
Use of Self-Assembled Colloidal Prodrug Nanoparticles for Controlled Drug Delivery of Anticancer, Antifibrotic and Antibacterial Mitomycin

 , ,
, ,
Abstract
:1. Introduction
2. Results
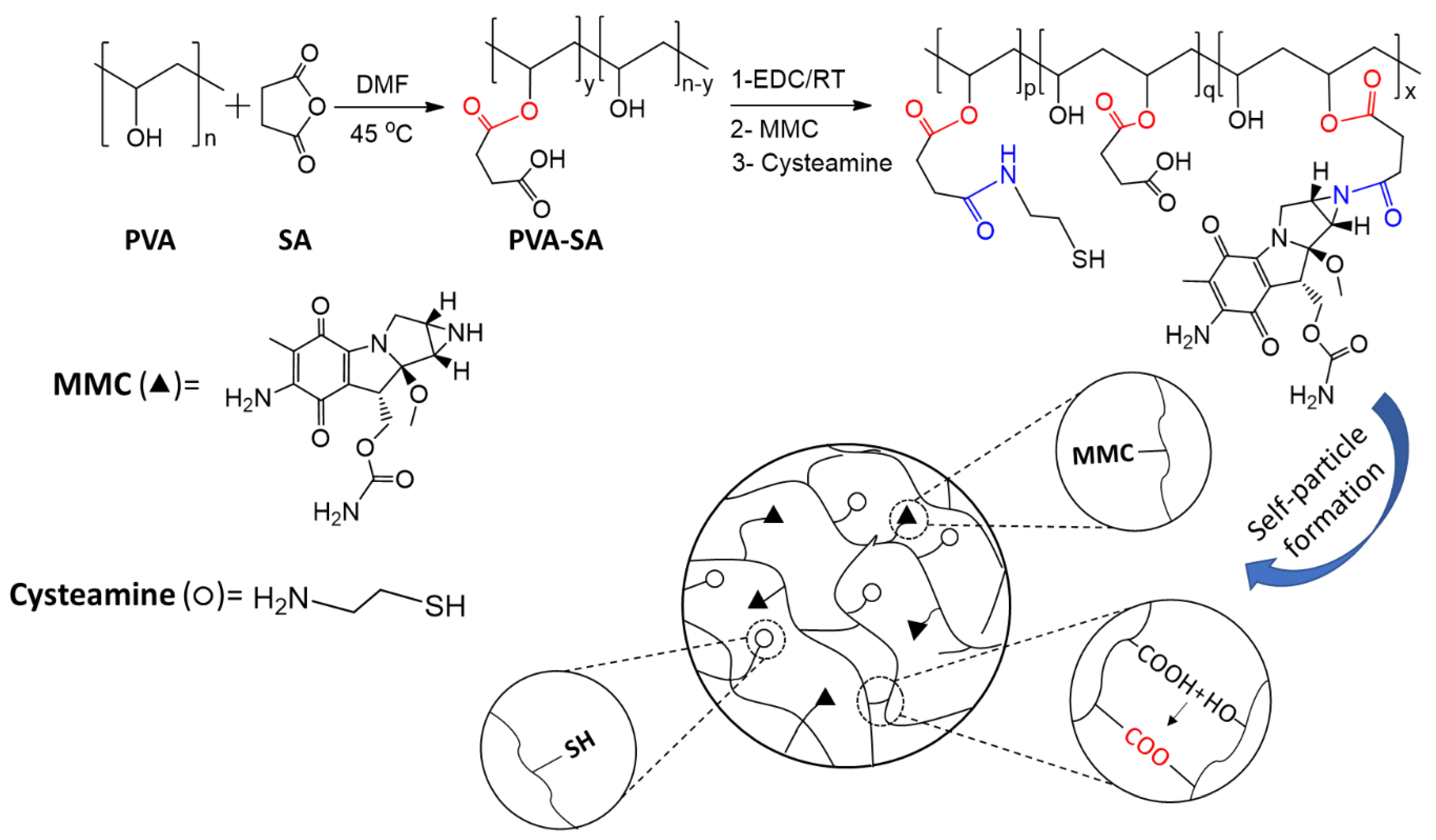
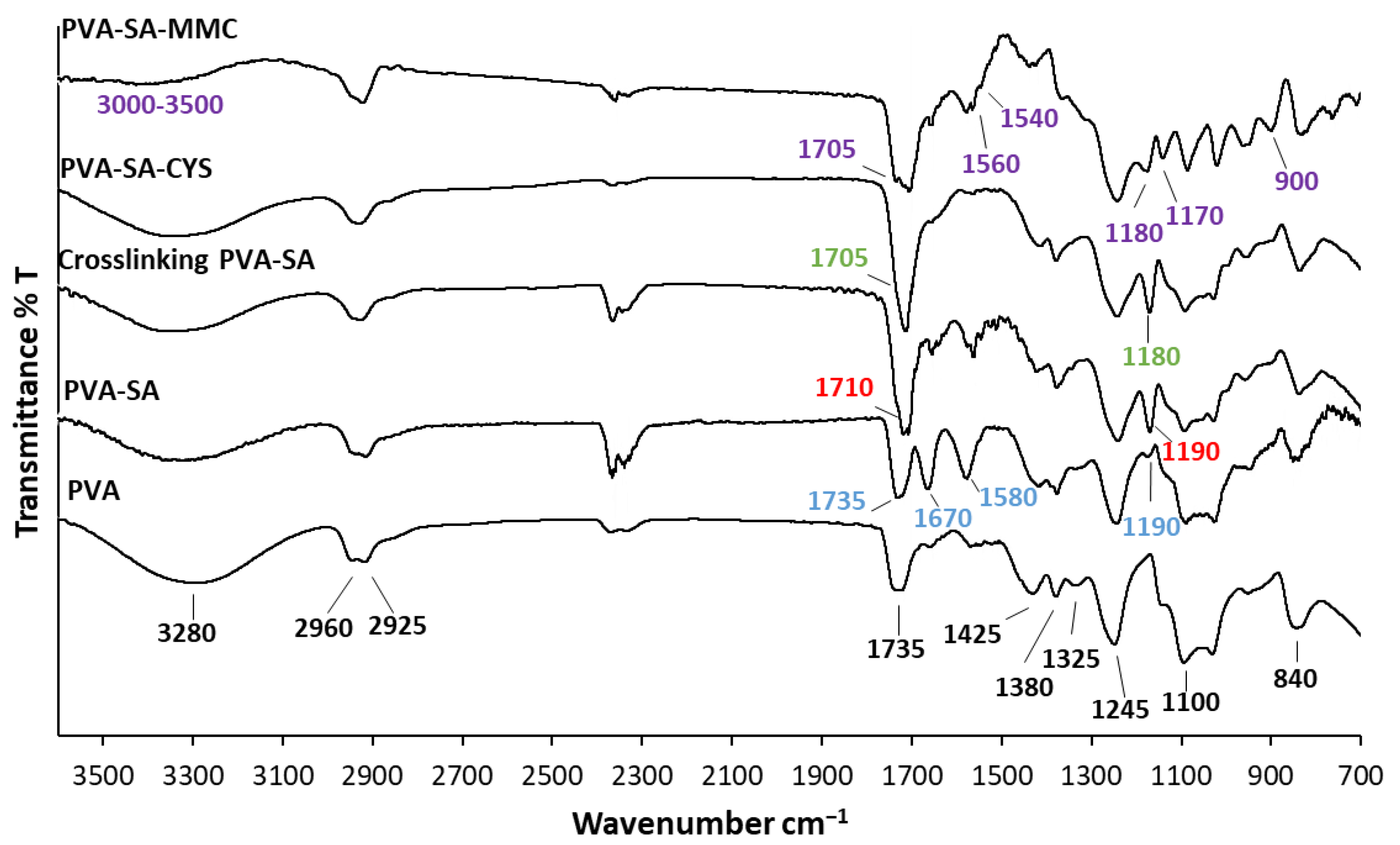
2.1. Structural Characterization of the Synthesized Samples
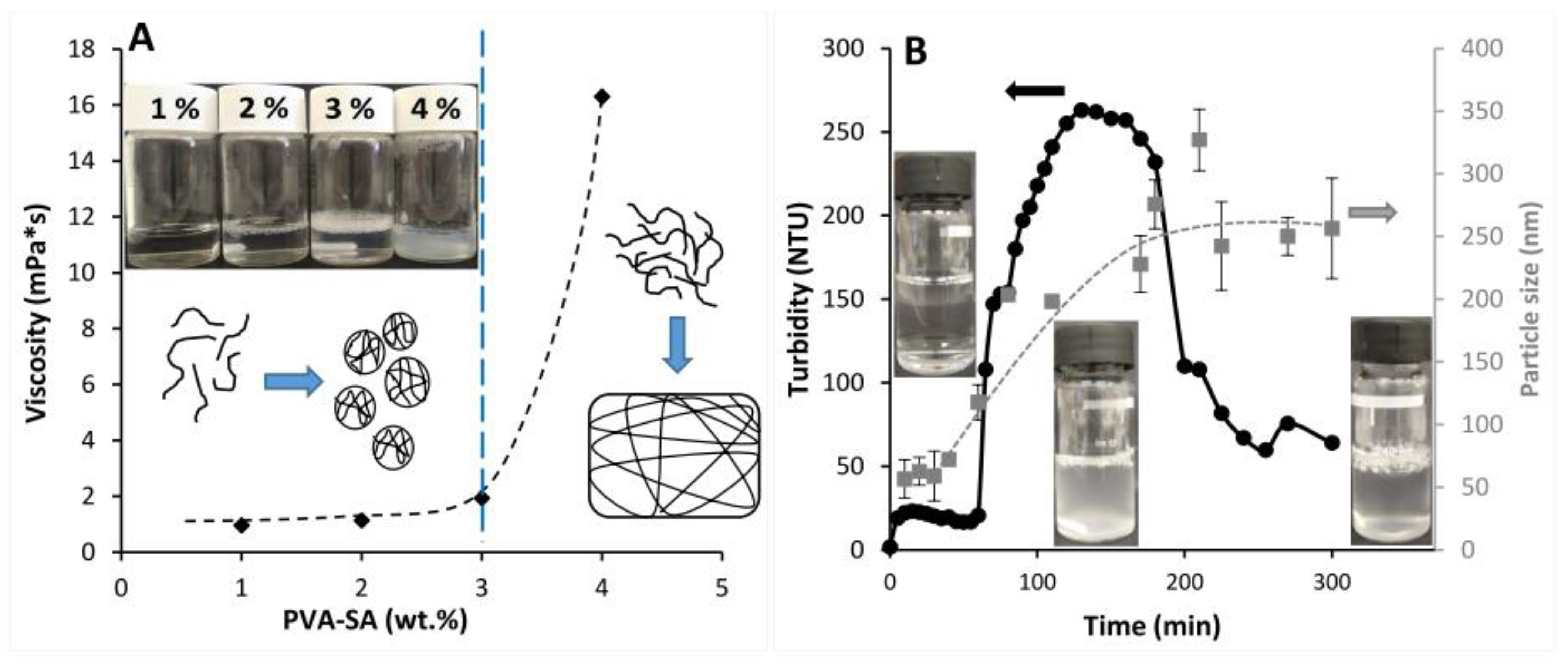
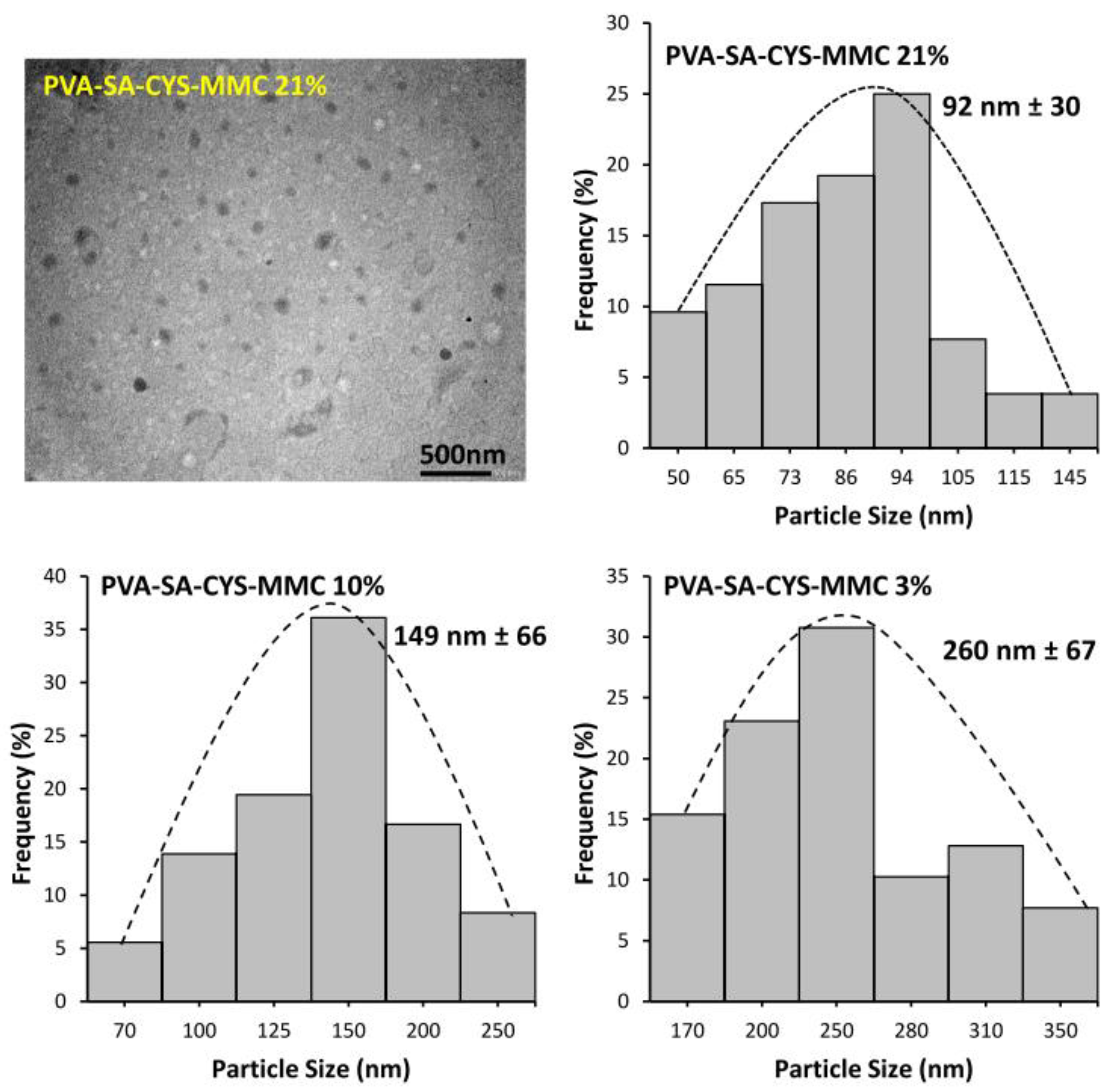
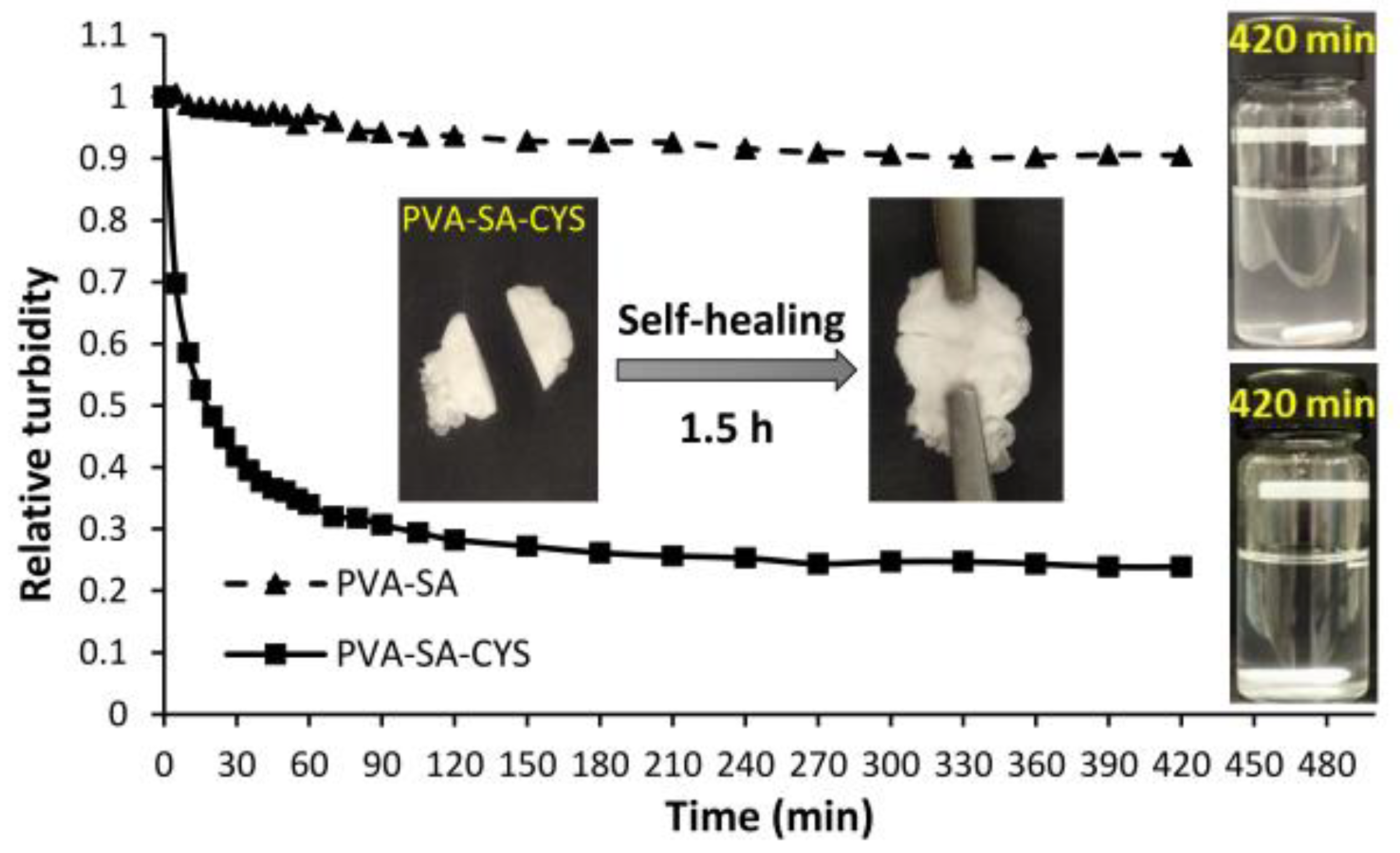
2.2. Self-Assembled Particle Formation
2.3. Mucoadhesive Properties of Cysteamine Polymeric Conjugate
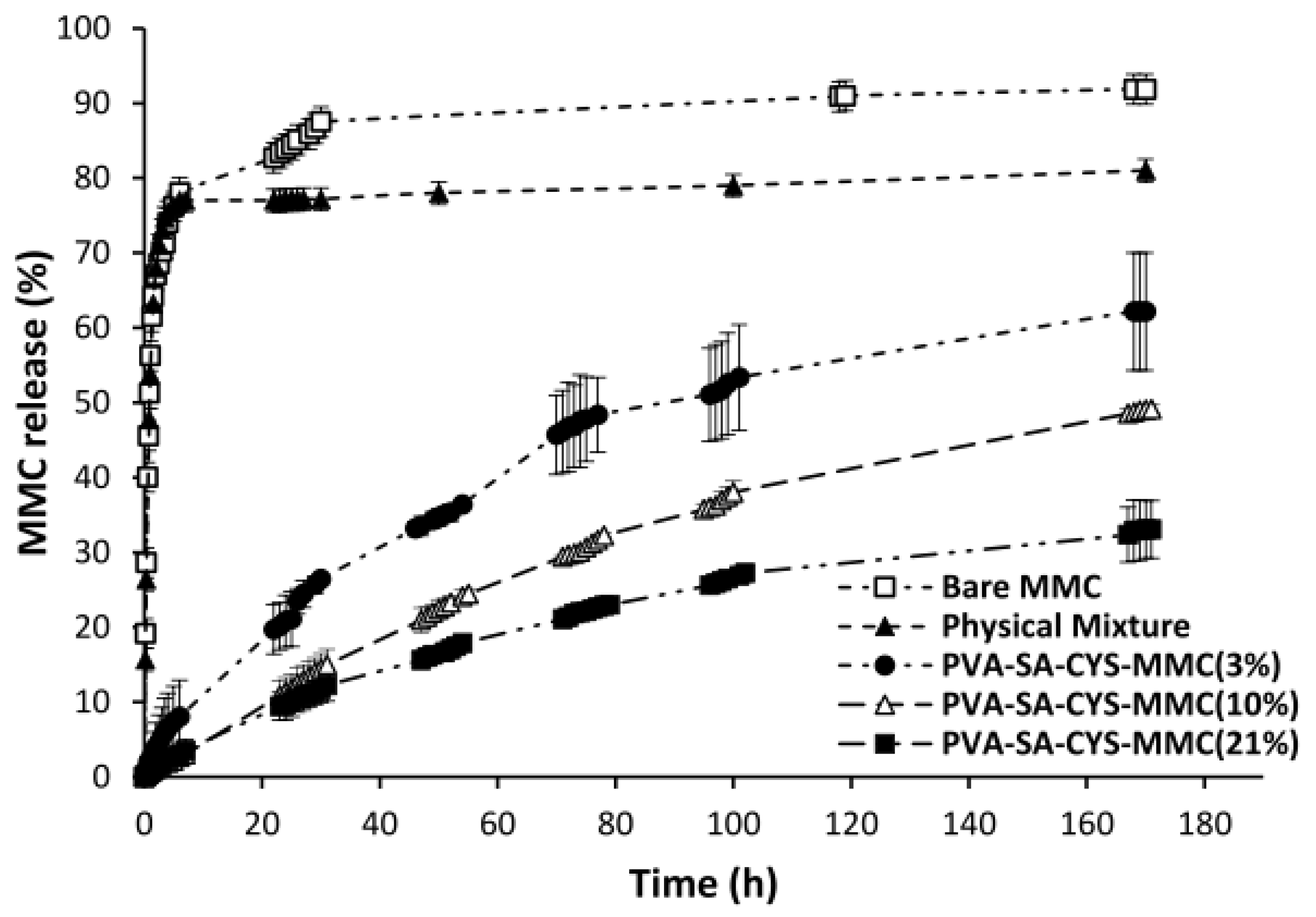
2.4. In Vitro Drug Release Measurement
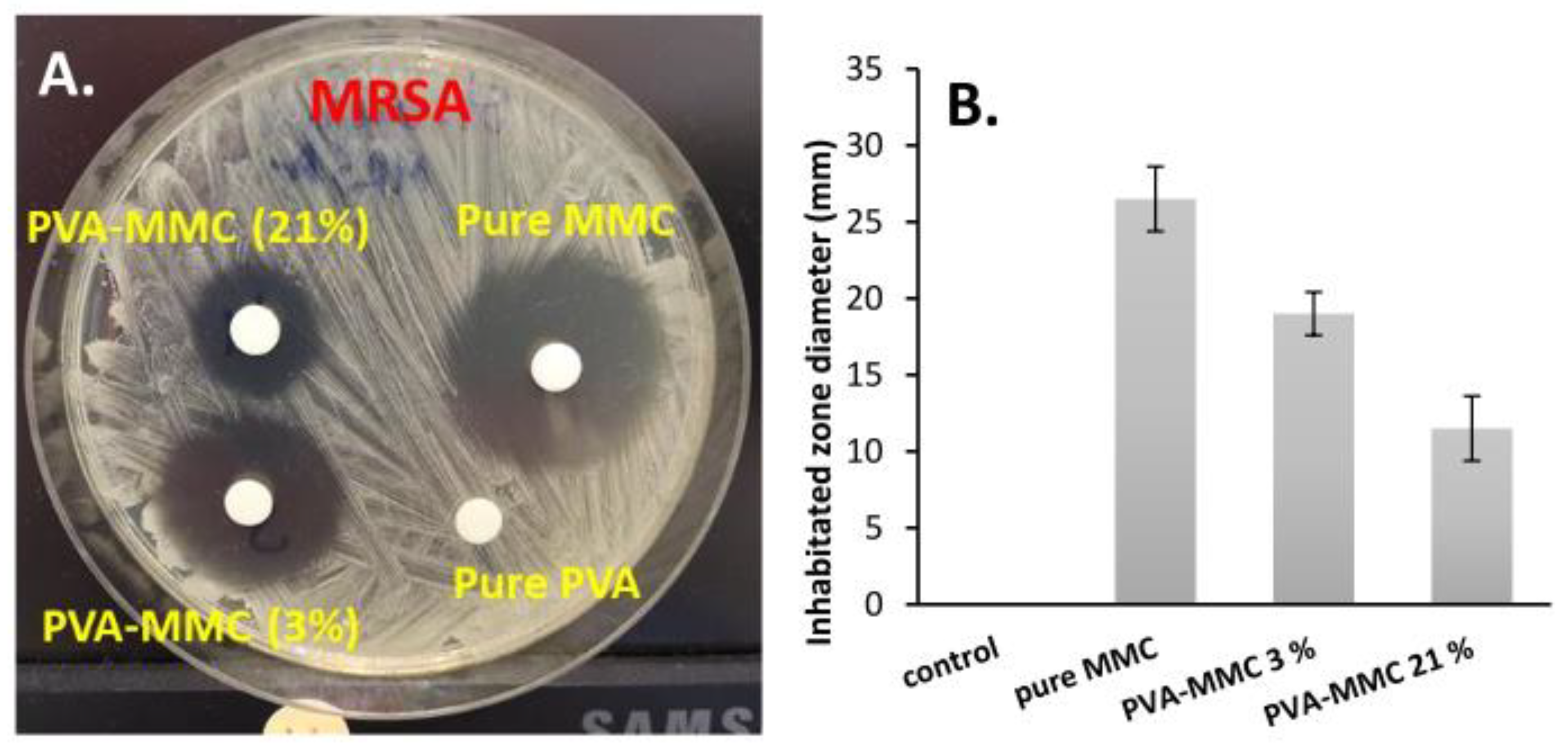
2.5. In Vitro Evaluation of the Anticancer and Antibacterial Activity of MMC Samples
3. Discussion
4. Materials and Methods
4.1. Preparation of PVA-SA-CYS-MMC by One-Pot Reaction
4.2. Methods of Characterization
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Merritt, S.R.; Velasquez, G.; von Recum, H.A. Adjustable release of mitomycin C for inhibition of scar tissue formation after filtration surgery. Exp. Eye Res. 2013, 116, 9–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mizuno, M.; Kawabata, S.; Hamaura, T.; Hashida, M.; Sezaki, H. Characterization of mitomycin C-induced gastrointestinal damage. I. In situ recirculation experiment. Toxicol. Appl. Pharmacol. 1989, 97, 415–423. [Google Scholar] [CrossRef]
- Molyneux, G.; Gibson, F.M.; Gordon-Smith, E.C.; Pilling, A.M.; Liu, K.C.; Rizzo, S.; Sulsh, S.; Turton, J.A. The haemotoxicity of mitomycin in a repeat dose study in the female CD-1 mouse. Int. J. Exp. Path. 2005, 86, 415–430. [Google Scholar] [CrossRef]
