yl)(pyridin-4-yl)methanone, [HD12]
2.4. Anti-Cancer Screening Methodology
2.5. Molecular Modelling
3. Results and Discussion
3.1. Chemical Synthesis
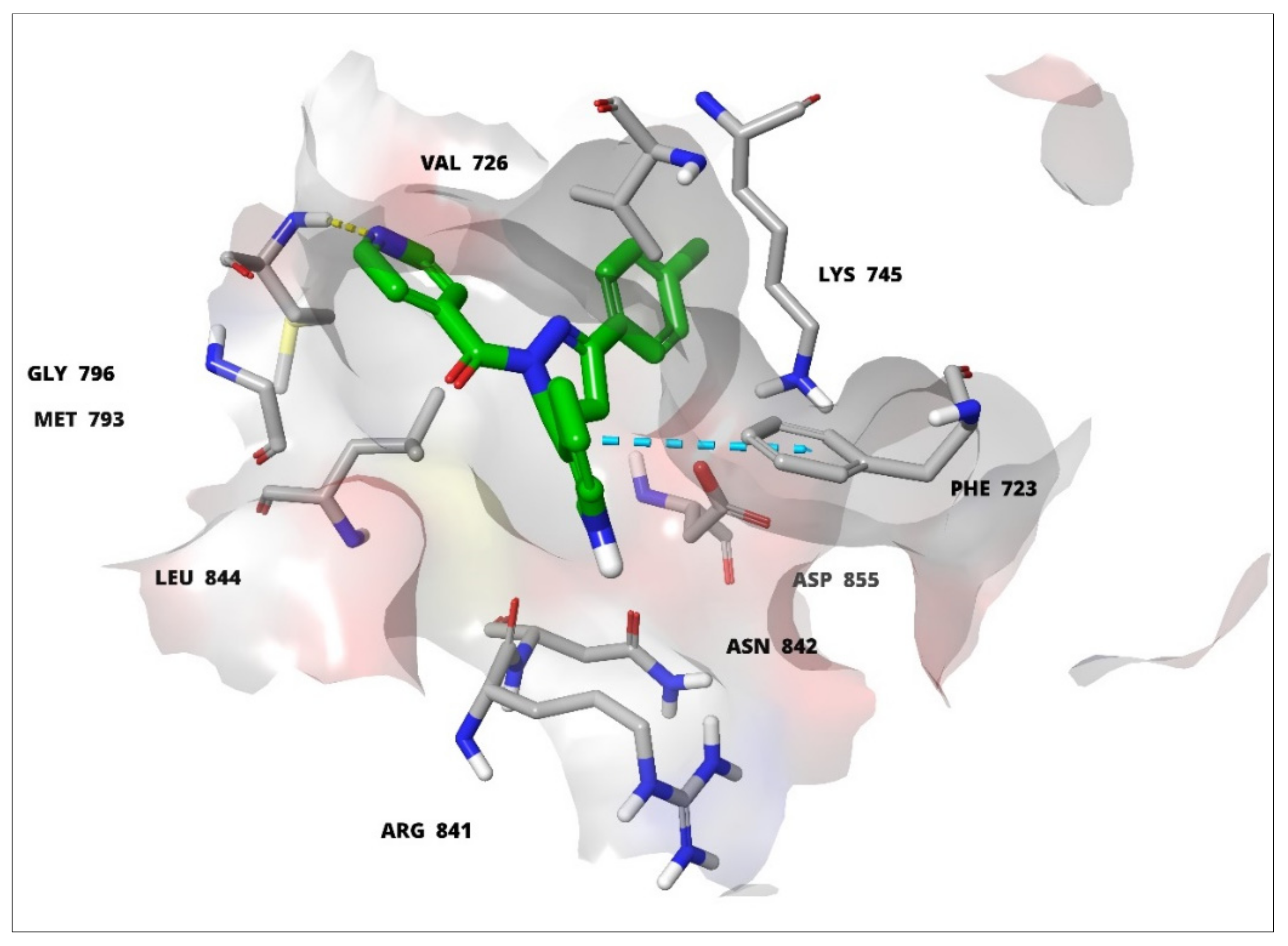
3.2. Molecular Modelling Studies
3.3. Cell-Line Based Anti-Cancers’ Biological Testings
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- World Health Organization. Available online: www.who.int/health-topics/cancer#tab=tab_1 (accessed on 15 April 2022).
