
Generalized Approach towards Secretion-Based Protein Production via Neutralization of Secretion-Preventing Cationic Substrate Residues

 , ,
, , 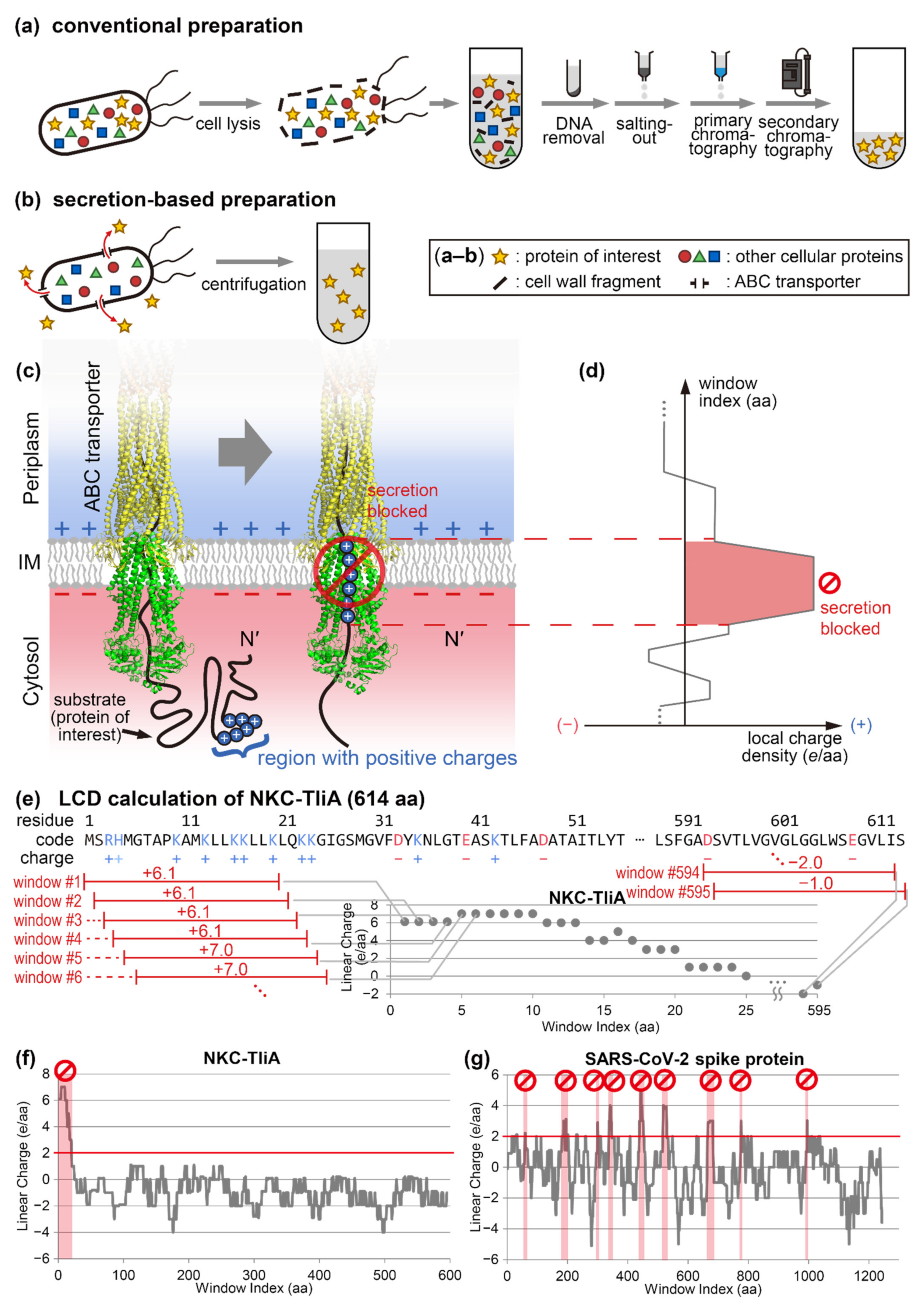{kind=link}
{kind=link}
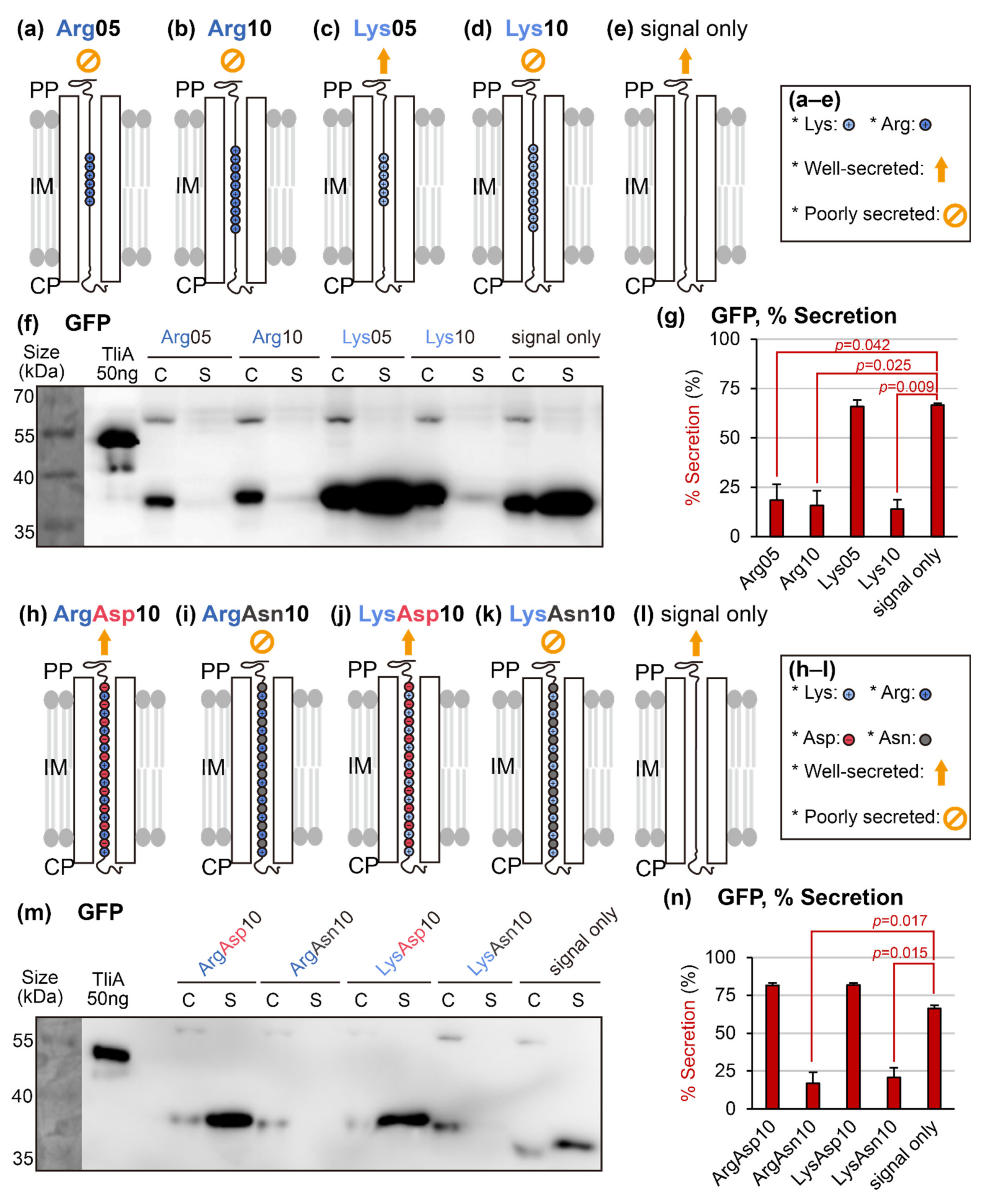{kind=link}
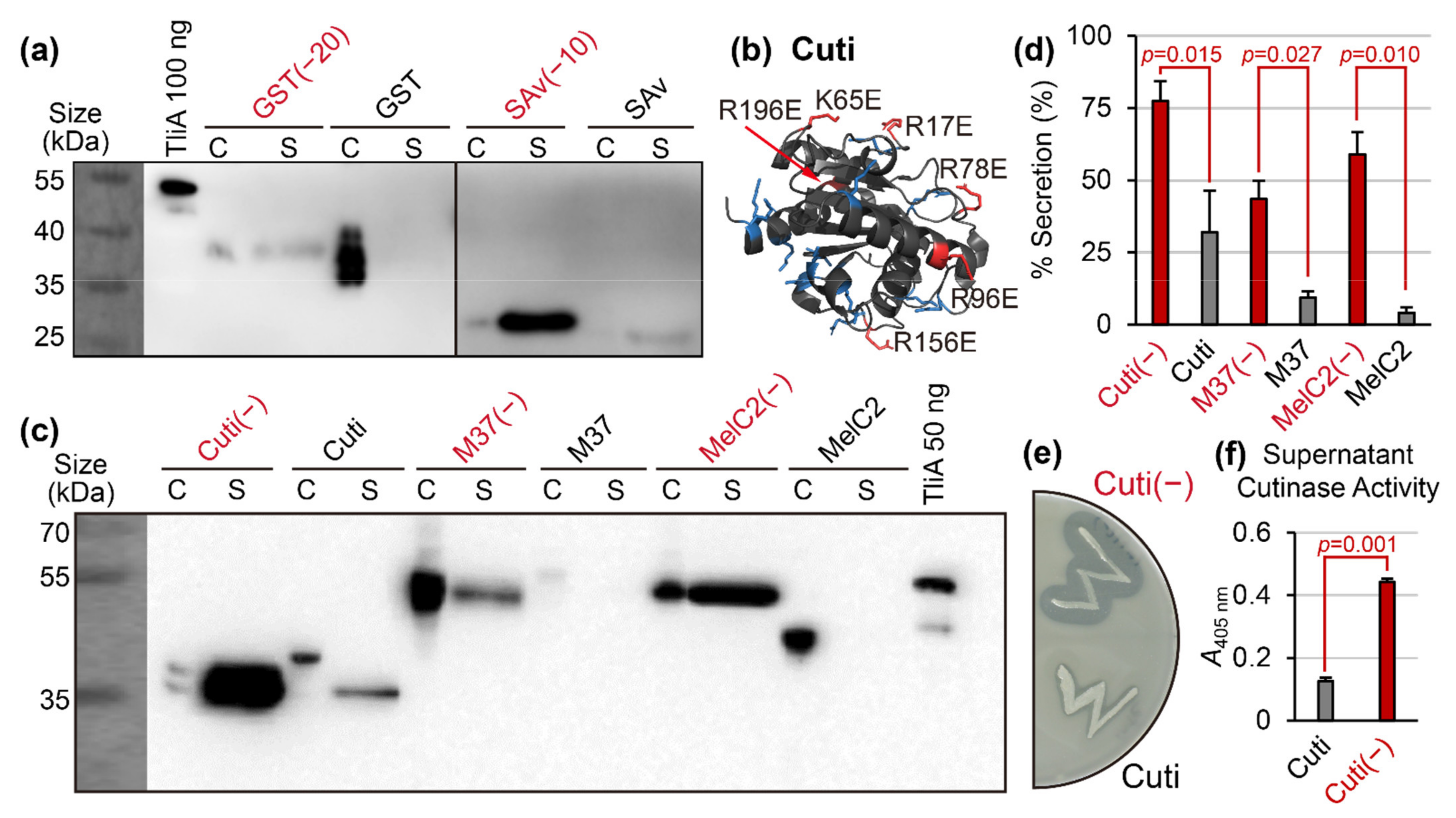{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Conversion of SARS-CoV-2 Spike Protein into Secretable Variants
2.2. Introducing Artificial “Cationic Supercharged Regions” to Substrate Proteins
2.3. Rescuing Secretion by Adding Negative Charges near the Positive Charges
2.4. “Negatively Supercharging” Poorly Secreted Proteins
2.5. “Super-Neutralization”: An Alternative Secretion-Rescuing with Less Structural Disturbance
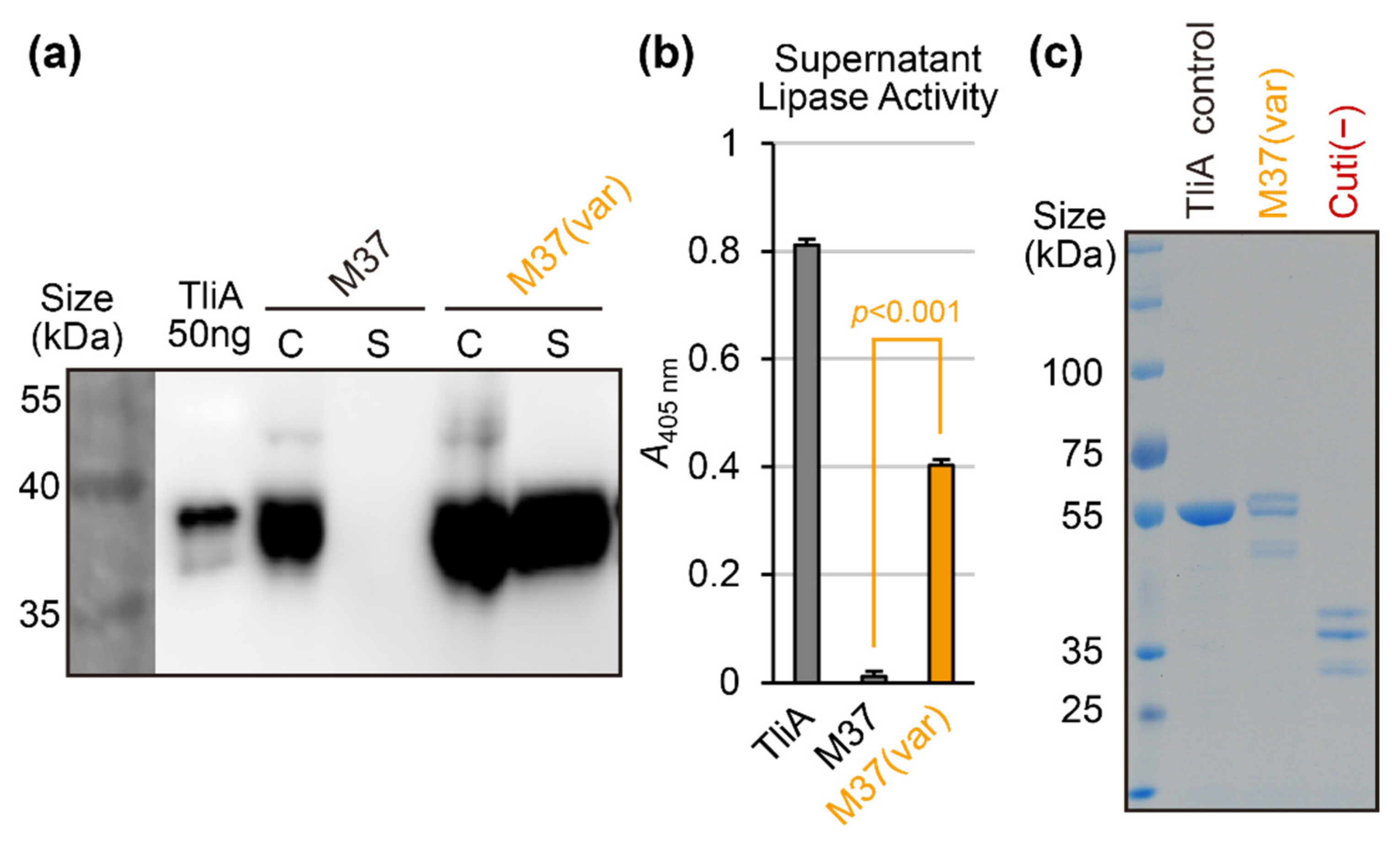
2.6. Random Mutation and Activity Screening: Avoiding Structure-Disrupting Mutations
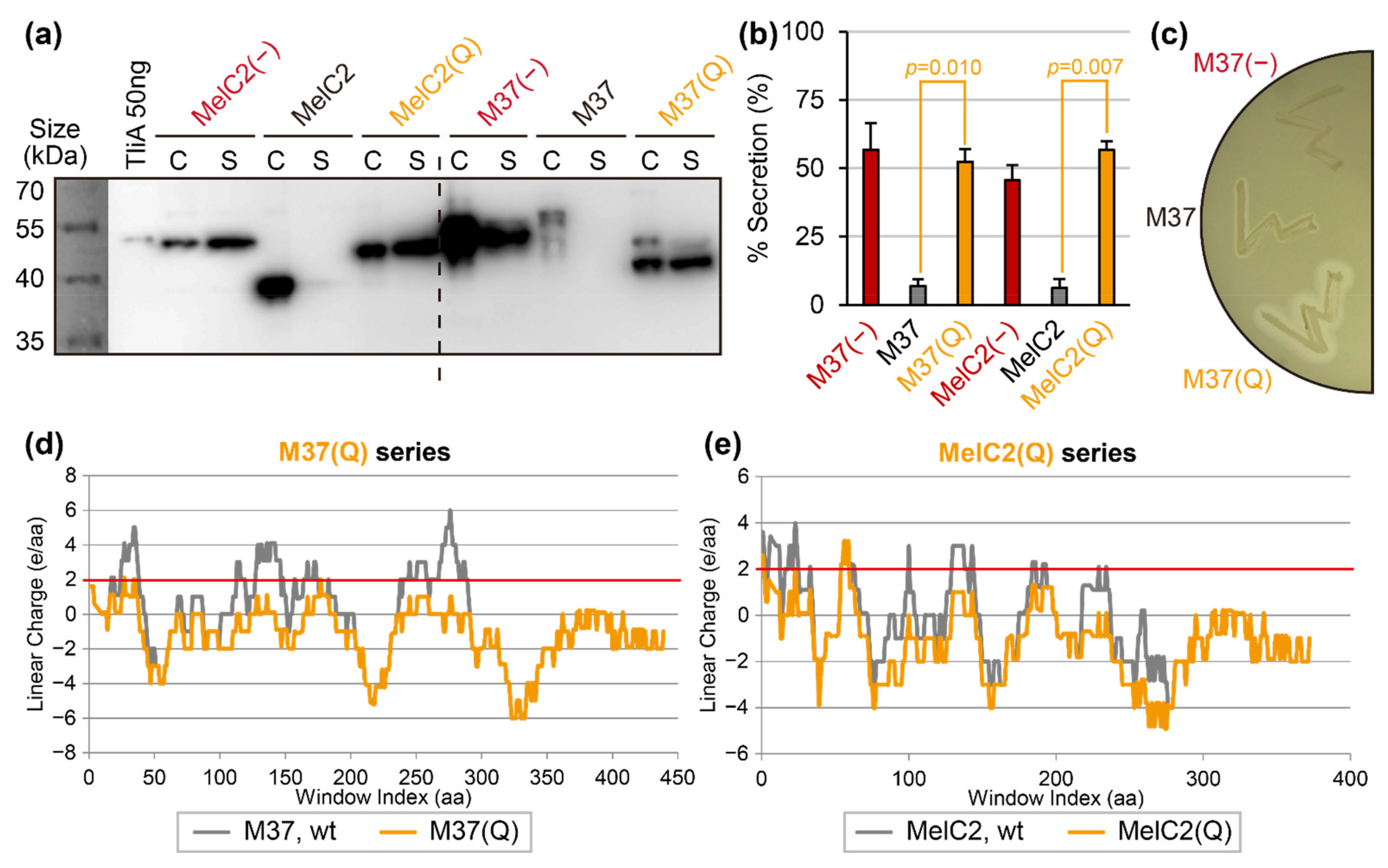
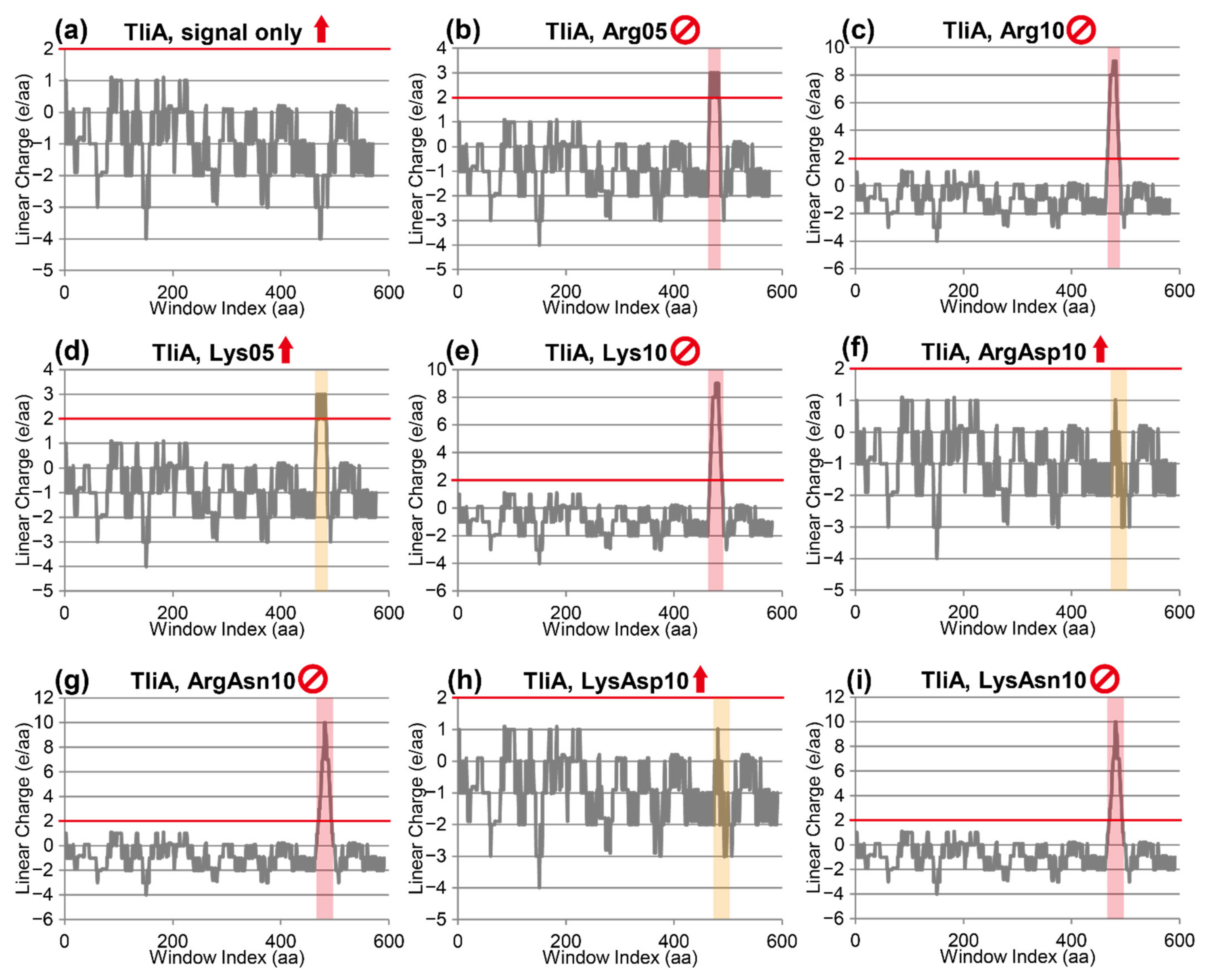
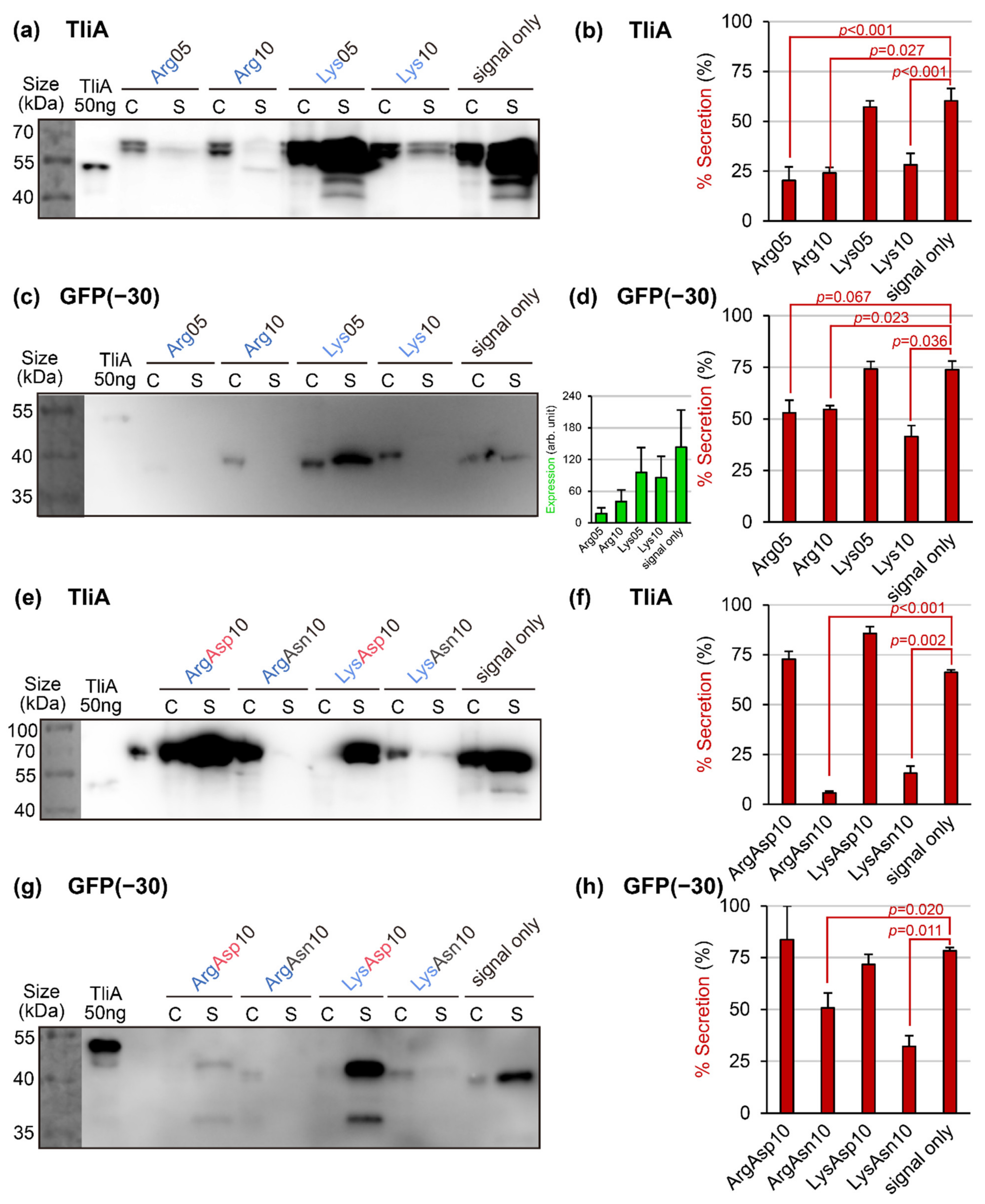
2.7. Linear Charge Density-Focused Rational Mutant Design
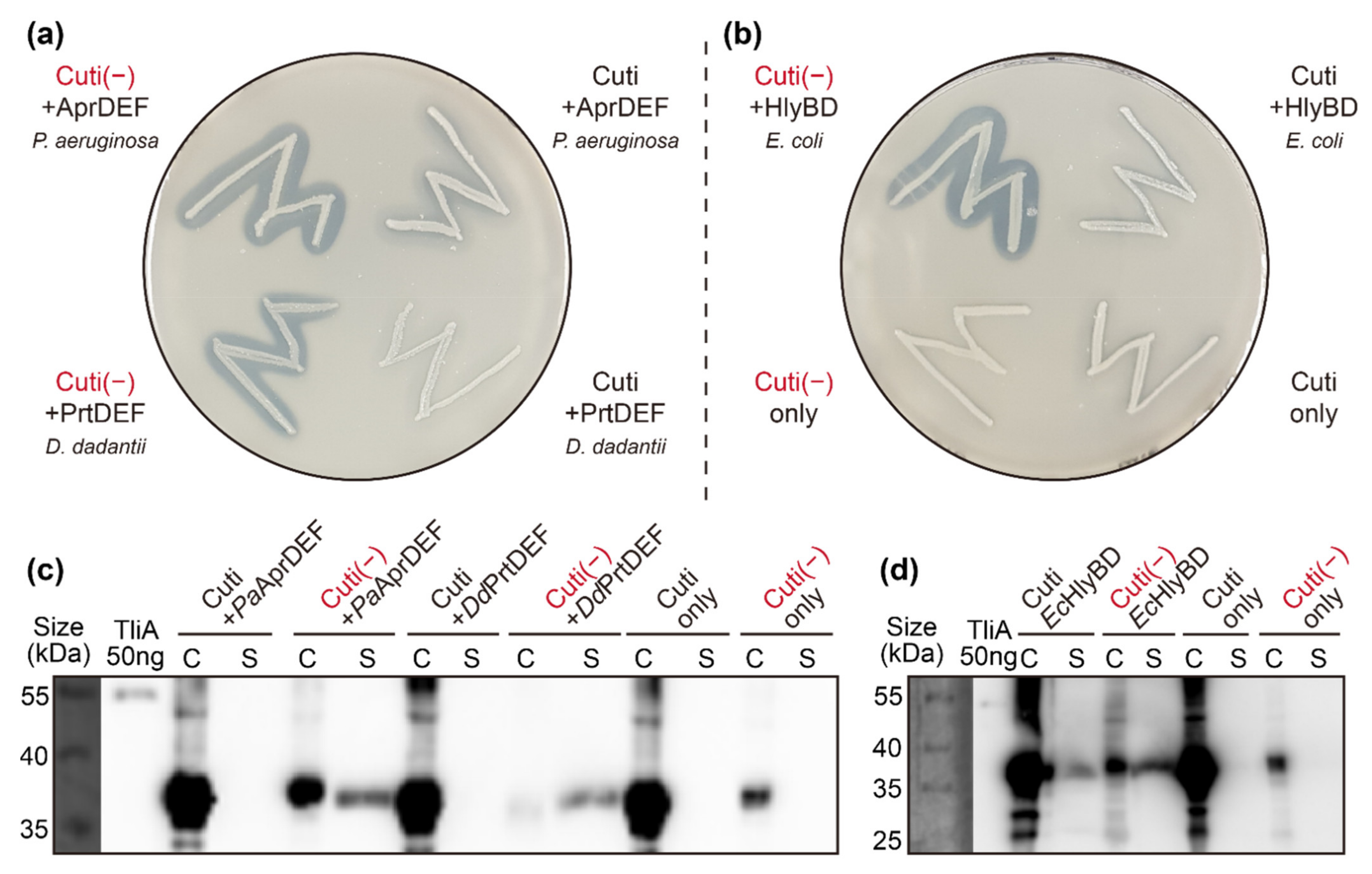
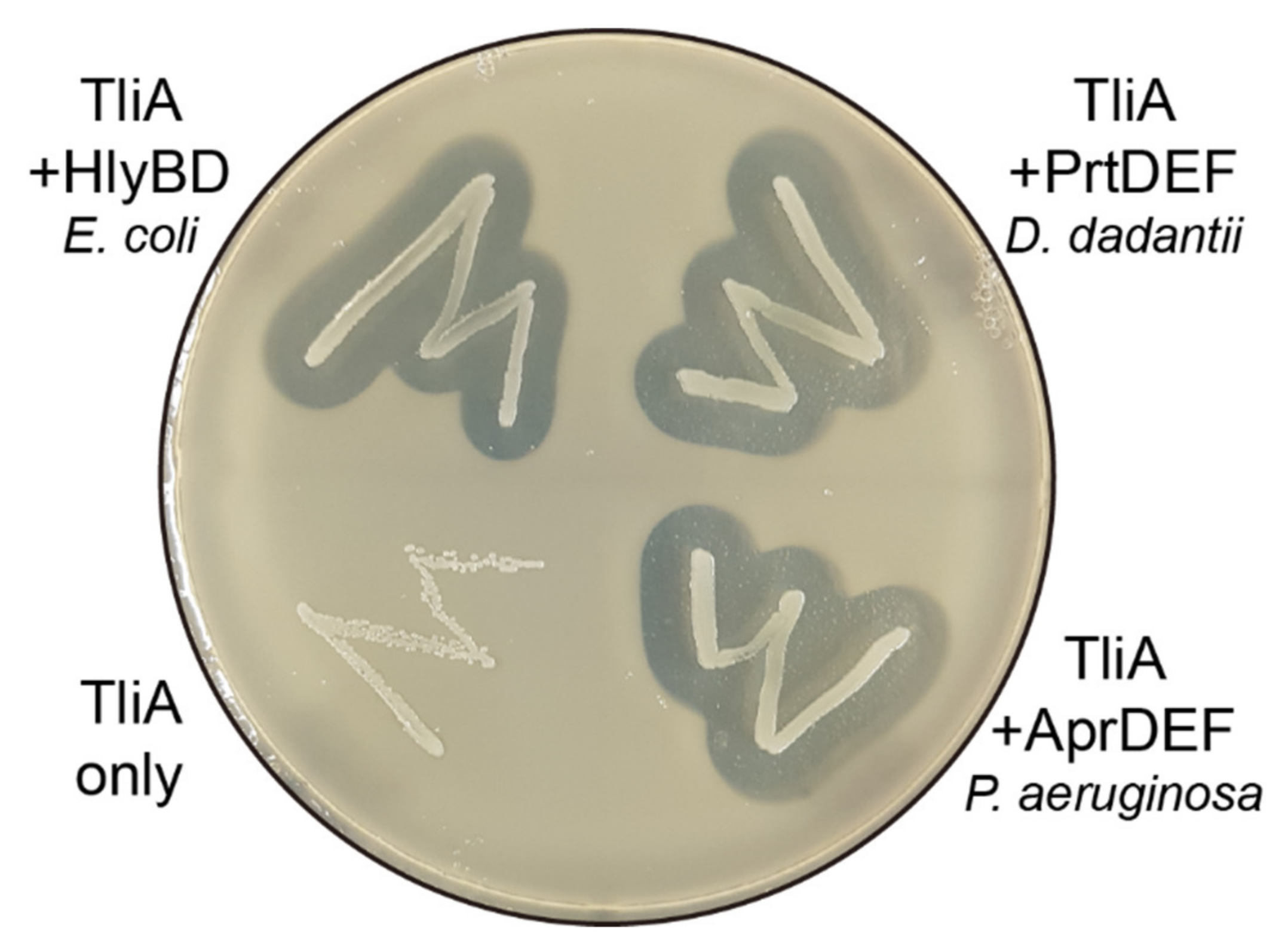
2.8. Other ABC Transporters Exhibited Similar Behaviors
3. Discussion
4. Materials and Methods
4.1. Bacterial Strains and Growth Conditions
4.2. Examining the Expression and Secretion of the Proteins
4.3. Examining the Enzymatic Activities of the Substrate Proteins
4.4. The Positively Charged Patch Experiments
4.5. Manual Mutagenesis
4.6. Computational Design of Supercharged Proteins
4.7. Synthesizing and Cloning the Genes of Supercharged Proteins
4.8. Various ABC Transporters Test (Double-Transformation)
4.9. Synthesizing and Cloning the Gene of Variationally Supercharged M37
4.10. Linear Charge Density Analyses
4.11. The linear Charge Density-Focused Supercharging
