
Role of miR-133/Dio3 Axis in the T3-Dependent Modulation of Cardiac mitoK-ATP Expression

 , , ,
, , ,
Abstract
:1. Introduction
2. Results
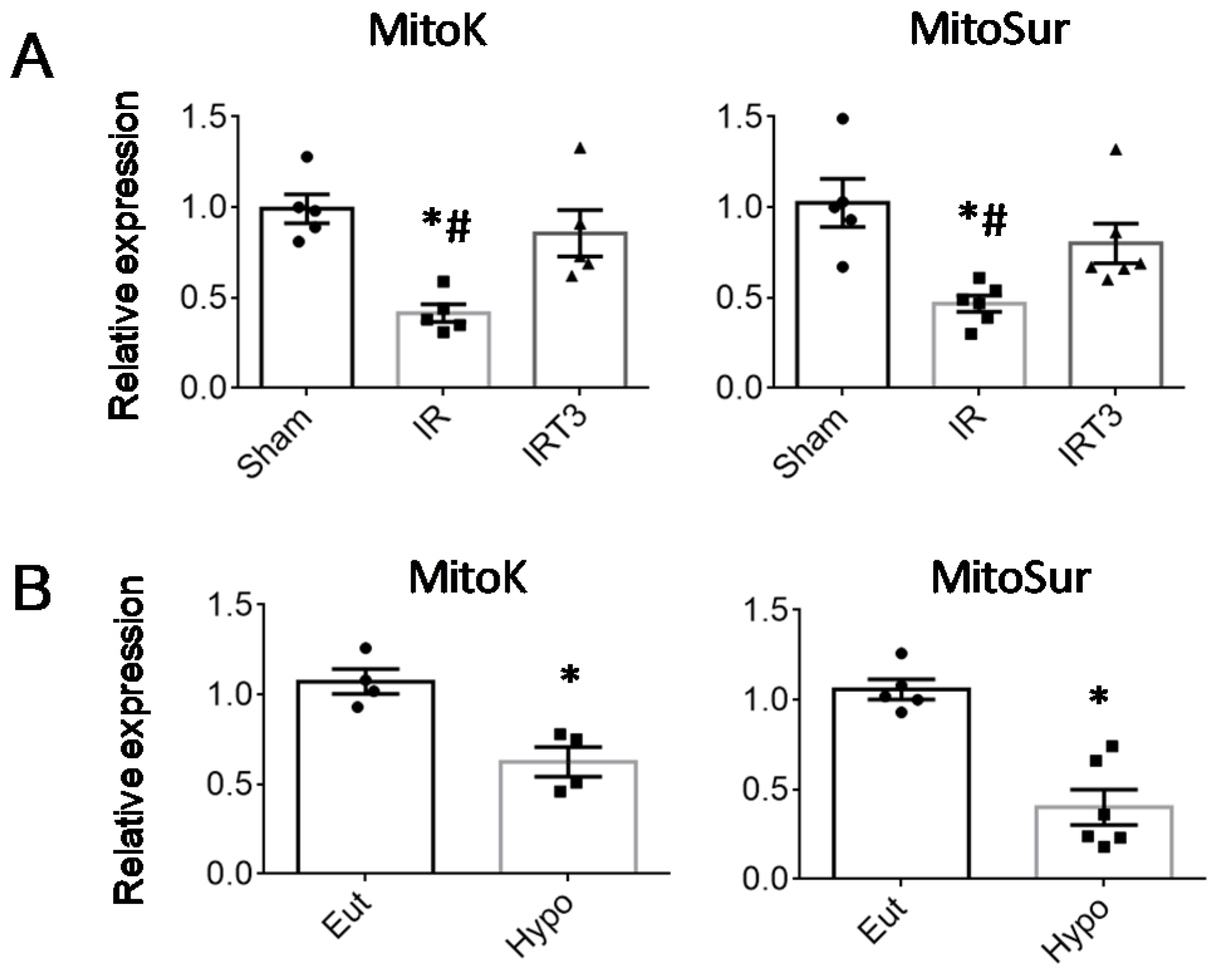
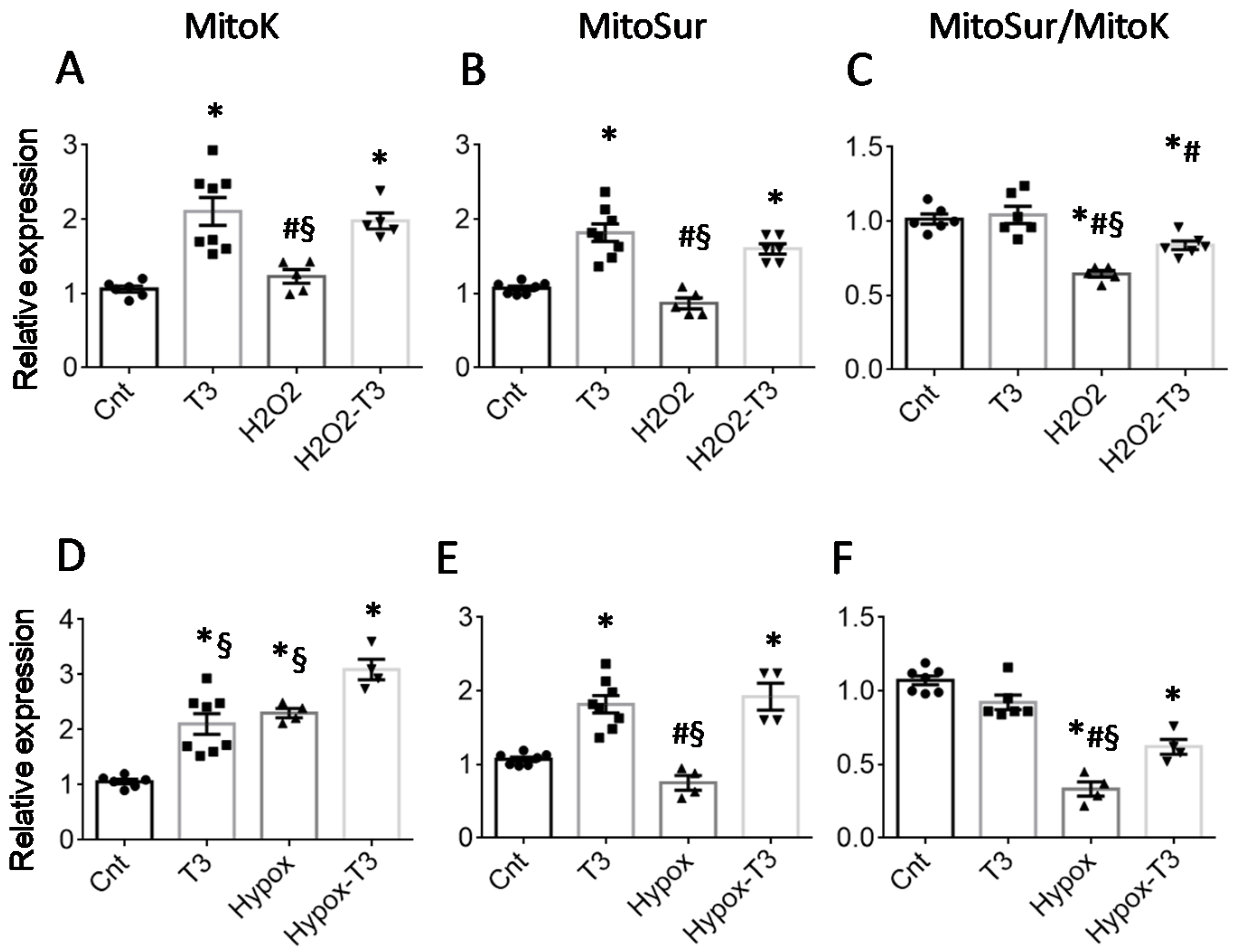
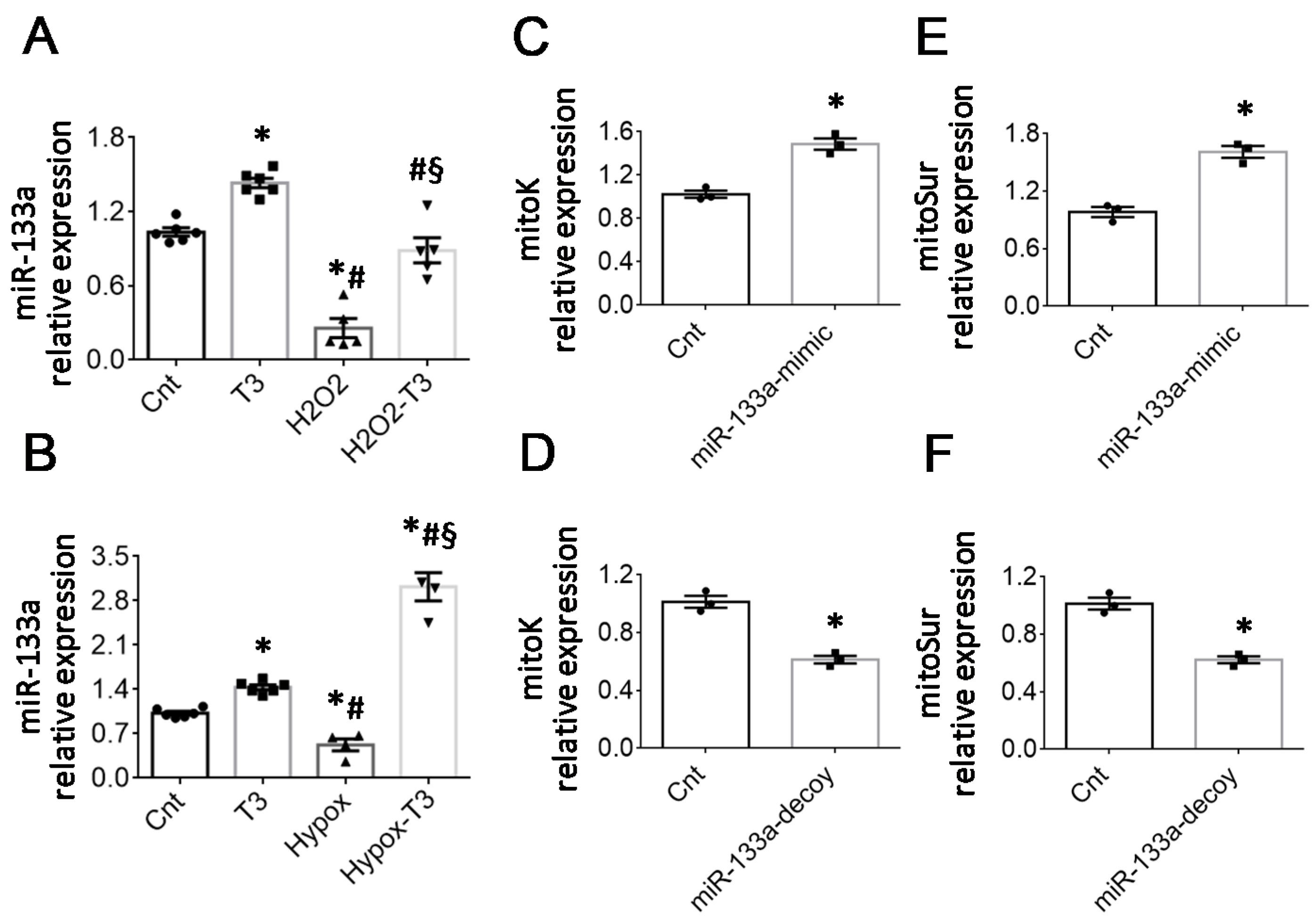
2.1. T3 Replacement Is Associated with the Restoration of mitoK and mitoSur Expression Levels in Injured Cardiac Tissue and Cardiomyocytes
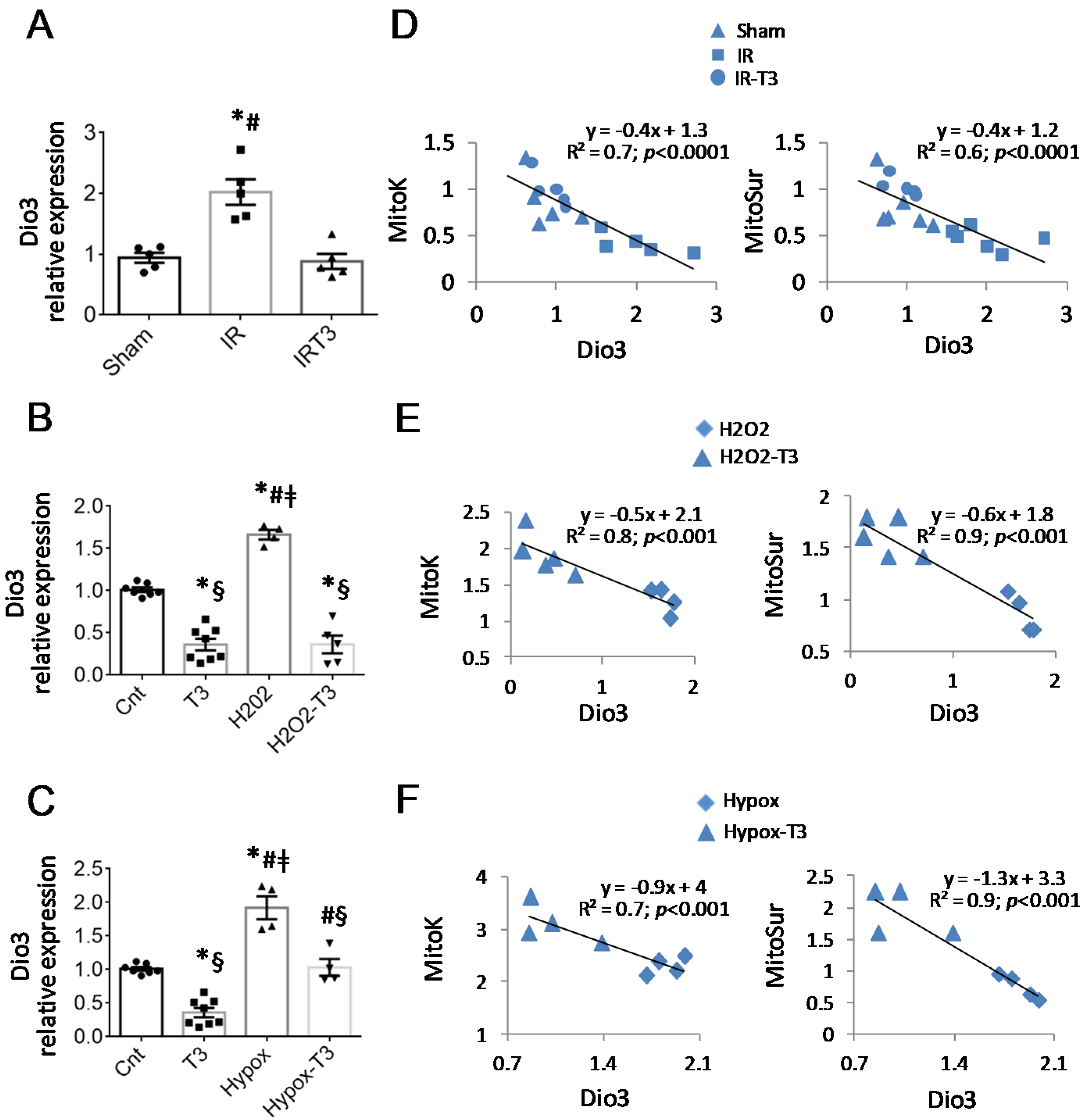
2.2. The Cardiac Expression of Deiodinase 3 Is Inversely Associated with That of mitoK and mitoSur
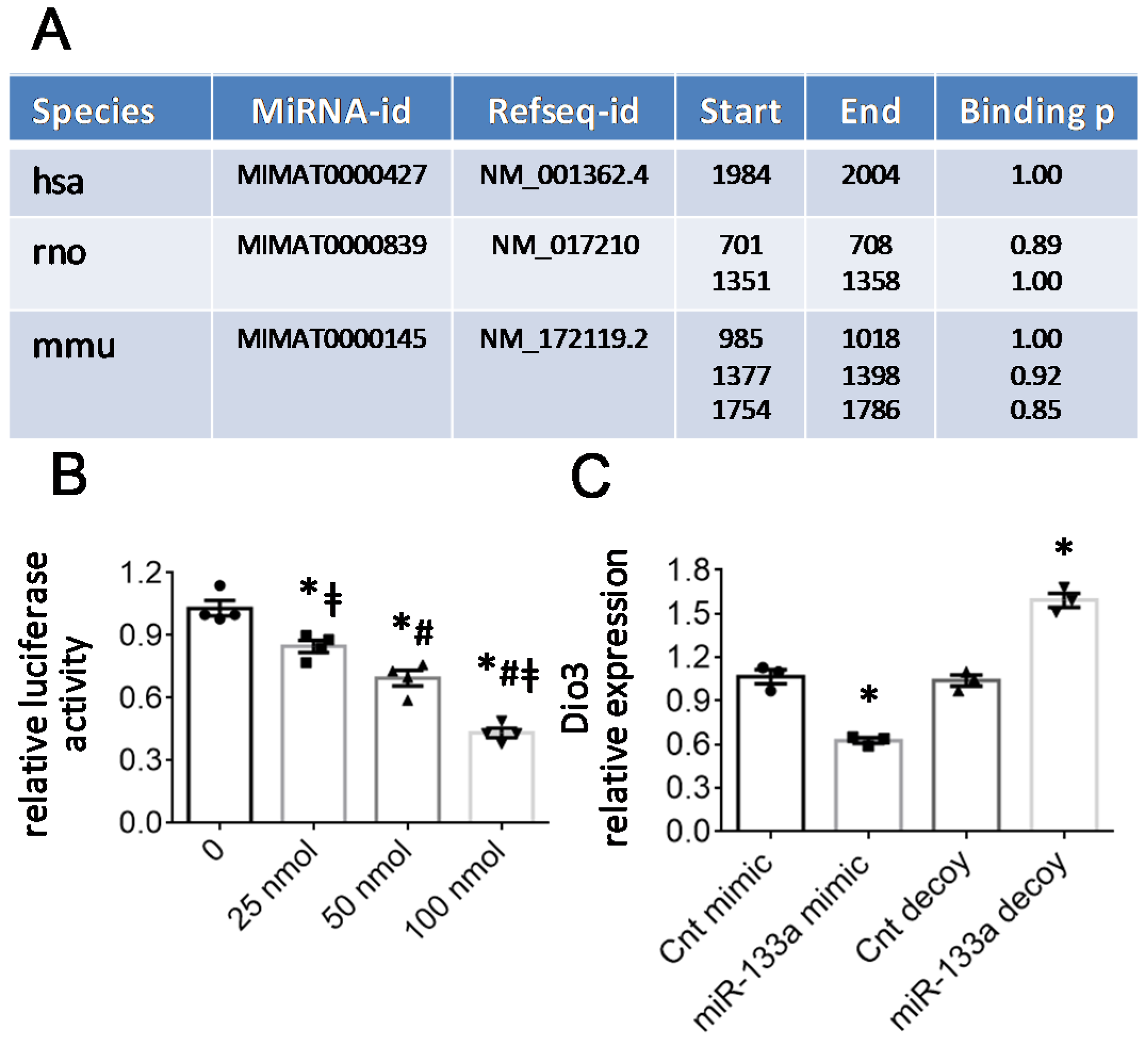
2.3. The T3-Dependent microRNA miR-133a Is Involved in the Activation of mitoK and mitoSur and the Post-Transcriptional Repression of Dio3
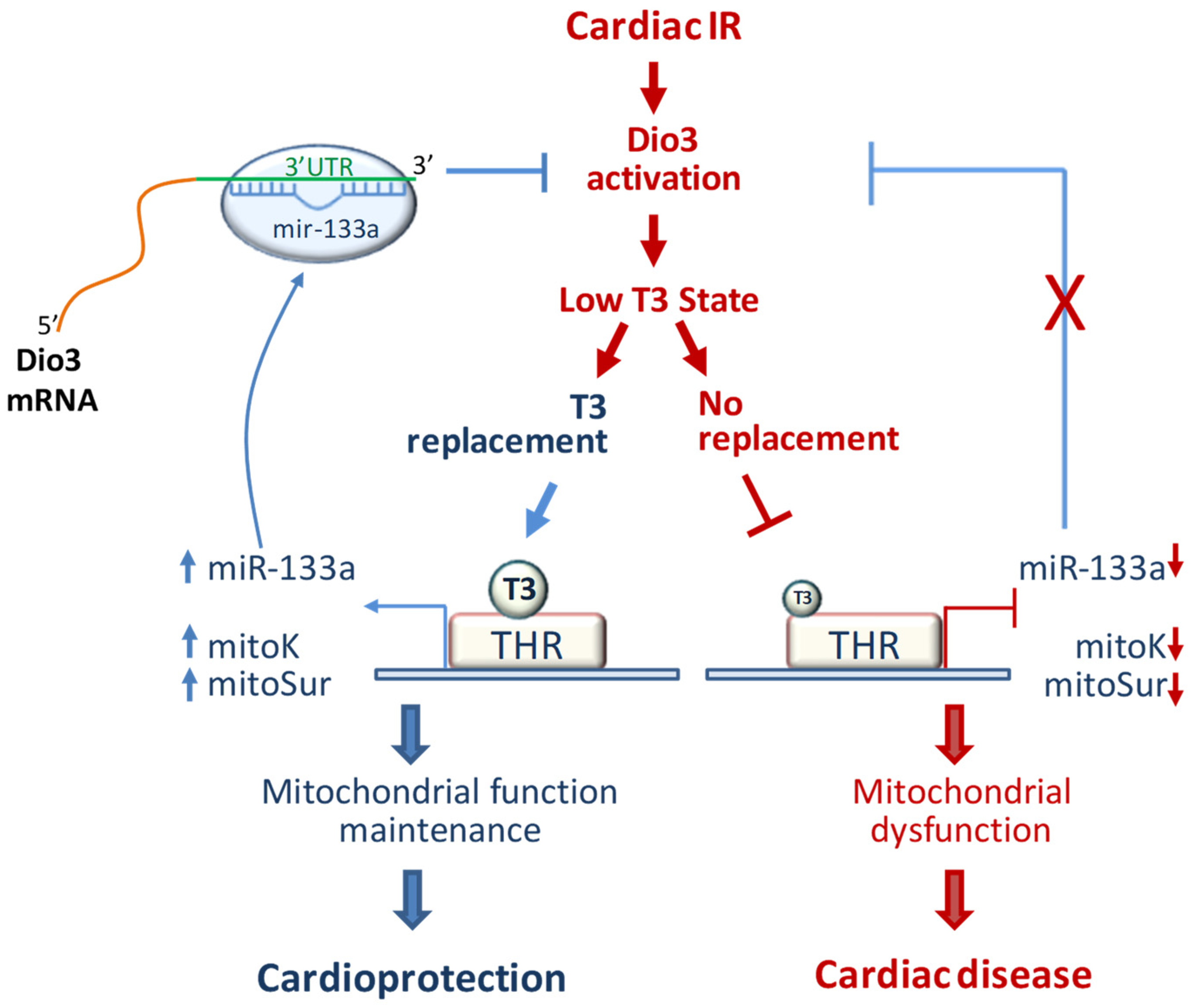
3. Discussion
4. Materials and Methods
4.1. Experimental Design and Animal Models
4.2. Isolation of Neonatal Rat Cardiomyocytes
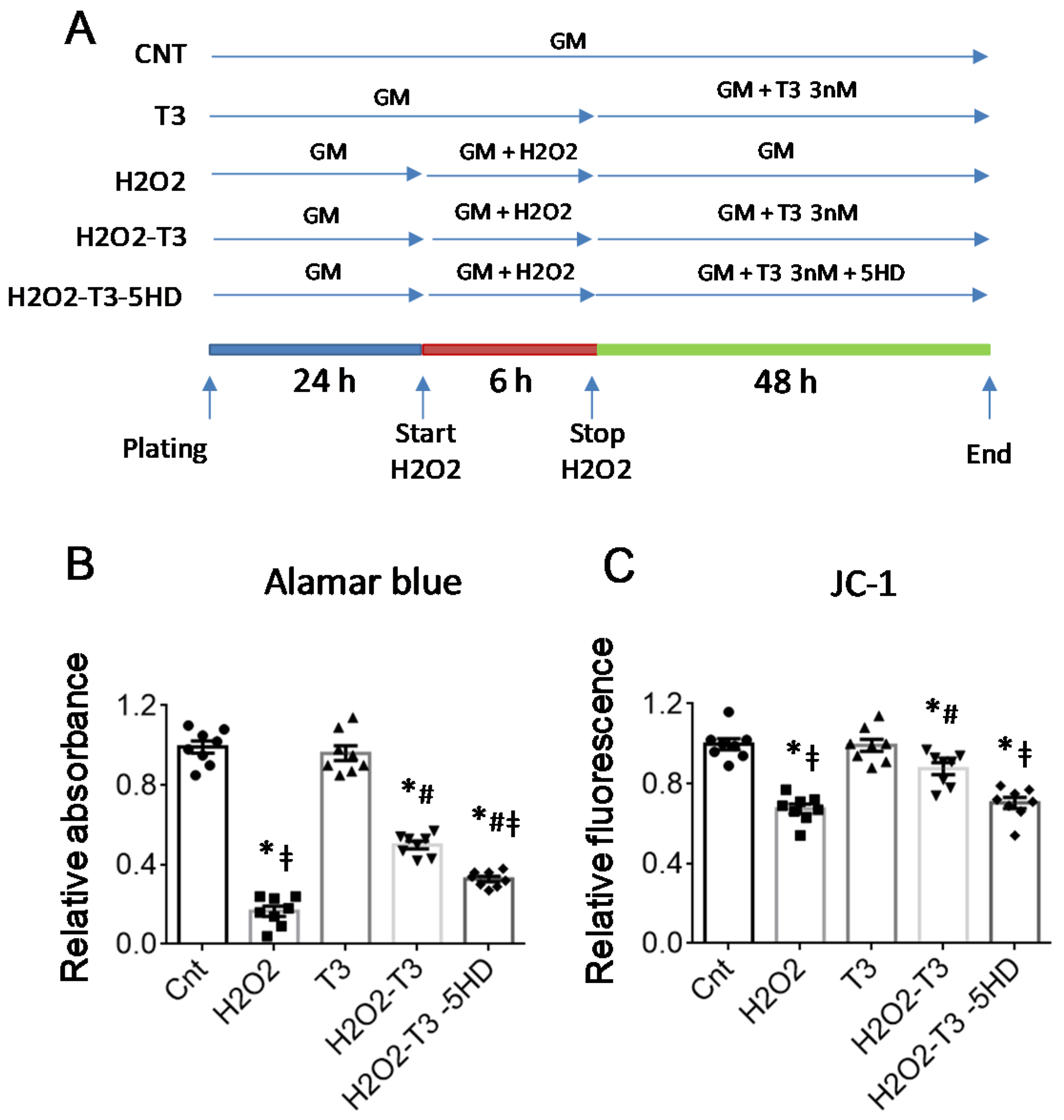
4.3. In Vitro Stress Protocols
4.4. Viability Assay
4.5. Measurement of the Inner Mitochondrial Membrane Potential
4.6. In Silico Analyses
4.7. Overexpression and Downregulation of miR-133a-3p in NRCM
4.8. Plasmid Construction and Reporter Assays
4.9. RNA Extraction and Real-Time PCR
4.10. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Roth, G.A.; Mensah, G.A.; Johnson, C.O.; Addolorato, G.; Ammirati, E.; Baddour, L.M.; Barengo, N.C.; Beaton, A.Z.; Benjamin, E.J.; Benziger, C.P.; et al. Global Burden of Cardiovascular Diseases and Risk Factors, 1990–2019: Update From the GBD 2019 Study. J. Am. Coll. Cardiol. 2020, 76, 2982–3021. [Google Scholar] [CrossRef] [PubMed]
- Hausenloy, D.J.; Yellon, D.M. Myocardial ischemia-reperfusion injury: A neglected therapeutic target. J. Clin. Investig. 2013, 123, 92–100. [Google Scholar] [CrossRef] [PubMed]