- Schwartz, R.N. Anemia in patients with cancer: Incidence, causes, impact, management, and use of treatment guidelines and protocols. Am. J. Health Pharm. 2007, 64, S5–S13. [Google Scholar] [CrossRef]
- Noolvi, M.N.; Patel, H.M.; Bhardwaj, V.; Chauhan, A. Synthesis and in vitro antitumor activity of substituted quinazoline and quinoxaline derivatives: Search for anticancer agent. Eur. J. Med. Chem. 2011, 46, 2327–2346. [Google Scholar] [CrossRef] [PubMed]
- Aydemir, N.; Bilaloğlu, R. Genotoxicity of two anticancer drugs, gemcitabine and topotecan, in mouse bone marrow in vivo. Mutat. Res. Toxicol. Environ. Mutagen. 2003, 537, 43–51. [Google Scholar] [CrossRef]
- Tok, F.; Abas, B.İ.; Çevik, Ö.; Koçyiğit-Kaymakçıoğlu, B. Design, synthesis and biological evaluation of some new 2-Pyrazoline derivatives as potential anticancer agents. Bioorg. Chem. 2020, 102, 104063. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Zheng, J.; Xu, W.; Chen, C.; Wei, D.; Ni, W.; Pan, Y. A new series of cytotoxic pyrazoline derivatives as potential anticancer agents that induce cell cycle arrest and apoptosis. Molecules 2017, 22, 1635. [Google Scholar] [CrossRef]
- Chouiter, M.I.; Boulebd, H.; Pereira, D.M.; Valentão, P.; Andrade, P.B.; Belfaitah, A.; Silva, A.M. New chalcone-type compounds and 2-pyrazoline derivatives: Synthesis and caspase-dependent anticancer activity. Future Med. Chem. 2020, 12, 493–509. [Google Scholar] [CrossRef]
- Pecoraro, C.; Parrino, B.; Cascioferro, S.; Puerta, A.; Avan, A.; Peters, G.J.; Diana, P.; Giovannetti, E.; Carbone, D. A New Oxadiazole-Based Topsentin Derivative Modulates Cyclin-Dependent Kinase 1 Expression and Exerts Cytotoxic Effects on Pancreatic Cancer Cells. Molecules 2021, 27, 19. [Google Scholar] [CrossRef]
- Carbone, D.; Vestuto, V.; Ferraro, M.R.; Ciaglia, T.; Pecoraro, C.; Sommella, E.; Cascioferro, S.; Salviati, E.; Novi, S.; Tecce, M.F.; et al. Metabolomics-assisted discovery of a new anticancer GLS-1 inhibitor chemotype from a nortopsentin-inspired library: From phenotype screening to target identification. Eur. J. Med. Chem. 2022, 234, 114233. [Google Scholar] [CrossRef]
- Song, Y.; Feng, S.; Feng, J.; Dong, J.; Yang, K.; Liu, Z.; Qiao, X. Synthesis and biological evaluation of novel pyrazoline derivatives containing indole skeleton as anti-cancer agents targeting topoisomerase II. Eur. J. Med. Chem. 2020, 200, 112459. [Google Scholar] [CrossRef]
- Parmar, S.S.; Pandey, B.R.; Dwivedi, C.; Harbison, R.D. Anticonvulsant activity and monoamine oxidase inhibitory properties of 1, 3, 5-trisubstituted pyrazolines. J. Pharm. Sci. 1974, 63, 1152–1155. [Google Scholar] [CrossRef]
