
The TRPC1 Channel Forms a PI3K/CaM Complex and Regulates Pancreatic Ductal Adenocarcinoma Cell Proliferation in a Ca2+-Independent Manner

 , , , , and
, , , , and
Abstract
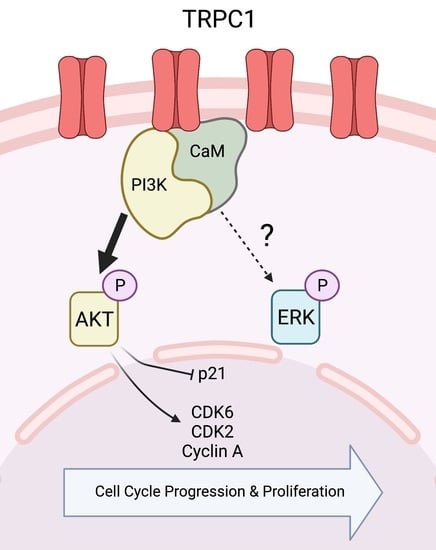
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
2.1. TRPC1 Upregulation Correlates with the Aggressive Basal-like Subtype of PDAC and with Low Overall Survival
2.2. TRPC1 Is Overexpressed in Cell Lines with an Aggressive Phenotype and Is Localized to the Plasma Membrane
2.3. The Knockdown of TPRC1 Inhibits PANC-1 Cell and Spheroid Growth
2.4. The Knockdown of TRPC1 Regulates Cell Cycle Progression by Reducing the Expression of CDK6, 2, and Cyclin A and Increasing the Expression of p21CIP1
2.5. The Knockdown of TRPC1 Does Not Affect Ca2+ Entry nor Store-Operated Ca2+ Entry and Decreases Cell Proliferation Independently of Extracellular Ca2+
2.6. TRPC1 Strongly Regulates AKT Phosphorylation in PANC-1 Cells
2.7. TRPC1 Interacts with PI3K, Which Is Attenuated upon the Knockdown of TRPC1
3. Discussion
4. Materials and Methods
4.1. Public Database Analysis
4.2. Immunohistochemistry in Human PDAC Samples
4.3. Cell Culture
4.4. Transient Transfections
4.5. 3D-Spheroid Growth and CellTiter Glo Assay
4.6. Western Blot Analysis
4.7. Co-Immunoprecipitation
4.8. Trypan Blue Assay
4.9. Flow Cytometry
4.10. Calcium Imaging
4.11. Immunofluorescence
4.12. Proximity Ligation Assay
4.13. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kleeff, J.; Korc, M.; Apte, M.; La Vecchia, C.; Johnson, C.D.; Biankin, A.V.; Neale, R.E.; Tempero, M.; Tuveson, D.A.; Hruban, R.H.; et al. Pancreatic cancer. Nat. Rev. Dis. Primers 2016, 2, 16022. [Google Scholar] [CrossRef]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2018. CA A Cancer J. Clin. 2018, 68, 7–30. [Google Scholar] [CrossRef]
- Quinonero, F.; Mesas, C.; Doello, K.; Cabeza, L.; Perazzoli, G.; Jimenez-Luna, C.; Rama, A.R.; Melguizo, C.; Prados, J. The challenge of drug resistance in pancreatic ductal adenocarcinoma: A current overview. Cancer Biol. Med. 2019, 16, 688–699. [Google Scholar] [CrossRef]
- Berridge, M.J.; Lipp, P.; Bootman, M.D. The versatility and universality of calcium signalling. Nat. Rev. Mol. Cell Biol. 2000, 1, 11–21. [Google Scholar] [CrossRef]
- Borowiec, A.S.; Bidaux, G.; Pigat, N.; Goffin, V.; Bernichtein, S.; Capiod, T. Calcium channels, external calcium concentration and cell proliferation. Eur. J. Pharmacol. 2014, 739, 19–25. [Google Scholar] [CrossRef]
- Hodeify, R.; Yu, F.; Courjaret, R.; Nader, N.; Dib, M.; Sun, L.; Adap, E.; Hubrack, S.; Machaca, K. Regulation and Role of Store-Operated Ca(2+) Entry in Cellular Proliferation. In Calcium Entry Channels in Non-Excitable Cells; Kozak, J.A., Putney, J.W., Jr., Eds.; CRC Press/Taylor & Francis: Boca Raton, FL, USA, 2018; pp. 215–240. [Google Scholar]
- Capiod, T. The need for calcium channels in cell proliferation. Recent Pat. Anticancer Drug Discov. 2013, 8, 4–17. [Google Scholar] [CrossRef]
- Tajada, S.; Villalobos, C. Calcium Permeable Channels in Cancer Hallmarks. Front. Pharmacol. 2020, 11, 968. [Google Scholar] [CrossRef]