- Whelan, R.S.; Kaplinskiy, V.; Kitsis, R.N. Cell Death in the Pathogenesis of Heart Disease: Mechanisms and Significance. Annu. Rev. Physiol. 2010, 72, 19–44. [Google Scholar] [CrossRef] [PubMed]
- Baines, C.P. The mitochondrial permeability transition pore and ischemia-reperfusion injury. Basic Res. Cardiol. 2009, 104, 181–188. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galluzzi, L.; Kepp, O.; Kroemer, G. Mitochondria: Master regulators of danger signalling. Nat. Rev. Mol. Cell Biol. 2012, 13, 780–788. [Google Scholar] [CrossRef]
- Li, J.; Zhou, W.; Chen, W.; Wang, H.; Zhang, Y.; Yu, T. Mechanism of the hypoxia inducible factor 1/hypoxic response element pathway in rat myocardial ischemia/diazoxide post conditioning. Mol. Med. Rep. 2020, 21, 1527–1536. [Google Scholar] [CrossRef] [Green Version]
- Pan, Y.; Wang, Y.; Shi, W.; Liu, Y.; Cao, S.; Yu, T. Mitochondrial proteomics alterations in rat hearts following ischemia/reperfusion and diazoxide post conditioning. Mol. Med. Rep. 2021, 23, 161. [Google Scholar] [CrossRef]
- Testai, L.; Rapposelli, S.; Martelli, A.; Breschi, M.C.; Calderone, V. Mitochondrial Potassium Channels as Pharmacological Target for Cardioprotective Drugs. Med. Res. Rev. 2015, 35, 520–553. [Google Scholar] [CrossRef]
- Henn, M.C.; Janjua, M.B.; Kanter, E.M.; Makepeace, C.M.; Schuessler, R.B.; Nichols, C.G.; Lawton, J.S.; Duan, P.; Wang, J.; Li, Y.; et al. Opening of mitoKATP improves cardiac function and inhibits apoptosis via the AKT-Foxo1 signaling pathway in diabetic cardiomyopathy. Int. J. Mol. Med. 2018, 42, 2709–2719. [Google Scholar]