- Crooke, S.T.; Bradner, W.T. Mitomycin C: A review. Cancer Treat. Rev. 1976, 3, 121–139. [Google Scholar] [CrossRef]
- Nomura, T.; Saikawa, A.; Morita, S.; Sakaeda Kakutani, T.; Yamashita, F.; Honda, K.; Takakura, Y.; Hashida, M. Pharmacokinetic characteristics and therapeutic effects of mitomycin C-dextran conjugates after intratumoural injection. J. Control. Release 1998, 52, 239–252. [Google Scholar] [CrossRef]
- Cheung, R.Y.; Ying, Y.; Rauth, A.M.; Marcon, N.; Wu, X.Y. Biodegradable dextran-based microspheres for delivery of anticancer drug mitomycin C. Biomaterials 2005, 26, 5375–5385. [Google Scholar] [CrossRef]
- Cumming, J.; Allan, L.; Smyth, J.F. Encapsulation of mitomycin C in albumin microspheres markedly alters pharmacokinetics, drug quinone reduction in tumour tissue and antitumour activity: Implications for the drugs’ in vivo mechanism of action. Biochem. Pharmacol. 1994, 47, 1345–1356. [Google Scholar] [CrossRef]
- Xi-Xiao, Y.; Jan-Hai, C.; Shi-Ting, L.; Dan, G.; Xv-Xin, Z. Polybutylcyanoacrylate nanoparticles as a carrier for mitomycin C in rabbits bearing VX2-liver tumor. Regul. Toxicol. Pharmacol. 2006, 46, 211–217. [Google Scholar] [CrossRef]
- Ishiki, N.; Onishi, H.; Machida, Y. Evaluation of antitumor and toxic side effects of mitomycin C-estradiol conjugates. Int. J. Pharm. 2004, 279, 81–93. [Google Scholar] [CrossRef]
- Liu, Y.C.; Li, H.; Shu, X.Z.; Gray, S.D.; Prestwich, G.D. Crosslinked hyaluronan hydrogels containing mitomycin C reduce postoperative abdominal adhesions. Fertil. Steril. 2005, 83, 1275–1283. [Google Scholar] [CrossRef]