- Budakoti, A.; Bhat, A.R.; Athar, F.; Azam, A. Syntheses and evaluation of 3-(3-bromo phenyl)-5-phenyl-1-(thiazolo [4, 5-b] quinoxaline-2-yl)-2-pyrazoline derivatives. Eur. J. Med. Chem. 2008, 43, 1749–1757. [Google Scholar] [CrossRef]
- Mendelsohn, J.; Baselga, J. The EGF receptor family as targets for cancer therapy. Oncogene 2000, 19, 6550–6565. [Google Scholar] [CrossRef]
- Manna, F.; Chimenti, F.; Fioravanti, R.; Bolasco, A.; Secci, D.; Chimenti, P.; Ferlini, C.; Scambia, G. Synthesis of some pyrazole derivatives and preliminary investigation of their affinity binding to P-glycoprotein. Bioorg. Med. Chem. Lett. 2005, 15, 4632–4635. [Google Scholar] [CrossRef]
- Sim, M.M.; Ng, S.B.; Buss, A.D.; Crasta, S.C.; Goh, K.L.; Lee, S.K. Benzylidene rhodanines as novel inhibitors of UDP-N-acetylmuramate/L-alanine ligase. Bioorg. Med. Chem. Lett. 2002, 12, 697–699. [Google Scholar] [CrossRef]
- Turan-Zitouni, G.; Chevallet, P.; Kilic, F.S.; Erol, K. Synthesis of some thiazolyl-pyrazoline derivatives and preliminary investigation of their hypotensive activity. Eur. J. Med. Chem. 2000, 35, 635–641. [Google Scholar] [CrossRef]
- Bilgin, A.A.; Palaska, E.; Sunal, R. Studies on the synthesis and antidepressant activity of some 1-thiocarbamoyl-3, 5-diphenyl-2-pyrazolines. Arzneimittelforschung 1993, 43, 1041–1044. [Google Scholar]
- Sharma, P.K.; Kumar, S.; Kumar, P.; Kaushik, P.; Kaushik, D.; Dhingra, Y.; Aneja, K.R. Synthesis and biological evaluation of some pyrazolylpyrazolines as anti-inflammatory–antimicrobial agents. Eur. J. Med. Chem. 2010, 45, 2650–2655. [Google Scholar] [CrossRef]
- Dawane, B.S.; Konda, S.G.; Mandawad, G.G.; Shaikh, B.M. Poly (ethylene glycol)(PEG-400) as an alternative reaction solvent for the synthesis of some new 1-(4-(4′-chlorophenyl)-2-thiazolyl)-3-aryl-5-(2-butyl-4-chloro-1H-imidazol-5yl)-2-pyrazolines and their in vitro antimicrobial evaluation. Eur. J. Med. Chem. 2010, 45, 387–392. [Google Scholar] [CrossRef]
- Yamaoka, T.; Ohba, M.; Ohmori, T. Molecular-Targeted Therapies for Epidermal Growth Factor Receptor and Its Resistance Mechanisms. Int. J. Mol. Sci. 2017, 18, 2420. [Google Scholar] [CrossRef]
- Monks, A.; Scudiero, D.; Skehan, P.; Shoemaker, R.; Paull, K.; Vistica, D.; Hose, C.; Langley, J.; Cronise, P.; Vaigro-Wolff, A.; et al. Feasibility of a high-flux anticancer drug screen using a diverse panel of cultured human tumor cell lines. J. Natl. Cancer Inst. 1991, 83, 757–766. [Google Scholar] [CrossRef]
