
Does G Protein-Coupled Estrogen Receptor 1 Contribute to Cisplatin-Induced Acute Kidney Injury in Male Mice?

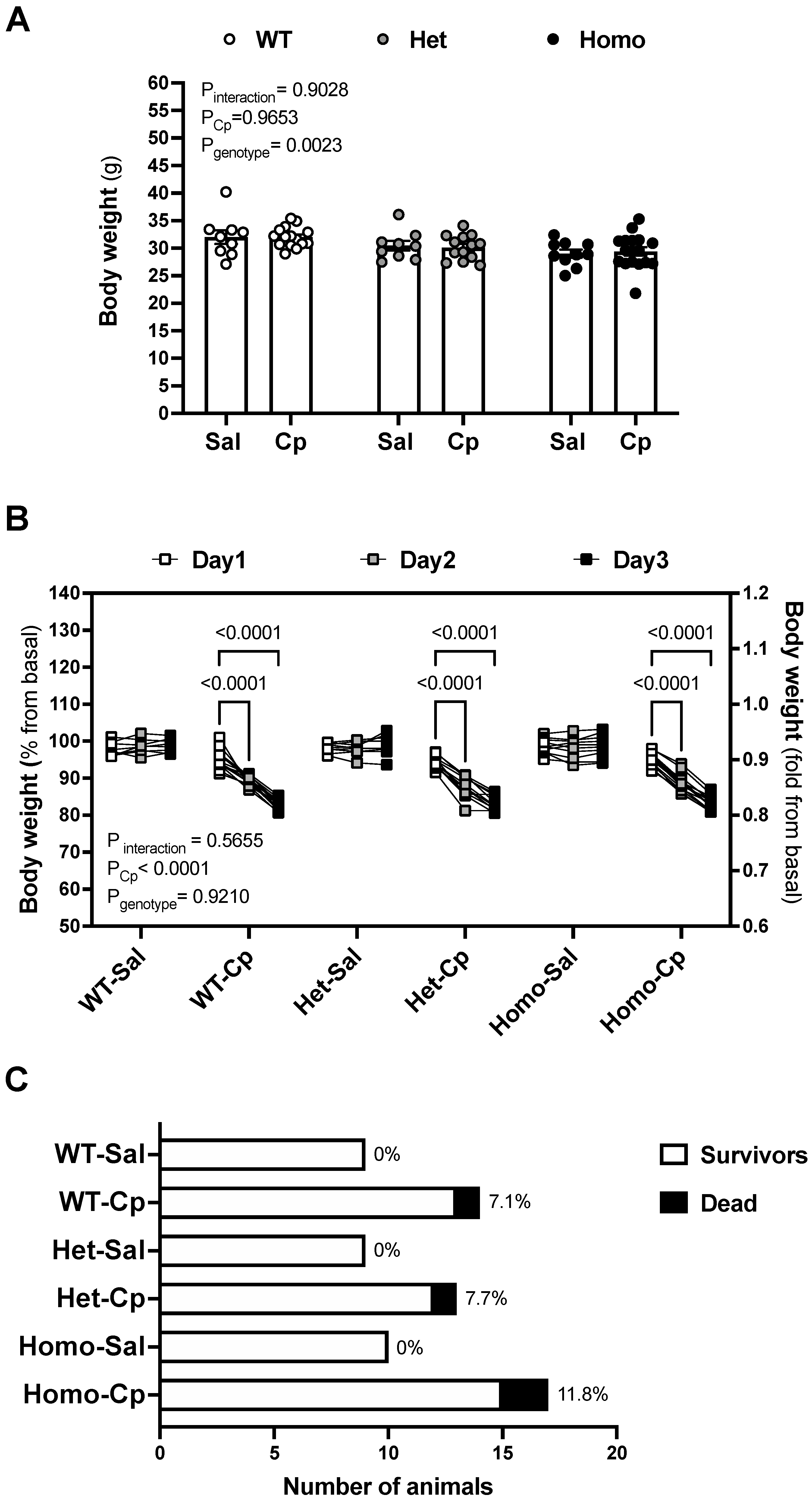{kind=link}
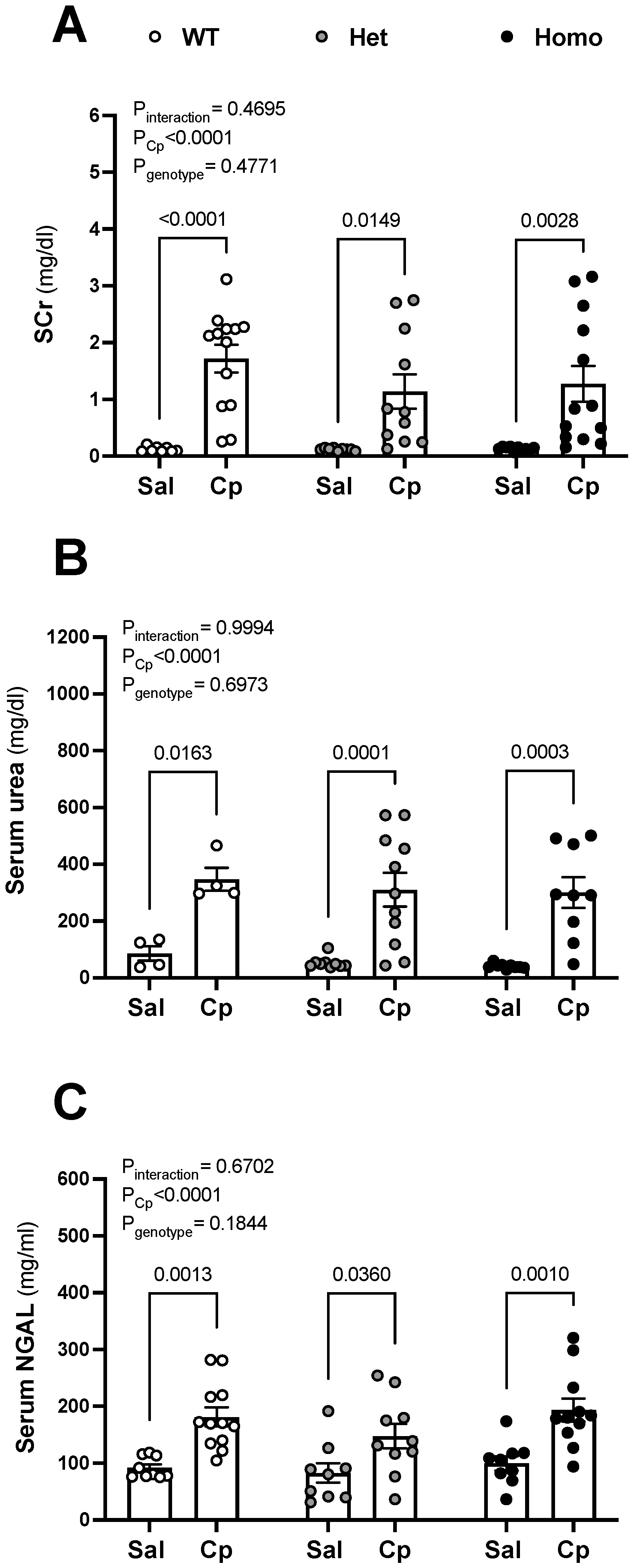{kind=link}
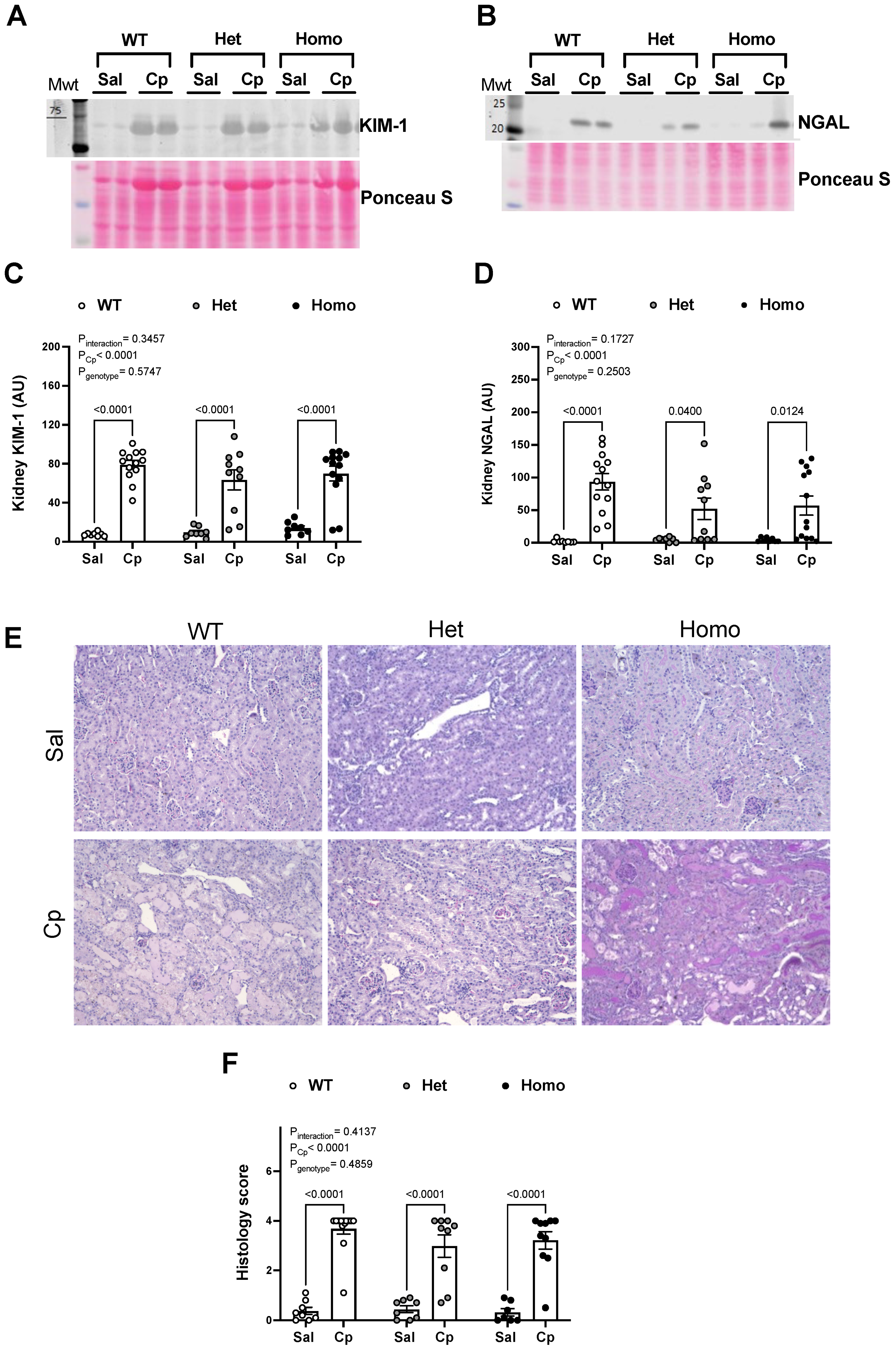{kind=link}
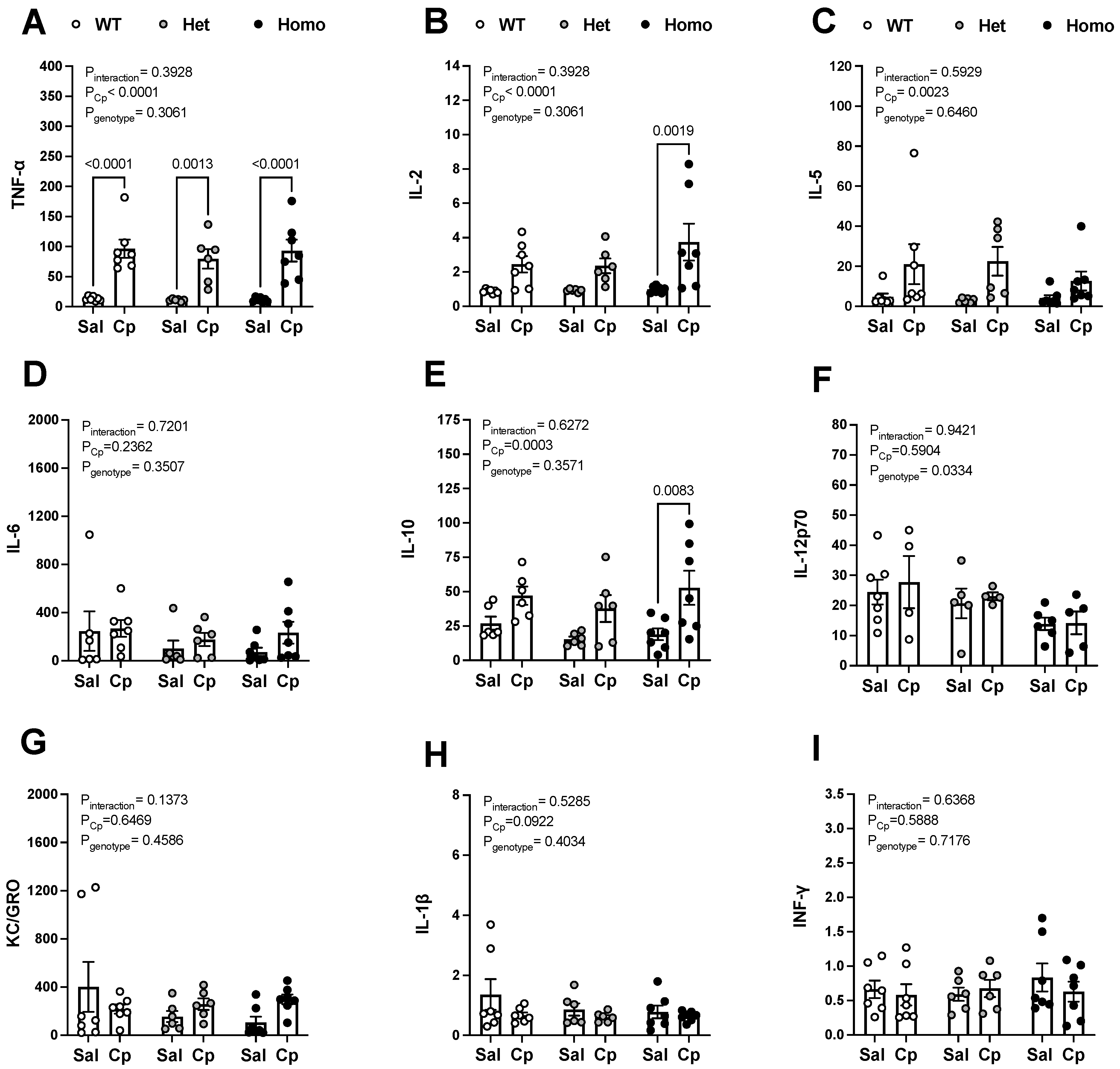{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
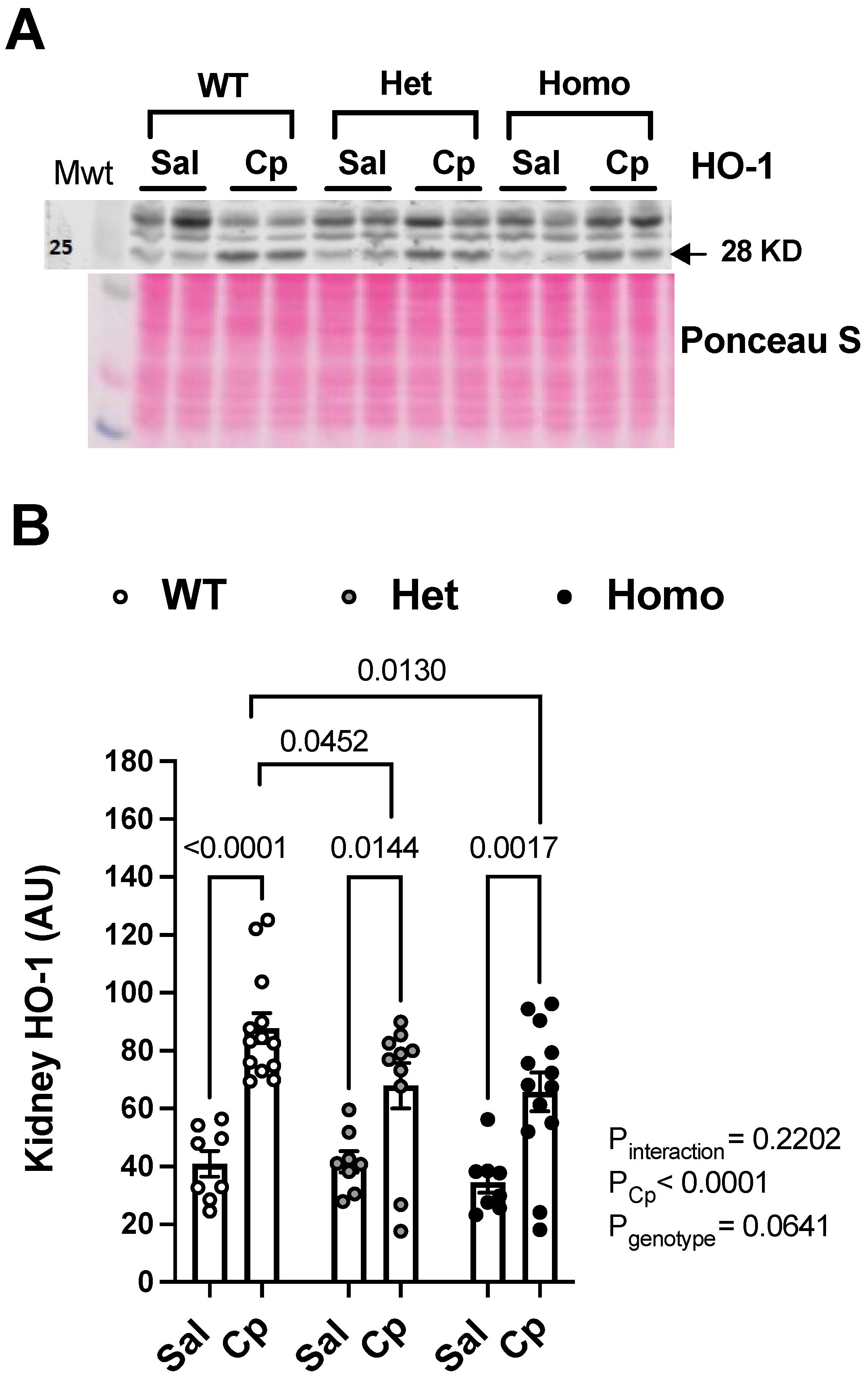
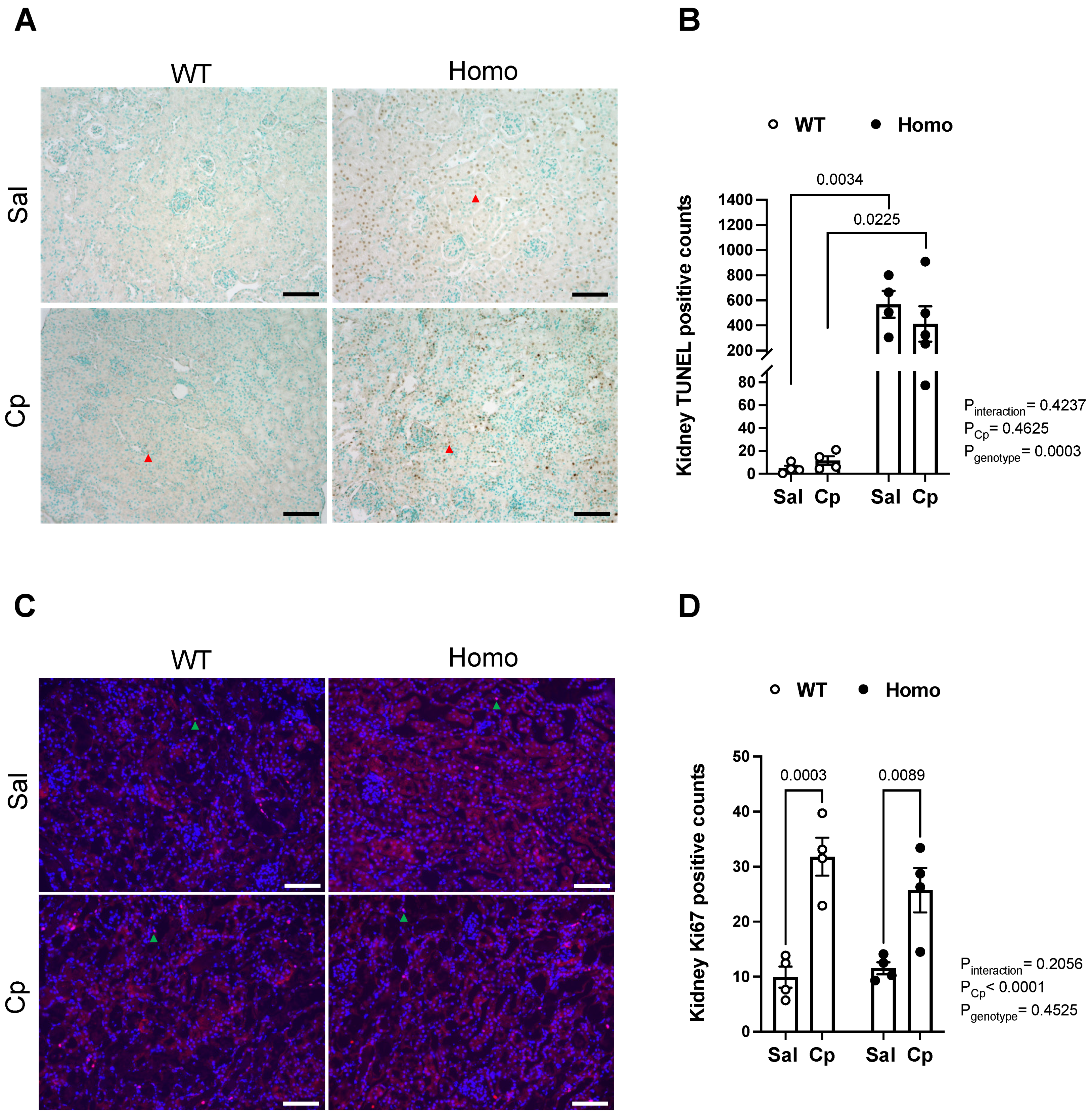
2. Results
3. Discussion
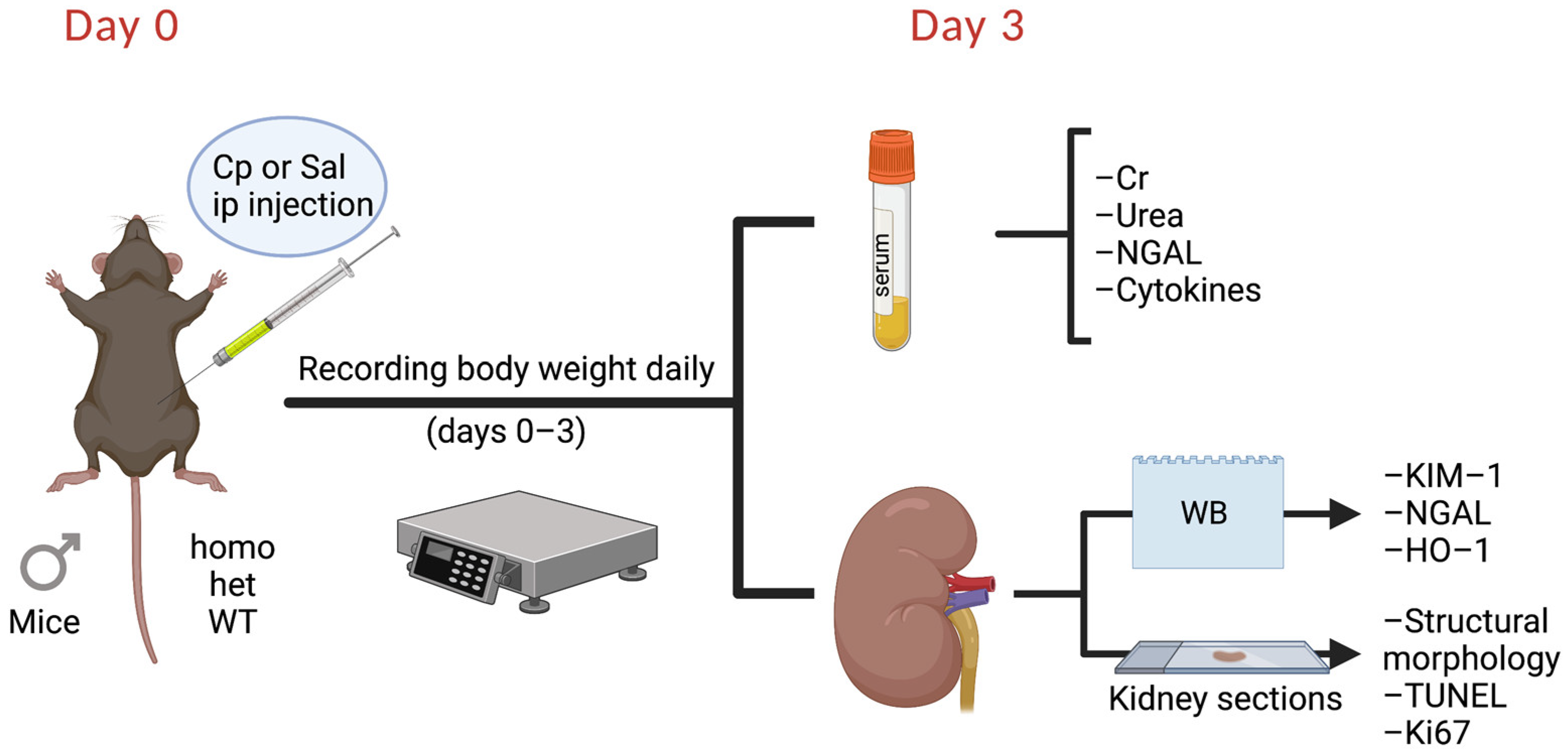
4. Materials and Methods
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Footnote
Abbreviations
| AKI | acute kidney injury |
| Cp | cisplatin |
| GPER1 | G protein-coupled estrogen receptor 1 |
| IL | interleukin |
| INF-γ | interferon-γ |
| HO-1 | heme oxygenase-1 |
| het | heterozygous knockout for GPER1 gene |
| homo | homozygous knockout for GPER1 gene |
| KC/GRO | keratinocyte chemoattractant-growth-regulated oncogene |
| KIM-1 | kidney injury molecule-1 |
| NGAL | neutrophil gelatinase-associated lipocalin |
| SEM | standard error of the mean |
| Sal | saline |
| SCr | serum creatinine |
| TNF-α | tumor necrosis factor-α |
| TUNEL | Terminal deoxynucleotidyl transferase-mediated dUTP nick-end labeling |
| WT | wild-type mice |
References
- Lebwohl, D.; Canetta, R. Clinical development of platinum complexes in cancer therapy: An historical perspective and an update. Eur. J. Cancer 1998, 34, 1522–1534. [Google Scholar] [CrossRef]
- Safirstein, R.; Winston, J.; Goldstein, M.; Moel, D.; Dikman, S.; Guttenplan, J. Cisplatin nephrotoxicity. Am. J. Kidney Dis. 1986, 8, 356–367. [Google Scholar] [CrossRef]