- Sarisozen, C.; Aktaş, Y.; Mungan, A.; Bilensoy, E. Bioadhesive coated poly-epsilon-caprolactone nanoparticles loaded with mitomycin C for the treatment of superficial bladder tumors. Eur. J. Pharm. Sci. 2007, 32, S36. [Google Scholar] [CrossRef]
- Ringsdorf, H. Structure and properties of pharmacologically active polymers. J. Polym. Sci. Polym. Symp. 1975, 51, 135–153. [Google Scholar] [CrossRef]
- Mahesh, S.; Tang, K.C.; Raj, M. Amide bond activation of biological molecules. Molecules 2018, 23, 2615. [Google Scholar] [CrossRef] [Green Version]
- Singh, I.; Rana, V. Techniques for the assessment of mucoadhesion in drug delivery systems: An overview. J. Adhes. Sci. Technol. 2012, 26, 2251–2267. [Google Scholar] [CrossRef]
- Leitner, V.; Walker, G.; Bernkop-Schnürch, A. Thiolated polymers: Evidence for the formation of disulphide bonds with mucus glycoproteins. Eur. J. Pharm. Biopharm. 2003, 56, 207–714. [Google Scholar] [CrossRef]
- Albrecht, K.; Zirm, E.J.; Palmberger, T.F.; Schlocker, W.; Bernkop-Schnürch, A. Preparation of thiomer microparticles and in vitro evaluation of parameters influencing their mucoadhesive properties. Drug Dev. Ind. Pharm. 2006, 32, 1149–1157. [Google Scholar] [CrossRef]
- Chiellini, E.; Corti, A.; D’Antone, S.; Solaro, R. Biodegradation of poly (vinyl alcohol) based materials. Prog. Polym. Sci. 2003, 28, 963–1014. [Google Scholar] [CrossRef]
- Moulay, S. Review: Poly(vinyl alcohol) functionalizations and applications. Polym. Plast. Technol. Eng. 2015, 54, 1289–1319. [Google Scholar] [CrossRef]