4.12. Protein Structure Prediction
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A





References
- Baneyx, F. Recombinant protein expression in Escherichia coli. Curr. Opin. Biotechnol. 1999, 10, 411–421. [Google Scholar] [CrossRef]
- Structural Genomics Consortium; China Structural Genomics Consortium; Northeast Structural Genomics Consortium; Gräslund, S.; Nordlund, P.; Weigelt, J.; Hallberg, B.M.; Bray, J.; Gileadi, O.; Knapp, S.; et al. Protein production and purification. Nat. Methods 2008, 5, 135–146. [Google Scholar]
- Chen, R. Bacterial expression systems for recombinant protein production: E. Coli and beyond. Biotechnol. Adv. 2012, 30, 1102–1107. [Google Scholar] [CrossRef] [PubMed]
- Ihling, N.; Uhde, A.; Scholz, R.; Schwarz, C.; Schmitt, L.; Büchs, J. Scale-up of a type i secretion system in e. Coli using a defined mineral medium. Biotechnol. Prog. 2020, 36, e2911. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burdette, L.A.; Leach, S.A.; Wong, H.T.; Tullman-Ercek, D. Developing gram-negative bacteria for the secretion of heterologous proteins. Microb. Cell Factories 2018, 17, 196. [Google Scholar] [CrossRef] [Green Version]
- Von Graevenitz, A.; Weinstein, J. Pathogenic significance of pseudomonas fluorescens and pseudomonas putida. Yale J. Biol. Med. 1971, 44, 265–273. [Google Scholar]
- Huang, K.-X.; Badger, M.; Haney, K.; Evans, S.L. Large scale production of bacillus thuringiensis ps149b1 insecticidal proteins cry34ab1 and cry35ab1 from pseudomonas fluorescens. Protein Expr. Purif. 2007, 53, 325–330. [Google Scholar] [CrossRef]
- Squires, C.H.; Retallack, D.M.; Chew, L.C.; Ramseier, T.M.; Schneider, J.C.; Talbot, H.W. Heterologous protein production in p. Fluorescens. BioProcess Int. 2004, 2004, 54–58. [Google Scholar]