- Lucas, A.M.B.; de Lacerda Alexandre, J.V.; Araújo, M.T.S.; David, C.E.B.; Ponte Viana, Y.I.; Coelho, B.N.; Caldas, F.R.L.; Varela, A.L.N.; Kowaltowski, A.J.; Facundo, H.T. Diazoxide Modulates Cardiac Hypertrophy by Targeting H2O2 Generation and Mitochondrial Superoxide Dismutase Activity. Curr Mol Pharmacol. 2020, 13, 76–83. [Google Scholar] [CrossRef]
- Forini, F.; Lionetti, V.; Ardehali, H.; Pucci, A.; Cecchetti, F.; Ghanefar, M.; Nicolini, G.; Ichikawa, Y.; Nannipieri, M.; Recchia, F.A.; et al. Early long-term L-T3 replacement rescues mitochondria and prevents ischemic cardiac remodelling in rats. J. Cell. Mol. Med. 2010, 15, 514–524. [Google Scholar] [CrossRef] [PubMed]
- Forini, F.; Nicolini, G.; Kusmic, C.; Iervasi, G. Protective Effects of Euthyroidism Restoration on Mitochondria Function and Quality Control in Cardiac Pathophysiology. Int. J. Mol. Sci. 2019, 20, 3377. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Forini, F.; Nicolini, G.; Kusmic, C.; D’Aurizio, R.; Rizzo, M.; Baumgart, M.; Groth, M.; Doccini, S.; Iervasi, G.; Pitto, L. Integrative analysis of differentially expressed genes and miRNAs predicts complex T3-mediated protective circuits in a rat model of cardiac ischemia reperfusion. Sci. Rep. 2018, 8, 13870. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nicolini, G.; Forini, F.; Kusmic, C.; Pitto, L.; Mariani, L.; Iervasi, G. Early and Short-term Triiodothyronine Supplementation Prevents Adverse Postischemic Cardiac Remodeling: Role of Transforming Growth Factor-β1 and Antifibrotic miRNA Signaling. Mol. Med. 2015, 21, 900–911. [Google Scholar] [CrossRef] [PubMed]
- Forini, F.; Kusmic, C.; Nicolini, G.; Mariani, L.; Zucchi, R.; Matteucci, M.; Iervasi, G.; Pitto, L. Triiodothyronine Prevents Cardiac Ischemia/Reperfusion Mitochondrial Impairment and Cell Loss by Regulating miR30a/p53 Axis. Endocrinology 2014, 155, 4581–4590. [Google Scholar] [CrossRef]