- Baker, M.I.; Walsh, S.P.; Schwartz, Z.; Boyan, B.D. A review of polyvinyl alcohol and its uses in cartilage and orthopedic applications. J. Biomed. Mater. Res. B Appl. Biomater. 2012, 100, 1451–1457. [Google Scholar] [CrossRef] [PubMed]
- Chong, S.F.; Smith, A.A.A.; Zelikin, A.N. Microstructured, Functional PVA hydrogels through bioconjugation with oligopeptides under physiological conditions. Small 2013, 9, 942–950. [Google Scholar] [CrossRef]
- Tadavarthy, S.M.; Moller, J.H.; Amplatz, K. Polyvinyl alcohol (Ivalon)—A new embolic material. Am. J. Roentgenol. 1975, 125, 609–616. [Google Scholar] [CrossRef] [Green Version]
- Greenwald, R.B.; Pendri, A.; Conover, C.D.; Zhao, H.; Choe, Y.H.; Martinez, A.; Shum, K.; Guan, S. Drug delivery systems employing 1,4- or 1,6-elimination: Poly(ethylene glycol) prodrugs of amine-containing compounds. J. Med. Chem. 1999, 42, 3657–3667. [Google Scholar] [CrossRef]
- Tsakos, M.; Schaffert, E.S.; Clement, L.L.; Villadsen, N.L.; Poulsen, T.B. Ester coupling reactions–an enduring challenge in the chemical synthesis of bioactive natural products. Nat. Prod. Rep. 2015, 32, 605–632. [Google Scholar] [CrossRef]
- Paarakh, M.P.; Ani Jose, P.; Setty, C.M.; Christoper, G.V.P. Release kinetics–concepts and applications. Int. J. Pharm. Res. Technol. 2018, 8, 12–20. [Google Scholar] [CrossRef]
- Takács, T.; Abdelghafour, M.M.; Deák, Á.; Szabó, D.; Sebők, D.; Dékány, I.; Rovó, L.; Kukovecz, Á.; Janovák, L. Surface wetting driven release of antifibrotic Mitomycin-C drug from modified biopolymer thin films. Eur. Polym. J. 2020, 139, 109995. [Google Scholar] [CrossRef]