- Ahn, J.H.; Pan, J.G.; Rhee, J.S. Identification of the tlidef abc transporter specific for lipase in pseudomonas fluorescens sik w1. J. Bacteriol. 1999, 181, 1847–1852. [Google Scholar] [CrossRef] [Green Version]
- Park, Y.; Moon, Y.; Ryoo, J.; Kim, N.; Cho, H.; Ahn, J.H. Identification of the minimal region in lipase abc transporter recognition domain of pseudomonas fluorescens for secretion and fluorescence of green fluorescent protein. Microb. Cell Factories 2012, 11, 60. [Google Scholar] [CrossRef] [Green Version]
- Son, M.; Moon, Y.; Oh, M.J.; Han, S.B.; Park, K.H.; Kim, J.-G.; Ahn, J.H. Lipase and protease double-deletion mutant of pseudomonas fluorescens suitable for extracellular protein production. Appl. Environ. Microbiol. 2012, 78, 8454–8462. [Google Scholar] [CrossRef] [Green Version]
- Ryu, J.; Lee, U.; Park, J.; Yoo, D.-H.; Ahn, J.H. A vector system for abc transporter-mediated secretion and purification of recombinant proteins in pseudomonas species. Appl. Environ. Microbiol. 2015, 81, 1744–1753. [Google Scholar] [CrossRef] [Green Version]
- Park, J.; Eom, G.T.; Young Oh, J.; Hyun Park, J.; Kim, S.C.; Kwang Song, J.; Hoon Ahn, J. High-level production of bacteriotoxic phospholipase a1 in bacterial host pseudomonas fluorescens via abc transporter-mediated secretion and inducible expression. Microorganisms 2020, 8, 239. [Google Scholar] [CrossRef] [PubMed]
- Byun, H.; Park, J.; Kim, S.C.; Ahn, J.H. A lower isoelectric point increases signal sequence-mediated secretion of recombinant proteins through a bacterial abc transporter. J. Biol. Chem. 2017, 292, 19782–19791. [Google Scholar] [CrossRef] [Green Version]