- Chen, X.; Sooch, G.; Demaree, I.S.; White, F.A.; Obukhov, A.G. Transient Receptor Potential Canonical (TRPC) Channels: Then and Now. Cells 2020, 9, 1983. [Google Scholar] [CrossRef]
- Wang, H.; Cheng, X.; Tian, J.; Xiao, Y.; Tian, T.; Xu, F.; Hong, X.; Zhu, M.X. TRPC channels: Structure, function, regulation and recent advances in small molecular probes. Pharmacol. Ther. 2020, 209, 107497. [Google Scholar] [CrossRef]
- Elzamzamy, O.M.; Penner, R.; Hazlehurst, L.A. The Role of TRPC1 in Modulating Cancer Progression. Cells 2020, 9, 388. [Google Scholar] [CrossRef] [Green Version]
- Faouzi, M.; Hague, F.; Geerts, D.; Ay, A.S.; Potier-Cartereau, M.; Ahidouch, A.; Ouadid-Ahidouch, H. Functional cooperation between KCa3.1 and TRPC1 channels in human breast cancer: Role in cell proliferation and patient prognosis. Oncotarget 2016, 7, 36419–36435. [Google Scholar] [CrossRef] [Green Version]
- Sun, Y.; Ye, C.; Tian, W.; Ye, W.; Gao, Y.Y.; Feng, Y.D.; Zhang, H.N.; Ma, G.Y.; Wang, S.J.; Cao, W.; et al. TRPC1 promotes the genesis and progression of colorectal cancer via activating CaM-mediated PI3K/AKT signaling axis. Oncogenesis 2021, 10, 67. [Google Scholar] [CrossRef]
- Zhang, Y.; Lun, X.; Guo, W. Expression of TRPC1 and SBEM protein in breast cancer tissue and its relationship with clinicopathological features and prognosis of patients. Oncol. Lett. 2020, 20, 392. [Google Scholar] [CrossRef]
- Wang, A.; Guo, H.; Long, Z. Integrative Analysis of Differently Expressed Genes Reveals a 17-Gene Prognosis Signature for Endometrial Carcinoma. Biomed. Res. Int. 2021, 2021, 4804694. [Google Scholar] [CrossRef]
- Mandavilli, S.; Singh, B.B.; Sahmoun, A.E. Serum calcium levels, TRPM7, TRPC1, microcalcifications, and breast cancer using breast imaging reporting and data system scores. Breast Cancer 2012, 2013, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Ibrahim, S.; Dakik, H.; Vandier, C.; Chautard, R.; Paintaud, G.; Mazurier, F.; Lecomte, T.; Gueguinou, M.; Raoul, W. Expression Profiling of Calcium Channels and Calcium-Activated Potassium Channels in Colorectal Cancer. Cancers 2019, 11, 561. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Zou, J.; Su, J.; Lu, Y.; Zhang, J.; Li, L.; Yin, F. Downregulation of transient receptor potential cation channel, subfamily C, member 1 contributes to drug resistance and high histological grade in ovarian cancer. Int. J. Oncol. 2016, 48, 243–252. [Google Scholar] [CrossRef] [Green Version]
- Chen, L.; Shan, G.; Ge, M.; Qian, H.; Xia, Y. Transient Receptor Potential Channel 1 Potentially Serves as a Biomarker Indicating T/TNM Stages and Predicting Long-Term Prognosis in Patients with Renal Cell Carcinoma. Front. Surg. 2022, 9, 853310. [Google Scholar] [CrossRef]
- Xu, Z.; Shao, Z.; Wang, M.; Thorndike, E.; Song, Y.; Shang, Z. Expression of transient receptor potential canonical 1 (TRPC1) in tongue squamous cell carcinoma and correlations with clinicopathological features and outcomes. Int. J. Clin. Exp. Pathol. 2017, 10, 1477–1487. [Google Scholar]