- Paggio, A.; Checchetto, V.; Campo, A.; Menabò, R.; Di Marco, G.; Di Lisa, F.; Szabo, I.; Rizzuto, R.; De Stefani, D. Identification of an ATP-sensitive potassium channel in mitochondria. Nature 2019, 572, 609–613. [Google Scholar] [CrossRef]
- Zhang, Y.; Liu, X.; Zhang, L.; Li, X.; Zhou, Z.; Jiao, L.; Shao, Y.; Li, M.; Leng, B.; Zhou, Y.; et al. Metformin Protects against H2O2-Induced Cardiomyocyte Injury by Inhibiting the miR-1a-3p/GRP94 Pathway. Mol. Ther. Nucleic Acids 2018, 13, 189–197. [Google Scholar] [CrossRef] [Green Version]
- Lindsey, M.L.; Bolli, R.; Canty, J.M., Jr.; Du, X.-J.; Frangogiannis, N.G.; Frantz, S.; Gourdie, R.G.; Holmes, J.W.; Jones, S.P.; Kloner, R.A.; et al. Guidelines for experimental models of myocardial ischemia and infarction. Am. J. Physiol. Heart Circ. Physiol. 2018, 314, H812–H838. [Google Scholar] [CrossRef]
- Forini, F.; Nicolini, G.; Pitto, L.; Iervasi, G. Novel Insight Into the Epigenetic and Post-transcriptional Control of Cardiac Gene Expression by Thyroid Hormone. Front. Endocrinol. 2019, 10, 601. [Google Scholar] [CrossRef] [Green Version]
- Poliseno, L.; Salmena, L.; Zhang, J.; Carver, B.; Haveman, W.J.; Pandolfi, P.P. A coding-independent function of gene and pseudogene mRNAs regulates tumour biology. Nature 2010, 465, 1033–1038. [Google Scholar] [CrossRef] [Green Version]
- Checchetto, V.; Leanza, L.; De Stefani, D.; Rizzuto, R.; Gulbins, E.; Szabo, I. Mitochondrial K+ channels and their implications for disease mechanisms. Pharmacol. Ther. 2021, 227, 107874. [Google Scholar] [CrossRef] [PubMed]