- Ries, F.; Klastersky, J. Nephrotoxicity Induced by Cancer Chemotherapy with Special Emphasis on Cisplatin Toxicity. Am. J. Kidney Dis. 1986, 8, 368–379. [Google Scholar] [CrossRef]
- Ozkok, A.; Edelstein, C.L. Pathophysiology of cisplatin-induced acute kidney injury. BioMed Res. Int. 2014, 2014, 967826. [Google Scholar] [CrossRef] [PubMed]
- Peres, L.A.B.; Júnior, A.D.D.C. Acute nephrotoxicity of cisplatin: Molecular mechanisms. J. Bras. Nefrol. 2013, 35, 332–340. [Google Scholar] [CrossRef] [PubMed]
- McSweeney, K.; Gadanec, L.; Qaradakhi, T.; Ali, B.; Zulli, A.; Apostolopoulos, V. Mechanisms of Cisplatin-Induced Acute Kidney Injury: Pathological Mechanisms, Pharmacological Interventions, and Genetic Mitigations. Cancers 2021, 13, 1572. [Google Scholar] [CrossRef]
- Boddu, R.; Fan, C.; Rangarajan, S.; Sunil, R.; Bolisetty, S.; Curtis, L.M. Unique sex- and age-dependent effects in protective pathways in acute kidney injury. Am. J. Physiol. Physiol. 2017, 313, F740–F755. [Google Scholar] [CrossRef] [Green Version]
- Neugarten, J.; Golestaneh, L.; Kolhe, N.V. Sex differences in acute kidney injury requiring dialysis. BMC Nephrol. 2018, 19, 131. [Google Scholar] [CrossRef]
- Mahani, F.D.; Khaksari, M.; Raji-Amirhasani, A. Renoprotective effects of estrogen on acute kidney injury: The role of SIRT1. Int. Urol. Nephrol. 2021, 53, 2299–2310. [Google Scholar] [CrossRef]
- Feng, J.-Y.; Liu, K.-T.; Abraham, E.; Chen, C.-Y.; Tsai, P.-Y.; Chen, Y.-C.; Lee, Y.-C.; Yang, K.-Y. Serum Estradiol Levels Predict Survival and Acute Kidney Injury in Patients with Septic Shock-A Prospective Study. PLoS ONE 2014, 9, e97967. [Google Scholar] [CrossRef] [Green Version]