- Zhou, Z.; Xu, W.; He, D.; Fan, J.; Yu, F.; Ren, F. Solid-state grafting of succinic anhydride onto poly(vinyl alcohol). J. Appl. Polym. Sci. 2007, 103, 848–852. [Google Scholar] [CrossRef]
- Bhat, N.V.; Nate, M.M.; Kurup, M.B.; Bambole, V.A.; Sabharwal, S. Effect of γ-radiation on the structure and morphology of polyvinyl alcohol films. Nucl. Instrum. Methods Phys. Res. B 2005, 237, 585–592. [Google Scholar] [CrossRef]
- Kharazmi, A.; Faraji, N.; Mat Hussin, R.; Saion, E.; Yunus, W.M.M.; Behzad, K. Structural, optical, opto-thermal and thermal properties of ZnS–PVA nanofluids synthesized through a radiolytic approach. Beilstein J. Nanotechnol. 2015, 6, 529–536. [Google Scholar] [CrossRef] [Green Version]
- Stipniece, L.; Salma-Ancane, K.; Rjabovs, V.; Juhnevica, I.; Turks, M.; Narkevica, I.; Berzina-Cimdina, L. Development of functionalized hydroxyapatite/poly (vinyl alcohol) composites. J. Cryst. Growth 2016, 444, 14–20. [Google Scholar] [CrossRef]
- Zhang, B.; Huang, Q.; Luo, F.X.; Fu, X.; Jiang, H.X.; Jane, J.L. Effects of octenylsuccinylation on the structure and properties of high-amylose maize starch. Carbohydr. Polym. 2011, 84, 1276–1281. [Google Scholar] [CrossRef]
- Guan, H.M.; Chung, T.S.; Huang, Z.; Mei, L.C.; Kulprathipanja, S. Poly(vinyl alcohol) multilayer mixed matrix membranes for the dehydration of ethanol-water mixture. J. Membr. Sci. 2006, 268, 113–122. [Google Scholar] [CrossRef]