- Alley, M.C.; Scudiero, D.A.; Monks, A.; Hursey, M.L.; Czerwinski, M.J.; Fine, D.L.; Abbott, B.J.; Mayo, J.G.; Shoemaker, R.H.; Boyd, M.R. Feasibility of drug screening with panels of human tumor cell lines using a microculture tetrazolium assay. Cancer Res. 1988, 48, 589–601. [Google Scholar]
- Boyd, M.R.; Paull, K.D. Some practical considerations and applications of the National Cancer Institute in vitro anticancer drug discovery screen. Drug Dev. Res. 1995, 34, 91–109. [Google Scholar] [CrossRef]
- Shoemaker, R.H. The NCI 60 human tumour cell line anticancer drug screen. Nat. Rev. Cancer 2006, 6, 813–823. [Google Scholar] [CrossRef]
- Blair, J.A.; Rauh, D.; Kung, C.; Yun, C.H.; Fan, Q.W.; Rode, H.; Zhang, C.; Eck, M.J.; Weiss, W.A.; Shokat, K.M. Structure-guided development of affinity probes for tyrosine kinases using chemical genetics. Nat. Chem. Biol. 2007, 4, 229–238. [Google Scholar] [CrossRef] [PubMed]
- Holla, B.S.; Udupa, K.V. Synthesis, spectral studies and biological activities of some N-bridged heterocycles derived from 3-arylaminomethyl-4-amino-5-mercapto-1,2,4-triazoles. Farmaco 1992, 47, 305–318. [Google Scholar] [CrossRef] [PubMed]
- Robinson, M.W.; Overmeyer, J.H.; Young, A.M.; Erhardt, P.W.; Maltese, W.A. Synthesis and evaluation of indole-based chalcones as inducers of methuosis, a novel type of nonapoptotic cell death. J. Med. Chem. 2012, 55, 1940–1956. [Google Scholar] [CrossRef] [PubMed]
- Karthikeyan, C.; SHNarayana Moorthy, N.; Ramasamy, S.; Vanam, U.; Manivannan, E.; Karunagaran, D.; Trivedi, P. Advances in chalcones with anticancer activities. Recent Pat. Anticancer Drug Discov. 2015, 10, 97–115. [Google Scholar] [CrossRef] [PubMed]
- Faritha, A.; Nasser, A.J.A.; Ahamed, A.P.; Thajuddin, N. Synthesis, characterization and biological activity of certain pyrazole derivatives. J. Chem. Pharm. Res. 2014, 6, 189–193. [Google Scholar]
- Karrouchi, K.; Radi, S.; Ramli, Y.; Taoufik, J.; Mabkhot, Y.N.; Al-Aizari, F.A.; Ansar, M.H. Synthesis and Pharmacological Activities of Pyrazole Derivatives: A Review. Molecules 2018, 23, 134. [Google Scholar] [CrossRef]
- Al-Anazi, M.; Al-Najjar, B.O.; Khairuddean, M. Structure-Based Drug Design Studies Toward the Discovery of Novel Chalcone Derivatives as Potential Epidermal Growth Factor Receptor (EGFR) Inhibitors. Molecules 2018, 23, 3203. [Google Scholar] [CrossRef]
- Zaharevitz, D.W.; Holbeck, S.L.; Bowerman, C.; Svetlik, P.A. COMPARE: A web accessible tool for investigating mechanisms of cell growth inhibition. J. Mol. Graph. Model. 2002, 20, 297–303. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Substitution Ar 1 | Substitution Ar |
|---|---|---|
| HD01 | 4-Hydroxyphenyl |  |
| HD02 * | 4-Toluyl | |
| HD03 | 2-Hydroxyphenyl | |
| HD04 | 4-Chlorophenyl | |