- Gohar, E.Y. G protein-coupled estrogen receptor 1 as a novel regulator of blood pressure. Am. J. Physiol. Physiol. 2020, 319, F612–F617. [Google Scholar] [CrossRef] [PubMed]
- Han, G.; Li, F.; Yu, X.; White, R.E. GPER: A novel target for non-genomic estrogen action in the cardiovascular system. Pharmacol. Res. 2013, 71, 53–60. [Google Scholar] [CrossRef] [PubMed]
- Prossnitz, E.R.; Barton, M. Estrogen biology: New insights into GPER function and clinical opportunities. Mol. Cell. Endocrinol. 2014, 389, 71–83. [Google Scholar] [CrossRef] [Green Version]
- Hutson, D.D.; Gurrala, R.; Ogola, B.O.; Zimmerman, M.A.; Mostany, R.; Satou, R.; Lindsey, S.H. Estrogen receptor profiles across tissues from male and female Rattus norvegicus. Biol. Sex Differ. 2019, 10, 4. [Google Scholar] [CrossRef] [Green Version]
- Cheng, S.-B.; Dong, J.; Pang, Y.; LaRocca, J.; Hixon, M.; Thomas, P.; Filardo, E.J. Anatomical location and redistribution of G protein-coupled estrogen receptor-1 during the estrus cycle in mouse kidney and specific binding to estrogens but not aldosterone. Mol. Cell. Endocrinol. 2013, 382, 950–959. [Google Scholar] [CrossRef] [PubMed]
- Lindsey, S.H.; Yamaleyeva, L.M.; Brosnihan, K.B.; Gallagher, P.E.; Chappell, M.C. Estrogen Receptor GPR30 Reduces Oxidative Stress and Proteinuria in the Salt-Sensitive Female mRen2.Lewis Rat. Hypertension 2011, 58, 665–671. [Google Scholar] [CrossRef] [Green Version]
- Gohar, E.Y.; Almutlaq, R.N.; Daugherty, E.M.; Butt, M.K.; Jin, C.; Pollock, J.S.; Pollock, D.M.; De Miguel, C. Activation of G protein-coupled estrogen receptor 1 ameliorates proximal tubular injury and proteinuria in Dahl salt-sensitive female rats. Am. J. Physiol. Integr. Comp. Physiol. 2021, 320, R297–R306. [Google Scholar] [CrossRef]