- Uzu, K.; Harada, Y.; Wakaki, S.; Yamada, Y. Studies on Mitomycins, Carcinostatic Antibiotics: Part I. Derivatives and Acid Hydrolysis of Mitomycin A and C Part II. Mitomycinone. Agric. Biol. Chem. 1964, 28, 388–402. [Google Scholar] [CrossRef]
- Janovák, L.; Varga, J.; Kemény, L.; Dékány, I. The effect of surface modification of layer silicates on the thermoanalytical properties of poly(NIPAAm-co-AAm) based composite hydrogels. J. Therm. Anal. Calorim. 2009, 98, 485–493. [Google Scholar] [CrossRef]
- Joyner, K.; Song, D.; Hawkins, R.F.; Silcott, R.D.; Duncan, G.A. A rational approach to form disulfide linked mucin hydrogels. Soft Matter 2019, 15, 9632–9639. [Google Scholar] [CrossRef]
- Cook, S.L.; Bull, S.P.; Methven, L.; Parker, J.K.; Khutoryanskiy, V.V. Mucoadhesion: A food perspective. Food Hydrocoll. 2017, 72, 281–296. [Google Scholar] [CrossRef]
- Korsmeyer, R.W.; Gurny, R.; Doelker, E.; Buri, P.; Peppas, N.A. Mechanisms of solute release from porous hydrophilic polymers. Int. J. Pharm. 1983, 15, 25–35. [Google Scholar] [CrossRef]
- Ritger, P.L.; Peppas, N.A. A simple equation for description of solute release. I. Fickian and non-Fickian release from non-swellable devices in the form of slabs, spheres, cylinders or discs. J. Control. Release 1987, 5, 23–36. [Google Scholar] [CrossRef]
- Rahim, H.; Khan, M.A.; Badshah, A.; Chishti, K.A.; Khan, S.; Junaid, M. Evaluation of prunus domestica gum as a novel tablet binder. Braz. J. Pharm. Sci. 2014, 50, 195–202. [Google Scholar] [CrossRef] [Green Version]
- Kumar, U.; Islam, M.S.; Halder, S.; Rouf, A.S. Assessment of once daily sustained release hydrophilic matrix tablet of carvedilol. Dhaka. Univ. J. Pharm. Sci. 2017, 16, 43–53. [Google Scholar] [CrossRef] [Green Version]