| HD05 * | 4-Chlorophenyl |  |
| HD06 | 4-Toluyl | |
| HD07 | 2-Hydroxyphenyl | |
| HD08 | 4-Hydroxyphenyl | |
| HD09 | 4-Hydroxyphenyl |  |
| HD10 | 4-Toluyl | |
| HD11 | 2-Hydroxyphenyl | |
| HD12 * | 4-Chlorophenyl |
| Structures |  |  |  | ||||
|---|---|---|---|---|---|---|---|
| Panels | Cell Lines | HD02 | HD05 | HD12 | |||
| GP + | %GI ++ | GP | %GI | GP | %GI | ||
| Leukemia | CCRF-CEM | 84.23 | 15.77 | 11.26 | 88.74 | 56.71 | 43.29 |
| K-562 | 94.06 | 5.94 | 71.25 | 28.75 | 88.34 | 11.66 | |
| MOLT-4 | 69.21 | 30.79 | −8.31 | 108.31 | 75.08 | 24.92 | |
| SR | 71.04 | 28.96 | 10.76 | 89.24 | 57.48 | 44.52 | |
| Non-Small Lung Cancer | A549/ATCC | 91.82 | 8.18 | 58.36 | 41.66 | 95.40 | 4.60 |
| HOP-62 | 78.86 | 21.14 | 66.58 | 33.42 | 75.10 | 24.90 | |
| HOP-92 | 125.23 | −25.23 | 62.13 | 37.87 | 94.69 | 5.31 | |
| NCI-H226 | 85.76 | 14.24 | 65.18 | 34.82 | 82.72 | 17.28 | |
| NCI-H23 | 82.32 | 17.68 | 76.06 | 23.96 | 85.92 | 14.08 | |
| NCI-H322M | 87.09 | 12.91 | 86.43 | 13.57 | 96.29 | 3.71 | |
| NCI-H460 | 97.31 | 2.69 | 47.85 | 52.15 | 96.81 | 3.19 | |
| NCI-H522 | 76.40 | 13.60 | 73.92 | 26.08 | 80.83 | 9.17 | |
| Colon Cancer | Colo205 | 100.95 | −0.95 | 86.89 | 13.11 | 97.86 | 2.14 |
| HCC-2998 | 105.08 | −5.08 | 102.59 | −2.59 | 105.27 | −5.27 | |
| HCT-116 | 87.46 | 12.54 | 68.50 | 31.50 | 94.28 | 5.72 | |
| HCT-15 | 80.91 | 19.09 | 62.13 | 37.87 | 93.56 | 6.44 | |
| HT29 | 93.44 | 6.56 | 90.38 | 9.62 | 101.08 | −1.08 | |
| KM12 | 91.57 | 8.43 | 74.05 | 25.95 | 96.75 | 3.25 | |
| SW-620 | 100.21 | −0.21 | 54.48 | 45.52 | 96.01 | 3.99 | |
| CNS Cancer | SF-268 | 87.31 | 12.69 | 71.91 | 28.09 | 94.19 | 5.81 |
| SF-295 | 88.67 | 11.33 | 92.08 | 7.92 | 96.97 | 3.03 | |
| SF-539 | 87.24 | 12.76 | 87.08 | 12.92 | 97.05 | 2.95 | |
| SNB-19 | 104.05 | −4.05 | 93.31 | 6.69 | 105.40 | −5.40 | |
| SNB-75 | 87.00 | 13.00 | 66.56 | 33.44 | 82.41 | 17.59 | |
| U251 | 87.64 | 12.36 | 58.04 | 41.96 | 93.37 | 6.63 | |
| Melanoma | LOX IMVI | 83.15 | 16.85 | 59.10 | 40.90 | 86.90 | 13.10 |
| MALME-3M | 86.75 | 13.25 | 74.10 | 25.90 | 96.65 | 3.35 | |
| M14 | 91.89 | 8.11 | 83.11 | 16.89 | 95.25 | 4.75 | |
| MDA-MB-435 | 106.49 | −6.49 | 93.82 | 6.18 | 99.89 | 0.11 | |
| SK-MEL-2 | 87.80 | 12.20 | 85.33 | 14.67 | 82.96 | 17.04 | |
| SK-MEL-28 | 112.91 | −12.91 | 103.52 | -3.52 | 107.05 | −7.05 | |
| SK-MEL-5 | 93.37 | 6.63 | 85.51 | 14.49 | 93.26 | 6.74 | |
| UACC-257 | 99.55 | 0.45 | 89.06 | 10.94 | 95.95 | 4.05 | |
| UACC-62 | 85.17 | 14.83 | 70.90 | 29.10 | 90.53 | 9.47 | |
| Ovarian Cancer | IGROV1 | 75.59 | 24.41 | 30.18 | 69.82 | 79.27 | 20.83 |