- Chang, Y.; Han, Z.; Zhang, Y.; Zhou, Y.; Feng, Z.; Chen, L.; Li, X.; Li, L.; Si, J.-Q. G protein-coupled estrogen receptor activation improves contractile and diastolic functions in rat renal interlobular artery to protect against renal ischemia reperfusion injury. Biomed. Pharmacother. 2019, 112, 108666. [Google Scholar] [CrossRef]
- Hess, A.R.; Cooke, P.S. Estrogen in the male: A historical perspective. Biol. Reprod. 2018, 99, 27–44. [Google Scholar] [CrossRef]
- Cooke, P.S.; Nanjappa, M.K.; Ko, C.; Prins, G.S.; Hess, R. Estrogens in Male Physiology. Physiol. Rev. 2017, 97, 995–1043. [Google Scholar] [CrossRef]
- Barut, O.; Seyithanoglu, M.; Kucukdurmaz, F.; Demir, B.T.; Olmez, C.; Dogan, N.T.; Resim, S. Relationship between the G protein-coupled oestrogen receptor and spermatogenesis, and its correlation with male infertility. Andrologia 2020, 52, e13779. [Google Scholar] [CrossRef] [PubMed]
- Debortoli, A.R.; Rouver, W.; Delgado, N.T.B.; Mengal, V.; Claudio, E.R.G.; Pernomian, L.; Bendhack, L.M.; Moysés, M.R.; Dos Santos, R.L. GPER modulates tone and coronary vascular reactivity in male and female rats. J. Mol. Endocrinol. 2017, 59, 171–180. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peixoto, P.; da Silva, J.F.; Aires, R.D.; Costa, E.D.; Lemos, V.S.; Bissoli, N.S.; dos Santos, R.L. Sex difference in GPER expression does not change vascular relaxation or reactive oxygen species generation in rat mesenteric resistance arteries. Life Sci. 2018, 211, 198–205. [Google Scholar] [CrossRef]