- Quindry, J.C.; Schreiber, L.; Hosick, P.; Wrieden, J.; Irwin, J.M.; Hoyt, E. Mitochondrial KATPchannel inhibition blunts arrhythmia protection in ischemic exercised hearts. Am. J. Physiol. Circ. Physiol. 2010, 299, H175–H183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rennison, J.H.; Li, L.; Lin, C.R.; Lovano, B.S.; Castel, L.; Wass, S.Y.; Cantlay, C.C.; McHale, M.; Gillinov, A.M.; Mehra, R.; et al. Atrial fibrillation rhythm is associated with marked changes in metabolic and myofibrillar protein expression in left atrial appendage. Pflügers Arch.-Eur. J. Physiol. 2021, 473, 461–475. [Google Scholar] [CrossRef] [PubMed]
- Razvi, S.; Jabbar, A.; Pingitore, A.; Danzi, S.; Biondi, B.; Klein, I.; Peeters, R.; Zaman, A.; Iervasi, G. Thyroid Hormones and Cardiovascular Function and Diseases. J. Am. Coll. Cardiol. 2018, 71, 1781–1796. [Google Scholar] [CrossRef] [PubMed]
- Pantos, C.; Mourouzis, I. Thyroid hormone receptor α1 as a novel therapeutic target for tissue repair. Ann. Transl. Med. 2018, 6, 254. [Google Scholar] [CrossRef]
- Wassen, F.W.J.S.; Schiel, A.E.; Kuiper, G.G.J.M.; Kaptein, E.; Bakker, O.; Visser, T.J.; Simonides, W.S. Induction of Thyroid Hormone-Degrading Deiodinase in Cardiac Hypertrophy and Failure. Endocrinology 2002, 143, 2812–2815. [Google Scholar] [CrossRef]
- Buermans, H.P.J.; Redout, E.M.; Schiel, A.E.; Musters, R.J.P.; Zuidwijk, M.; Eijk, P.P.; van Hardeveld, C.; Kasanmoentalib, S.; Visser, F.C.; Ylstra, B.; et al. Microarray analysis reveals pivotal divergent mRNA expression profiles early in the development of either compensated ventricular hypertrophy or heart failure. Physiol. Genom. 2005, 21, 314–323. [Google Scholar] [CrossRef] [Green Version]