- Lawrence, M.S.; Phillips, K.J.; Liu, D.R. Supercharging proteins can impart unusual resilience. J. Am. Chem. Soc. 2007, 129, 10110–10112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Delepelaire, P. Type i secretion in gram-negative bacteria. Biochim. Biophys. Acta 2004, 1694, 149–161. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bakkes, P.J.; Jenewein, S.; Smits, S.H.J.; Holland, I.B.; Schmitt, L. The rate of folding dictates substrate secretion by the escherichia coli hemolysin type 1 secretion system. J. Biol. Chem. 2010, 285, 40573–40580. [Google Scholar] [CrossRef] [Green Version]
- Schwarz, C.K.W.; Lenders, M.H.H.; Smits, S.H.J.; Schmitt, L. Secretion of slow-folding proteins by a type 1 secretion system. Bioengineered 2012, 3, 289–292. [Google Scholar] [CrossRef]
- Bumba, L.; Masin, J.; Macek, P.; Wald, T.; Motlova, L.; Bibova, I.; Klimova, N.; Bednarova, L.; Veverka, V.; Kachala, M.; et al. Calcium-driven folding of rtx domain beta-rolls ratchets translocation of rtx proteins through type i secretion ducts. Mol. Cell 2016, 62, 47–62. [Google Scholar] [CrossRef] [Green Version]
- Berg, J.M.; Tymoczko, J.L.; Stryer, L. Biochemistry, 6th ed.; W.H. Freeman: New York, NY, USA, 2007. [Google Scholar]
- Booth, I.R. Regulation of cytoplasmic ph in bacteria. Microbiol. Rev. 1985, 49, 359–378. [Google Scholar] [CrossRef]
- Wilks, J.C.; Slonczewski, J.L. Ph of the cytoplasm and periplasm of escherichia coli: Rapid measurement by green fluorescent protein fluorimetry. J. Bacteriol. 2007, 189, 5601–5607. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arce-Rodríguez, A.; Volke, D.C.; Bense, S.; Häussler, S.; Nikel, P.I. Non-invasive, ratiometric determination of intracellular ph in pseudomonas species using a novel genetically encoded indicator. Microb. Biotechnol. 2019, 12, 799–813. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Panahi, A.; Brooks, C.L. Membrane environment modulates the pka values of transmembrane helices. J. Phys. Chem. B 2015, 119, 4601–4607. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andersson, H.; von Heijne, G. Positively charged residues influence the degree of seca dependence in protein translocation across the e. Coli inner membrane. FEBS Lett. 1994, 347, 169–172. [Google Scholar] [CrossRef] [Green Version]
- Summers, R.G.; Knowles, J.R. Illicit secretion of a cytoplasmic protein into the periplasm of escherichia coli requires a signal peptide plus a portion of the cognate secreted protein. Demarcation of the critical region of the mature protein. J. Biol. Chem. 1989, 264, 20074–20081. [Google Scholar] [CrossRef]
- Summers, R.G.; Harris, C.R.; Knowles, J.R. A conservative amino acid substitution, arginine for lysine, abolishes export of a hybrid protein in escherichia coli. Implications for the mechanism of protein secretion. J. Biol. Chem. 1989, 264, 20082–20088. [Google Scholar] [CrossRef]
- Boyd, D.; Beckwith, J. The role of charged amino acids in the localization of secreted and membrane proteins. Cell 1990, 62, 1031–1033. [Google Scholar] [CrossRef]
- Allen, W.J.; Corey, R.A.; Watkins, D.W.; Pereira, G.C.; Oliveira, A.S.F.; Hards, K.; Cook, G.M.; Collinson, I. Rate-limiting transport of positive charges through the Sec-machinery is integral to the mechanism of protein transport. bioRxiv 2022. [Google Scholar] [CrossRef] [Green Version]
- Simon, S.M.; Peskin, C.S.; Oster, G.F. What drives the translocation of proteins? Proc. Natl. Acad. Sci. USA 1992, 89, 3770–3774. [Google Scholar] [CrossRef] [Green Version]
- Allen, W.J.; Corey, R.A.; Oatley, P.; Sessions, R.B.; Baldwin, S.A.; Radford, S.E.; Tuma, R.; Collinson, I. Two-way communication between secy and seca suggests a brownian ratchet mechanism for protein translocation. eLife 2016, 5, e15598. [Google Scholar] [CrossRef]