- Meyer, M.R.; Amann, K.; Field, A.S.; Hu, C.; Hathaway, H.J.; Kanagy, N.L.; Walker, M.K.; Barton, M.; Prossnitz, E.R. Deletion of G Protein–Coupled Estrogen Receptor Increases Endothelial Vasoconstriction. Hypertension 2012, 59, 507–512. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deschamps, A.; Murphy, E. Activation of a novel estrogen receptor, GPER, is cardioprotective in male and female rats. Am. J. Physiol. Circ. Physiol. 2009, 297, H1806–H1813. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Francesco, E.M.; Angelone, T.; Pasqua, T.; Pupo, M.; Cerra, M.C.; Maggiolini, M. GPER Mediates Cardiotropic Effects in Spontaneously Hypertensive Rat Hearts. PLoS ONE 2013, 8, e69322. [Google Scholar] [CrossRef] [Green Version]
- Alencar, A.K.; Montes, G.C.; Montagnoli, T.; Silva, A.M.; Martinez, S.T.; Fraga, A.G.; Wang, H.; Groban, L.; Sudo, R.T.; Zapata-Sudo, G. Activation of GPER ameliorates experimental pulmonary hypertension in male rats. Eur. J. Pharm. Sci. 2016, 97, 208–217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharma, G.; Hu, C.; Staquicini, D.I.; Brigman, J.L.; Liu, M.; Mauvais-Jarvis, F.; Pasqualini, R.; Arap, W.; Arterburn, J.B.; Hathaway, H.J.; et al. Preclinical efficacy of the GPER-selective agonist G-1 in mouse models of obesity and diabetes. Sci. Transl. Med. 2020, 12. [Google Scholar] [CrossRef] [PubMed]
- Sharma, G.; Hu, C.; Brigman, J.L.; Zhu, G.; Hathaway, H.J.; Prossnitz, E.R. GPER Deficiency in Male Mice Results in Insulin Resistance, Dyslipidemia, and a Proinflammatory State. Endocrinology 2013, 154, 4136–4145. [Google Scholar] [CrossRef] [Green Version]