| OVCAR-3 | 92.32 | 7.68 | 77.86 | 22.14 | 95.48 | 4.52 | |
| OVCAR-5 | 91.79 | 8.21 | 112.11 | −12.11 | 105.36 | −5.36 | |
| OVCAR-8 | 93.68 | 6.32 | 70.43 | 29.57 | 98.46 | 1.54 | |
| NCI/ADR-RES | 92.96 | 7.04 | 83.00 | 17.00 | 96.98 | 3.02 | |
| SK-OV-3 | 84.72 | 13.28 | 81.39 | 18.61 | 84.19 | 13.81 | |
| Renal cancer | 786-0 | 103.27 | −3.27 | 71.02 | 28.98 | 104.02 | −4.02 |
| A498 | 75.77 | 24.23 | 72.07 | 27.93 | 80.54 | 19.46 | |
| ACHN | 89.39 | 10.61 | 60.75 | 39.25 | 94.83 | 5.17 | |
| CAKI-1 | 93.03 | 6.97 | 46.03 | 53.97 | 90.21 | 9.79 | |
| RXF 393 | 108.82 | −8.82 | 103.23 | −3.23 | 111.61 | −11.61 | |
| SN12C | 93.05 | 6.95 | 62.69 | 37.31 | 92.95 | 7.05 | |
| TK-10 | 110.87 | −10.87 | 93.59 | 6.41 | 107.04 | −7.04 | |
| UO-31 | 46.20 | 53.80 | 25.70 | 74.30 | 45.17 | 54.83 | |
| Prostate Cancer | PC-3 | 76.57 | 23.43 | 63.22 | 36.78 | 82.77 | 17.23 |
| DU-145 | 101.58 | −1.58 | 91.97 | 8.03 | 102.22 | −2.22 | |
| Breast cancer | MCF7 | 45.44 | 54.56 | 39.74 | 60.26 | 70.15 | 29.85 |
| MDA-MB-231/ATCC | 70.51 | 29.49 | 47.73 | 52.27 | 71.35 | 28.65 | |
| HS 578T | 93.97 | 6.03 | 93.73 | 6.27 | 100.69 | −0.69 | |
| BT-549 | 74.40 | 25.60 | 71.34 | 28.66 | 92.75 | 7.25 | |
| T-47D | 60.60 | 39.40 | 51.42 | 48.58 | 74.80 | 25.20 | |
| MDA-MB-468 | 73.17 | 26.83 | 85.72 | 14.28 | 102.09 | −2.90 | |
| Mean | 88.21 | 11.89 | 69.80 | 30.20 | 90.55 | 9.45 | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Khalilullah, H.; Agarwal, D.K.; Ahsan, M.J.; Jadav, S.S.; Mohammed, H.A.; Khan, M.A.; Mohammed, S.A.A.; Khan, R. Synthesis and Anti-Cancer Activity of New Pyrazolinyl-Indole Derivatives: Pharmacophoric Interactions and Docking Studies for Identifying New EGFR Inhibitors. Int. J. Mol. Sci. 2022, 23, 6548. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23126548
Khalilullah H, Agarwal DK, Ahsan MJ, Jadav SS, Mohammed HA, Khan MA, Mohammed SAA, Khan R. Synthesis and Anti-Cancer Activity of New Pyrazolinyl-Indole Derivatives: Pharmacophoric Interactions and Docking Studies for Identifying New EGFR Inhibitors. International Journal of Molecular Sciences. 2022; 23(12):6548. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23126548
Chicago/Turabian StyleKhalilullah, Habibullah, Deepak K. Agarwal, Mohamed J. Ahsan, Surender S. Jadav, Hamdoon A. Mohammed, Masood Alam Khan, Salman A. A. Mohammed, and Riaz Khan. 2022. "Synthesis and Anti-Cancer Activity of New Pyrazolinyl-Indole Derivatives: Pharmacophoric Interactions and Docking Studies for Identifying New EGFR Inhibitors" International Journal of Molecular Sciences 23, no. 12: 6548. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23126548