- Seidman, C.E.; Struhl, K.; Sheen, J.; Jessen, T. Introduction of plasmid DNA into cells. In Current Protocols in Molecular Biology; Ausubel, M.F., Brent, R., Kingston, R.E., Moore, D.D., Seidman, J.G., Smith, J.A., Struhl, K., Eds.; John Wiley & Sons, Inc.: New York, NY, USA, 1987; Volume 1. [Google Scholar]
- Waterhouse, A.; Bertoni, M.; Bienert, S.; Studer, G.; Tauriello, G.; Gumienny, R.; Heer, F.T.; de Beer, T.A.P.; Rempfer, C.; Bordoli, L. Swiss-model: Homology modelling of protein structures and complexes. Nucleic Acids Res. 2018, 46, W296–W303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Der, B.S.; Kluwe, C.; Miklos, A.E.; Jacak, R.; Lyskov, S.; Gray, J.J.; Georgiou, G.; Ellington, A.D.; Kuhlman, B. Alternative computational protocols for supercharging protein surfaces for reversible unfolding and retention of stability. PLoS ONE 2013, 8, e64363. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chew, L.C.; Ramseier, T.M.; Retallack, D.M.; Schneider, J.C.; Squires, C.H.; Talbot, H.W. Pseudomonas fluorescens. In Production of Recombinant Proteins; Gellissen, G., Ed.; Wiley-VCH: Weinheim, Germany, 2005; pp. 45–62. [Google Scholar]
- Hoover, D.M.; Lubkowski, J. Dnaworks: An automated method for designing oligonucleotides for pcr-based gene synthesis. Nucleic Acids Res. 2002, 30, e43. [Google Scholar] [CrossRef] [PubMed]
- Xiong, A.-S.; Yao, Q.-H.; Peng, R.-H.; Li, X.; Fan, H.-Q.; Cheng, Z.-M.; Li, Y. A simple, rapid, high-fidelity and cost-effective pcr-based two-step DNA synthesis method for long gene sequences. Nucleic Acids Res. 2004, 32, e98. [Google Scholar] [CrossRef] [Green Version]