- Bopassa, J.C.; Eghbali, M.; Toro, L.; Stefani, E. A novel estrogen receptor GPER inhibits mitochondria permeability transition pore opening and protects the heart against ischemia-reperfusion injury. Am. J. Physiol. Circ. Physiol. 2010, 298, H16–H23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Randeva, H.S.; Patel, V.H.; Chen, J.; Ramanjaneya, M.; Karteris, E.; Zachariades, E.; Thomas, P.; Been, M. G-Protein coupled estrogen receptor 1 expression in rat and human heart: Protective role during ischaemic stress. Int. J. Mol. Med. 2010, 26, 193–199. [Google Scholar] [CrossRef]
- Feng, Y.; Madungwe, N.; Junho, C.V.C.; Bopassa, J.C. Activation of G protein-coupled oestrogen receptor 1 at the onset of reperfusion protects the myocardium against ischemia/reperfusion injury by reducing mitochondrial dysfunction and mitophagy. J. Cereb. Blood Flow Metab. 2017, 174, 4329–4344. [Google Scholar] [CrossRef]
- Ibañez, A.M.; Arbeláez, L.F.G.; Pardo, A.C.; Mosca, S.; Lofeudo, J.M.; Rueda, J.O.V.; Aiello, E.A.; De Giusti, V.C. Chronic GPER activation prevents ischemia/reperfusion injury in ovariectomized rats. Biochim. Biophys. Acta (BBA)—Gen. Subj. 2021, 1866, 130060. [Google Scholar] [CrossRef]
- Si, J.-Q.; Li, L.; Han, Z.-W.; Chang, Y.-C.; Zhou, Y.; Zhang, H.; Chen, L. GPER agonist G1 suppresses neuronal apoptosis mediated by endoplasmic reticulum stress after cerebral ischemia/reperfusion injury. Neural Regen. Res. 2019, 14, 1221–1229. [Google Scholar] [CrossRef]
- Zhao, T.; Ding, Q.; Hu, J.; He, S.; Shi, F.; Ma, L. GPER expressed on microglia mediates the anti-inflammatory effect of estradiol in ischemic stroke. Brain Behav. 2016, 6, e00449. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Z.; Chen, L.; Chu, H.; Wang, W.; Yang, L. Estrogen alleviates hepatocyte necroptosis depending on GPER in hepatic ischemia reperfusion injury. J. Physiol. Biochem. 2021, 78, 125–137. [Google Scholar] [CrossRef] [PubMed]