- Simonides, W.S.; Mulcahey, M.A.; Redout, E.M.; Muller, A.; Zuidwijk, M.J.; Visser, T.J.; Wassen, F.W.; Crescenzi, A.; Da-Silva, W.S.; Harney, J.; et al. Hypoxia-inducible factor induces local thyroid hormone inactivation during hypoxic-ischemic disease in rats. J. Clin. Investig. 2008, 118, 975–983. [Google Scholar] [CrossRef] [Green Version]
- Trivieri, M.G.; Oudit, G.Y.; Sah, R.; Kerfant, B.-G.; Sun, H.; Gramolini, A.O.; Pan, Y.; Wickenden, A.D.; Croteau, W.; de Escobar, G.M.; et al. Cardiac-specific elevations in thyroid hormone enhance contractility and prevent pressure overload-induced cardiac dysfunction. Proc. Natl. Acad. Sci. USA 2006, 103, 6043–6048. [Google Scholar] [CrossRef] [Green Version]
- Pol, C.J.; Muller, A.; Zuidwijk, M.J.; Van Deel, E.D.; Kaptein, E.; Saba, A.; Marchini, M.; Zucchi, R.; Visser, T.J.; Paulus, W.J.; et al. Left-Ventricular Remodeling After Myocardial Infarction Is Associated with a Cardiomyocyte-Specific Hypothyroid Condition. Endocrinology 2010, 152, 669–679. [Google Scholar] [CrossRef] [Green Version]
- Olivares, E.L.; Marassi, M.P.; Fortunato, R.; Da Silva, A.C.M.; Costa-E-Sousa, R.H.; Araújo, I.G.; Mattos, E.C.; Masuda, M.O.; Mulcahey, M.A.; Huang, S.A.; et al. Thyroid Function Disturbance and Type 3 Iodothyronine Deiodinase Induction after Myocardial Infarction in Rats—A Time Course Study. Endocrinology 2007, 148, 4786–4792. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weltman, N.Y.; Ojamaa, K.; Schlenker, E.H.; Chen, Y.-F.; Zucchi, R.; Saba, A.; Colligiani, D.; Rajagopalan, V.; Pol, C.J.; Gerdes, A.M. Low-Dose T3 Replacement Restores Depressed Cardiac T3 Levels, Preserves Coronary Microvasculature and Attenuates Cardiac Dysfunction in Experimental Diabetes Mellitus. Mol. Med. 2014, 20, 302–312. [Google Scholar] [CrossRef] [PubMed]