- Ashkenazy, H.; Abadi, S.; Martz, E.; Chay, O.; Mayrose, I.; Pupko, T.; Ben-Tal, N. Consurf 2016: An improved methodology to estimate and visualize evolutionary conservation in macromolecules. Nucleic Acids Res. 2016, 44, W344–W350. [Google Scholar] [CrossRef] [Green Version]
- Landau, M.; Mayrose, I.; Rosenberg, Y.; Glaser, F.; Martz, E.; Pupko, T.; Ben-Tal, N. Consurf 2005: The projection of evolutionary conservation scores of residues on protein structures. Nucleic Acids Res. 2005, 33, W299–W302. [Google Scholar] [CrossRef]
- Murata, D.; Okano, H.; Angkawidjaja, C.; Akutsu, M.; Tanaka, S.I.; Kitahara, K.; Yoshizawa, T.; Matsumura, H.; Kado, Y.; Mizohata, E.; et al. Structural basis for the serratia marcescens lipase secretion system: Crystal structures of the membrane fusion protein and nucleotide-binding domain. Biochemistry 2017, 56, 6281–6291. [Google Scholar] [CrossRef]
- Baek, M.; Park, T.; Heo, L.; Park, C.; Seok, C. Galaxyhomomer: A web server for protein homo-oligomer structure prediction from a monomer sequence or structure. Nucleic Acids Res. 2017, 45, W320–W324. [Google Scholar] [CrossRef] [Green Version]
- Yang, K.-S.; Sung, B.-H.; Park, M.-K.; Lee, J.-H.; Lim, K.-J.; Park, S.-C.; Kim, S.-J.; Kim, H.-K.; Sohn, J.-H.; Kim, H.-M.; et al. Recombinant Lipase Engineered with Amphipathic and Coiled-Coil Peptides. ACS Catal. 2015, 5, 5016–5025. [Google Scholar] [CrossRef]
- Kim, D.; Jeon, C.; Kim, J.-H.; Kim, M.-S.; Yoon, C.-H.; Choi, I.-S.; Kim, S.-H.; Bae, Y.-S. Cytoplasmic transduction peptide (CTP): New approach for the delivery of biomolecules into cytoplasm in vitro and in vivo. Exp. Cell Res. 2006, 312, 1277–1288. [Google Scholar] [CrossRef]