- Chai, S.; Liu, K.; Feng, W.; Liu, T.; Wang, Q.; Zhou, R.; Chen, S.; Wang, L.; Chen, G.; Ming, T.; et al. Activation of G protein–coupled estrogen receptor protects intestine from ischemia/reperfusion injury in mice by protecting the crypt cell proliferation. Clin. Sci. 2019, 133, 449–464. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Guo, Y.; Wu, H.-J.; Zhang, Z.-G.; Guo, M.-Y. Apoptosis inducing factor mediates cisplatin-induced apoptosis of renal tubular epithelial cells. Chin. J. Oncol. 2010, 32, 173–178. [Google Scholar]
- Ma, S.K.; Joo, S.Y.; Choi, H.-I.; Bae, E.H.; Nam, K.I.; Lee, J.; Kim, S.W. Activation of G-protein-coupled receptor 40 attenuates the cisplatin-induced apoptosis of human renal proximal tubule epithelial cells. Int. J. Mol. Med. 2014, 34, 1117–1123. [Google Scholar] [CrossRef] [Green Version]
- Gohar, E.Y.; Daugherty, E.M.; Aceves, J.O.; Sedaka, R.; Obi, I.E.; Allan, J.M.; Soliman, R.H.; Jin, C.; De Miguel, C.; Lindsey, S.; et al. Evidence for G-Protein–Coupled Estrogen Receptor as a Pronatriuretic Factor. J. Am. Heart Assoc. 2020, 9, e015110. [Google Scholar] [CrossRef] [PubMed]
- Hutchens, M.P.; Kosaka, Y.; Zhang, W.; Fujiyoshi, T.; Murphy, S.; Alkayed, N.; Anderson, S. Estrogen-Mediated Renoprotection following Cardiac Arrest and Cardiopulmonary Resuscitation Is Robust to GPR30 Gene Deletion. PLoS ONE 2014, 9, e99910. [Google Scholar] [CrossRef] [PubMed]
- Silva Barbosa, A.C.; Zhou, D.; Xie, Y.; Choi, Y.-J.; Tung, H.-C.; Chen, X.; Xu, M.; Gibbs, R.B.; Poloyac, S.M.; Liu, S.; et al. Inhibition of Estrogen Sulfotransferase (SULT1E1/EST) Ameliorates Ischemic Acute Kidney Injury in Mice. J. Am. Soc. Nephrol. 2020, 31, 1496–1508. [Google Scholar] [CrossRef]