- Freichel, O.L.; Lippold, B.C. A new oral erosion controlled drug delivery system with a late burst in the release profile. Eur. J. Pharm. Biopharm. 2000, 50, 345–351. [Google Scholar] [CrossRef]
- Sackett, C.K.; Narasimhan, B. Mathematical modeling of polymer erosion: Consequences for drug delivery. Int. J. Pharm. 2011, 418, 104–114. [Google Scholar] [CrossRef]
- Fieser, L.F. Experiments in Organic Chemistry, 2nd ed.; New Yorks Heath: New York, NY, USA, 1941; p. 104. [Google Scholar]
- Sanchez-Chaves, M.; Arranz, F.; Cortazar, M. Poly(vinyl alcohol)functionalized by monosuccinate groups. Coupling of bioactive amino compounds. Polymer 1998, 39, 2751–2757. [Google Scholar] [CrossRef]
- Gupta, B.; Anjum, S.; Ikram, S. Preparation of thiolated polyvinyl alcohol hydrogels. J. Appl. Polym. Sci. 2013, 129, 815–821. [Google Scholar] [CrossRef]
- Matuschek, E.; Brown, D.F.; Kahlmeter, G. Development of the EUCAST disk diffusion antimicrobial susceptibility testing method and its implementation in routine microbiology laboratories. Clin. Microbiol. Infect. 2014, 20, O255–O266. [Google Scholar] [CrossRef] [Green Version]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | G′ (kPa) | G″ (kPa) | G* (kPa) |
|---|---|---|---|
| 1% PVA-CYS | 24.7 | 7.55 | 32.25 |
| 1% Mucin | 12.4 | 0.78 | 13.18 |
| 1% PVA-CYS-Mucin | 158.3 | 23.2 | 181.5 |
| Sample | 1st Day | 3rd Day | 5th Day | 7th Day | ||||
|---|---|---|---|---|---|---|---|---|