- Ke, C.; Long, S. Dysregulated transient receptor potential channel 1 expression and its correlation with clinical features and survival profile in surgical non-small-cell lung cancer patients. J. Clin. Lab. Anal. 2022, 36, e24229. [Google Scholar] [CrossRef]
- Bollimuntha, S.; Singh, B.B.; Shavali, S.; Sharma, S.K.; Ebadi, M. TRPC1-mediated inhibition of 1-methyl-4-phenylpyridinium ion neurotoxicity in human SH-SY5Y neuroblastoma cells. J. Biol. Chem. 2005, 280, 2132–2140. [Google Scholar] [CrossRef] [Green Version]
- Bomben, V.C.; Sontheimer, H. Disruption of transient receptor potential canonical channel 1 causes incomplete cytokinesis and slows the growth of human malignant gliomas. Glia 2010, 58, 1145–1156. [Google Scholar] [CrossRef] [Green Version]
- Asghar, M.Y.; Magnusson, M.; Kemppainen, K.; Sukumaran, P.; Lof, C.; Pulli, I.; Kalhori, V.; Tornquist, K. Transient Receptor Potential Canonical 1 (TRPC1) Channels as Regulators of Sphingolipid and VEGF Receptor Expression: Implications for Thyroid Cancer Cell Migration and Proliferation. J. Biol. Chem. 2015, 290, 16116–16131. [Google Scholar] [CrossRef] [Green Version]
- Kaemmerer, E.; Turner, D.; Peters, A.A.; Roberts-Thomson, S.J.; Monteith, G.R. An automated epifluorescence microscopy imaging assay for the identification of phospho-AKT level modulators in breast cancer cells. J. Pharmacol. Toxicol. Methods 2018, 92, 13–19. [Google Scholar] [CrossRef] [Green Version]
- El Hiani, Y.; Ahidouch, A.; Lehen’kyi, V.; Hague, F.; Gouilleux, F.; Mentaverri, R.; Kamel, S.; Lassoued, K.; Brule, G.; Ouadid-Ahidouch, H. Extracellular signal-regulated kinases 1 and 2 and TRPC1 channels are required for calcium-sensing receptor-stimulated MCF-7 breast cancer cell proliferation. Cell Physiol. Biochem. 2009, 23, 335–346. [Google Scholar] [CrossRef]
- El Hiani, Y.; Lehen’kyi, V.; Ouadid-Ahidouch, H.; Ahidouch, A. Activation of the calcium-sensing receptor by high calcium induced breast cancer cell proliferation and TRPC1 cation channel over-expression potentially through EGFR pathways. Arch. Biochem. Biophys. 2009, 486, 58–63. [Google Scholar] [CrossRef]
- Tajeddine, N.; Gailly, P. TRPC1 protein channel is major regulator of epidermal growth factor receptor signaling. J. Biol. Chem. 2012, 287, 16146–16157. [Google Scholar] [CrossRef] [Green Version]
- Selli, C.; Erac, Y.; Kosova, B.; Erdal, E.S.; Tosun, M. Silencing of TRPC1 regulates store-operated calcium entry and proliferation in Huh7 hepatocellular carcinoma cells. Biomed. Pharmacother. 2015, 71, 194–200. [Google Scholar] [CrossRef]
- Selli, C.; Pearce, D.A.; Sims, A.H.; Tosun, M. Differential expression of store-operated calcium- and proliferation-related genes in hepatocellular carcinoma cells following TRPC1 ion channel silencing. Mol. Cell Biochem. 2016, 420, 129–140. [Google Scholar] [CrossRef] [Green Version]
- Zeng, B.; Yuan, C.; Yang, X.; Atkin, S.L.; Xu, S.Z. TRPC channels and their splice variants are essential for promoting human ovarian cancer cell proliferation and tumorigenesis. Curr. Cancer Drug Targets 2013, 13, 103–116. [Google Scholar] [CrossRef]