- Radaev, S.; Zou, Z.; Huang, T.; Lafer, E.M.; Hinck, A.P.; Sun, P.D. Ternary complex of transforming growth factor-beta1 reveals isoform-specific ligand recognition and receptor recruitment in the superfamily. J. Biol. Chem. 2010, 285, 14806–14814. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brown, A.; Robinson, C.J.; Gallagher, J.T.; Blundell, T.L. Cooperative heparin-mediated oligomerization of fibroblast growth factor-1 (FGF1) precedes recruitment of FGFR2 to ternary complexes. Biophys. J. 2013, 104, 1720–1730. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mori, S.; Wu, C.-Y.; Yamaji, S.; Saegusa, J.; Shi, B.; Ma, Z.; Kuwabara, Y.; Lam, K.S.; Isseroff, R.R.; Takada, Y.K.; et al. Direct Binding of Integrin αvβ3 to FGF1 Plays a Role in FGF1 Signaling. J. Biol. Chem. 2008, 283, 18066–18075. [Google Scholar] [CrossRef] [PubMed] [Green Version]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Byun, H.; Park, J.; Fabia, B.U.; Bingwa, J.; Nguyen, M.H.; Lee, H.; Ahn, J.H. Generalized Approach towards Secretion-Based Protein Production via Neutralization of Secretion-Preventing Cationic Substrate Residues. Int. J. Mol. Sci. 2022, 23, 6700. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23126700
Byun H, Park J, Fabia BU, Bingwa J, Nguyen MH, Lee H, Ahn JH. Generalized Approach towards Secretion-Based Protein Production via Neutralization of Secretion-Preventing Cationic Substrate Residues. International Journal of Molecular Sciences. 2022; 23(12):6700. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23126700
Chicago/Turabian StyleByun, Hyunjong, Jiyeon Park, Benedict U. Fabia, Joshua Bingwa, Mihn Hieu Nguyen, Haeshin Lee, and Jung Hoon Ahn. 2022. "Generalized Approach towards Secretion-Based Protein Production via Neutralization of Secretion-Preventing Cationic Substrate Residues" International Journal of Molecular Sciences 23, no. 12: 6700. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23126700