| Conc (mg/mL) | IC50 (mg/mL) | Conc (mg/mL) | IC50 (mg/mL) | Conc (mg/mL) | IC50 (mg/mL) | Conc (mg/mL) | IC50 (mg/mL) | |
| Pure MMC | 0.254 | 0.029 ± 0.001 | 0.270 | 0.041 ± 0.002 | 0.273 | 0.026 ± 0.006 | 0.275 | 0.05 ± 0.045 |
| Control | 0.000 | 0.000 | 0.000 | 0.000 | 0.000 | 0.000 | 0.000 | 0.000 |
| PVA-MMC (3%) | 0.063 | 0.014 ± 0.001 | 0.140 | 0.022 ± 0.007 | 0.173 | 0.043 ± 0.025 | 0.188 | 0.054 ± 0.029 |
| PVA-MMC (21%) | 0.029 | 0.037 ± 0.004 | 0.065 | 0.038 ± 0.01 | 0.089 | 0.039 ± 0.007 | 0.097 | 0.029 ± 0.004 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Abdelghafour, M.M.; Deák, Á.; Szabó, D.; Dékány, I.; Rovó, L.; Janovák, L. Use of Self-Assembled Colloidal Prodrug Nanoparticles for Controlled Drug Delivery of Anticancer, Antifibrotic and Antibacterial Mitomycin. Int. J. Mol. Sci. 2022, 23, 6807. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23126807
Abdelghafour MM, Deák Á, Szabó D, Dékány I, Rovó L, Janovák L. Use of Self-Assembled Colloidal Prodrug Nanoparticles for Controlled Drug Delivery of Anticancer, Antifibrotic and Antibacterial Mitomycin. International Journal of Molecular Sciences. 2022; 23(12):6807. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23126807
Chicago/Turabian StyleAbdelghafour, Mohamed M., Ágota Deák, Diána Szabó, Imre Dékány, László Rovó, and László Janovák. 2022. "Use of Self-Assembled Colloidal Prodrug Nanoparticles for Controlled Drug Delivery of Anticancer, Antifibrotic and Antibacterial Mitomycin" International Journal of Molecular Sciences 23, no. 12: 6807. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23126807