- Zeng, Y.Z.; Zhang, Y.Q.; Chen, J.Y.; Zhang, L.Y.; Gao, W.L.; Lin, X.Q.; Huang, S.M.; Zhang, F.; Wei, X.L. TRPC1 Inhibits Cell Proliferation/Invasion and Is Predictive of a Better Prognosis of Esophageal Squamous Cell Carcinoma. Front. Oncol. 2021, 11, 627713. [Google Scholar] [CrossRef]
- Zhang, L.Y.; Zhang, Y.Q.; Zeng, Y.Z.; Zhu, J.L.; Chen, H.; Wei, X.L.; Liu, L.J. TRPC1 inhibits the proliferation and migration of estrogen receptor-positive Breast cancer and gives a better prognosis by inhibiting the PI3K/AKT pathway. Breast Cancer Res. Treat 2020, 182, 21–33. [Google Scholar] [CrossRef]
- Selli, C.; Erac, Y.; Tosun, M. Simultaneous measurement of cytosolic and mitochondrial calcium levels: Observations in TRPC1-silenced hepatocellular carcinoma cells. J. Pharmacol. Toxicol. Methods 2015, 72, 29–34. [Google Scholar] [CrossRef]
- Madsen, C.P.; Klausen, T.K.; Fabian, A.; Hansen, B.J.; Pedersen, S.F.; Hoffmann, E.K. On the role of TRPC1 in control of Ca2+ influx, cell volume, and cell cycle. Am. J. Physiol. Cell Physiol. 2012, 303, C625–C634. [Google Scholar] [CrossRef]
- Davis, F.M.; Peters, A.A.; Grice, D.M.; Cabot, P.J.; Parat, M.O.; Roberts-Thomson, S.J.; Monteith, G.R. Non-stimulated, agonist-stimulated and store-operated Ca2+ influx in MDA-MB-468 breast cancer cells and the effect of EGF-induced EMT on calcium entry. PLoS ONE 2012, 7, e36923. [Google Scholar] [CrossRef] [Green Version]
- El Boustany, C.; Bidaux, G.; Enfissi, A.; Delcourt, P.; Prevarskaya, N.; Capiod, T. Capacitative calcium entry and transient receptor potential canonical 6 expression control human hepatoma cell proliferation. Hepatology 2008, 47, 2068–2077. [Google Scholar] [CrossRef]
- Lepannetier, S.; Zanou, N.; Yerna, X.; Emeriau, N.; Dufour, I.; Masquelier, J.; Muccioli, G.; Tajeddine, N.; Gailly, P. Sphingosine-1-phosphate-activated TRPC1 channel controls chemotaxis of glioblastoma cells. Cell Calcium 2016, 60, 373–383. [Google Scholar] [CrossRef]
- Rychkov, G.; Barritt, G.J. TRPC1 Ca(2+)-permeable channels in animal cells. Handb. Exp. Pharmacol. 2007, 179, 23–52. [Google Scholar] [CrossRef]
- Hong, J.H.; Li, Q.; Kim, M.S.; Shin, D.M.; Feske, S.; Birnbaumer, L.; Cheng, K.T.; Ambudkar, I.S.; Muallem, S. Polarized but differential localization and recruitment of STIM1, Orai1 and TRPC channels in secretory cells. Traffic 2011, 12, 232–245. [Google Scholar] [CrossRef] [Green Version]
- Kim, M.H.; Seo, J.B.; Burnett, L.A.; Hille, B.; Koh, D.S. Characterization of store-operated Ca2+ channels in pancreatic duct epithelia. Cell Calcium 2013, 54, 266–275. [Google Scholar] [CrossRef] [Green Version]
- Dong, H.; Shim, K.N.; Li, J.M.; Estrema, C.; Ornelas, T.A.; Nguyen, F.; Liu, S.; Ramamoorthy, S.L.; Ho, S.; Carethers, J.M.; et al. Molecular mechanisms underlying Ca2+-mediated motility of human pancreatic duct cells. Am. J. Physiol. Cell Physiol. 2010, 299, C1493–C1503. [Google Scholar] [CrossRef] [Green Version]
- Ravi, M.; Paramesh, V.; Kaviya, S.R.; Anuradha, E.; Solomon, F.D. 3D cell culture systems: Advantages and applications. J. Cell Physiol. 2015, 230, 16–26. [Google Scholar] [CrossRef]