- Jayawardena, T.M.; Egemnazarov, B.; Finch, E.A.; Zhang, L.; Payne, J.A.; Pandya, K.; Zhang, Z.; Rosenberg, P.; Mirotsou, M.; Dzau, V.J. MicroRNA-Mediated In Vitro and In Vivo Direct Reprogramming of Cardiac Fibroblasts to Cardiomyocytes. Circ. Res. 2012, 110, 1465–1473. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matkovich, S.J.; Wang, W.; Tu, Y.; Eschenbacher, W.H.; Dorn, L.E.; Condorelli, G.; Diwan, A.; Nerbonne, J.M.; Dorn, G.W. MicroRNA-133a Protects Against Myocardial Fibrosis and Modulates Electrical Repolarization Without Affecting Hypertrophy in Pressure-Overloaded Adult Hearts. Circ. Res. 2010, 106, 166–175. [Google Scholar] [CrossRef] [Green Version]
- Danowski, N.; Manthey, I.; Jakob, H.G.; Siffert, W.; Peters, J.; Frey, U.H. Decreased Expression of miR-133a but Not of miR-1 is Associated with Signs of Heart Failure in Patients Undergoing Coronary Bypass Surgery. Cardiology 2013, 125, 125–130. [Google Scholar] [CrossRef]
- Boštjančič, E.; Brandner, T.; Zidar, N.; Glavač, D.; Štajer, D. Down-regulation of miR-133a/b in patients with myocardial infarction correlates with the presence of ventricular fibrillation. Biomed. Pharmacother. 2018, 99, 65–71. [Google Scholar] [CrossRef]
- Li, N.; Zhou, H.; Tang, Q. miR-133: A Suppressor of Cardiac Remodeling? Front. Pharmacol. 2018, 9, 903. [Google Scholar] [CrossRef]
- He, B.; Xiao, J.; Ren, A.-J.; Zhang, Y.-F.; Zhang, H.; Chen, M.; Xie, B.; Gao, X.-G.; Wang, Y.-W. Role of miR-1 and miR-133a in myocardial ischemic postconditioning. J. Biomed. Sci. 2011, 18, 22. [Google Scholar] [CrossRef] [Green Version]
- Donzelli, R.; Colligiani, D.; Kusmic, C.; Sabatini, M.; Lorenzini, L.; Accorroni, A.; Nannipieri, M.; Saba, A.; Iervasi, G.; Zucchi, R. Effect of Hypothyroidism and Hyperthyroidism on Tissue Thyroid Hormone Concentrations in Rat. Eur. Thyroid J. 2016, 5, 27–34. [Google Scholar] [CrossRef] [Green Version]