- Ambudkar, I.S.; de Souza, L.B.; Ong, H.L. TRPC1, Orai1, and STIM1 in SOCE: Friends in tight spaces. Cell Calcium 2017, 63, 33–39. [Google Scholar] [CrossRef] [Green Version]
- Sobradillo, D.; Hernandez-Morales, M.; Ubierna, D.; Moyer, M.P.; Nunez, L.; Villalobos, C. A reciprocal shift in transient receptor potential channel 1 (TRPC1) and stromal interaction molecule 2 (STIM2) contributes to Ca2+ remodeling and cancer hallmarks in colorectal carcinoma cells. J. Biol. Chem. 2014, 289, 28765–28782. [Google Scholar] [CrossRef] [Green Version]
- Saez-Rodriguez, J.; MacNamara, A.; Cook, S. Modeling Signaling Networks to Advance New Cancer Therapies. Annu. Rev. Biomed. Eng. 2015, 17, 143–163. [Google Scholar] [CrossRef]
- Azimi, I.; Milevskiy, M.J.G.; Kaemmerer, E.; Turner, D.; Yapa, K.; Brown, M.A.; Thompson, E.W.; Roberts-Thomson, S.J.; Monteith, G.R. TRPC1 is a differential regulator of hypoxia-mediated events and Akt signalling in PTEN-deficient breast cancer cells. J. Cell Sci. 2017, 130, 2292–2305. [Google Scholar] [CrossRef] [Green Version]
- Fabian, A.; Bertrand, J.; Lindemann, O.; Pap, T.; Schwab, A. Transient receptor potential canonical channel 1 impacts on mechanosignaling during cell migration. Pflug. Arch. 2012, 464, 623–630. [Google Scholar] [CrossRef]
- Fels, B.; Bulk, E.; Petho, Z.; Schwab, A. The Role of TRP Channels in the Metastatic Cascade. Pharmaceuticals 2018, 11, 48. [Google Scholar] [CrossRef] [Green Version]
- Park, W.; Chawla, A.; O’Reilly, E.M. Pancreatic Cancer: A Review. JAMA 2021, 326, 851–862. [Google Scholar] [CrossRef]
- Perrouin-Verbe, M.A.; Schoentgen, N.; Talagas, M.; Garlantezec, R.; Uguen, A.; Doucet, L.; Rosec, S.; Marcorelles, P.; Potier-Cartereau, M.; Vandier, C.; et al. Overexpression of certain transient receptor potential and Orai channels in prostate cancer is associated with decreased risk of systemic recurrence after radical prostatectomy. Prostate 2019, 79, 1793–1804. [Google Scholar] [CrossRef]
- Perrouin Verbe, M.A.; Bruyere, F.; Rozet, F.; Vandier, C.; Fromont, G. Expression of store-operated channel components in prostate cancer: The prognostic paradox. Hum. Pathol. 2016, 49, 77–82. [Google Scholar] [CrossRef]
- Bonelli, P.; Tuccillo, F.M.; Borrelli, A.; Schiattarella, A.; Buonaguro, F.M. CDK/CCN and CDKI alterations for cancer prognosis and therapeutic predictivity. Biomed. Res. Int. 2014, 2014, 361020. [Google Scholar] [CrossRef]
- De Luca, A.; Maiello, M.R.; D’Alessio, A.; Pergameno, M.; Normanno, N. The RAS/RAF/MEK/ERK and the PI3K/AKT signalling pathways: Role in cancer pathogenesis and implications for therapeutic approaches. Expert Opin. Ther. Targets 2012, 16 (Suppl. S2), S17–S27. [Google Scholar] [CrossRef]
- Roy, S.K.; Srivastava, R.K.; Shankar, S. Inhibition of PI3K/AKT and MAPK/ERK pathways causes activation of FOXO transcription factor, leading to cell cycle arrest and apoptosis in pancreatic cancer. J. Mol. Signal 2010, 5, 10. [Google Scholar] [CrossRef] [Green Version]
- Nussinov, R.; Wang, G.; Tsai, C.J.; Jang, H.; Lu, S.; Banerjee, A.; Zhang, J.; Gaponenko, V. Calmodulin and PI3K Signaling in KRAS Cancers. Trends Cancer 2017, 3, 214–224. [Google Scholar] [CrossRef] [Green Version]