- Balzan, S.; Nicolini, G.; Forini, F.; Boni, G.; Del Carratore, R.; Nicolini, A.; Carpi, A.; Iervasi, G. Presence of a functional TSH receptor on human erythrocytes. Biomed. Pharmacother. 2007, 61, 463–467. [Google Scholar] [CrossRef]
- Forini, F.; Nicolini, G.; Kusmic, C.; D’Aurizio, R.; Mercatanti, A.; Iervasi, G.; Pitto, L. T3 Critically Affects the Mhrt/Brg1 Axis to Regulate the Cardiac MHC Switch: Role of an Epigenetic Cross-Talk. Cells 2020, 9, 2155. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sham | IR | IRT3 | |
|---|---|---|---|
| FT3 (pg/mL) | 3.0 ± 0.2 | 2.0 ± 0.01 *# | 2.9 ± 0.06 |
| FT4 (pg/mL) | 11.9 ± 1.05 | 12.6 ± 1.35 | 10.4 ± 1.5 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Canale, P.; Nicolini, G.; Pitto, L.; Kusmic, C.; Rizzo, M.; Balzan, S.; Iervasi, G.; Forini, F. Role of miR-133/Dio3 Axis in the T3-Dependent Modulation of Cardiac mitoK-ATP Expression. Int. J. Mol. Sci. 2022, 23, 6549. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23126549
Canale P, Nicolini G, Pitto L, Kusmic C, Rizzo M, Balzan S, Iervasi G, Forini F. Role of miR-133/Dio3 Axis in the T3-Dependent Modulation of Cardiac mitoK-ATP Expression. International Journal of Molecular Sciences. 2022; 23(12):6549. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23126549
Chicago/Turabian StyleCanale, Paola, Giuseppina Nicolini, Letizia Pitto, Claudia Kusmic, Milena Rizzo, Silvana Balzan, Giorgio Iervasi, and Francesca Forini. 2022. "Role of miR-133/Dio3 Axis in the T3-Dependent Modulation of Cardiac mitoK-ATP Expression" International Journal of Molecular Sciences 23, no. 12: 6549. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23126549