- Zanou, N.; Schakman, O.; Louis, P.; Ruegg, U.T.; Dietrich, A.; Birnbaumer, L.; Gailly, P. Trpc1 ion channel modulates phosphatidylinositol 3-kinase/Akt pathway during myoblast differentiation and muscle regeneration. J. Biol. Chem. 2012, 287, 14524–14534. [Google Scholar] [CrossRef] [Green Version]
- Chaudhuri, P.; Rosenbaum, M.A.; Sinharoy, P.; Damron, D.S.; Birnbaumer, L.; Graham, L.M. Membrane translocation of TRPC6 channels and endothelial migration are regulated by calmodulin and PI3 kinase activation. Proc. Natl. Acad. Sci. USA 2016, 113, 2110–2115. [Google Scholar] [CrossRef] [Green Version]
- Jonckheere, N.; Van Seuningen, I. Integrative analysis of the cancer genome atlas and cancer cell lines encyclopedia large-scale genomic databases: MUC4/MUC16/MUC20 signature is associated with poor survival in human carcinomas. J. Transl. Med. 2018, 16, 259. [Google Scholar] [CrossRef]
- Radoslavova, S.; Folcher, A.; Lefebvre, T.; Kondratska, K.; Guenin, S.; Dhennin-Duthille, I.; Gautier, M.; Prevarskaya, N.; Ouadid-Ahidouch, H. Orai1 Channel Regulates Human-Activated Pancreatic Stellate Cell Proliferation and TGFbeta1 Secretion through the AKT Signaling Pathway. Cancers 2021, 13, 2395. [Google Scholar] [CrossRef]
- Chamlali, M.; Kouba, S.; Rodat-Despoix, L.; Todesca, L.M.; Petho, Z.; Schwab, A.; Ouadid-Ahidouch, H. Orai3 Calcium Channel Regulates Breast Cancer Cell Migration through Calcium-Dependent and -Independent Mechanisms. Cells 2021, 10, 3487. [Google Scholar] [CrossRef]
- Pfaffl, M.W.; Horgan, G.W.; Dempfle, L. Relative expression software tool (REST) for group-wise comparison and statistical analysis of relative expression results in real-time PCR. Nucleic Acids Res. 2002, 30, e36. [Google Scholar] [CrossRef]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Schnipper, J.; Kouba, S.; Hague, F.; Girault, A.; Rybarczyk, P.; Telliez, M.-S.; Guénin, S.; Tebbakha, R.; Sevestre, H.; Ahidouch, A.; et al. The TRPC1 Channel Forms a PI3K/CaM Complex and Regulates Pancreatic Ductal Adenocarcinoma Cell Proliferation in a Ca2+-Independent Manner. Int. J. Mol. Sci. 2022, 23, 7923. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23147923
Schnipper J, Kouba S, Hague F, Girault A, Rybarczyk P, Telliez M-S, Guénin S, Tebbakha R, Sevestre H, Ahidouch A, et al. The TRPC1 Channel Forms a PI3K/CaM Complex and Regulates Pancreatic Ductal Adenocarcinoma Cell Proliferation in a Ca2+-Independent Manner. International Journal of Molecular Sciences. 2022; 23(14):7923. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23147923
Chicago/Turabian StyleSchnipper, Julie, Sana Kouba, Frédéric Hague, Alban Girault, Pierre Rybarczyk, Marie-Sophie Telliez, Stéphanie Guénin, Riad Tebbakha, Henri Sevestre, Ahmed Ahidouch, and et al. 2022. "The TRPC1 Channel Forms a PI3K/CaM Complex and Regulates Pancreatic Ductal Adenocarcinoma Cell Proliferation in a Ca2+-Independent Manner" International Journal of Molecular Sciences 23, no. 14: 7923. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23147923