- Wu, C.-C.; Chang, C.-Y.; Chang, S.-T.; Chen, S.-H. 17β-Estradiol Accelerated Renal Tubule Regeneration in Male Rats After Ischemia/Reperfusion-Induced Acute Kidney Injury. Shock 2016, 46, 158–163. [Google Scholar] [CrossRef]
- Singh, A.P.; Singh, N.; Pathak, D.; Singh Bedi, P.M. Estradiol attenuates ischemia reperfusion-induced acute kidney injury through PPAR-gamma stimulated eNOS activation in rats. Mol. Cell. Biochem. 2019, 453, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Buléon, M.; Cuny, M.; Grellier, J.; Charles, P.-Y.; Belliere, J.; Casemayou, A.; Arnal, J.-F.; Schanstra, J.-P.; Tack, I. A single dose of estrogen during hemorrhagic shock protects against Kidney Injury whereas estrogen restoration in ovariectomized mice is ineffective. Sci. Rep. 2020, 10, 17240. [Google Scholar] [CrossRef] [PubMed]
- Kurt, A.H.; Bozkus, F.; Uremis, N.; Uremis, M.M. The protective role of G protein-coupled estrogen receptor 1 (GPER-1) on methotrexate-induced nephrotoxicity in human renal epithelium cells. Ren. Fail. 2016, 38, 686–692. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eiam-Ong, S.; Chaipipat, M.; Manotham, K. Aldosterone rapidly activates p-PKC delta and GPR30 but suppresses p-PKC epsilon protein levels in rat kidney. Endocr. Regul. 2019, 53, 154–164. [Google Scholar] [CrossRef] [Green Version]
- Gozzelino, R.; Jeney, V.; Soares, M.P. Mechanisms of Cell Protection by Heme Oxygenase-1. Annu. Rev. Pharmacol. Toxicol. 2010, 50, 323–354. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lever, J.M.; Boddu, R.; George, J.F.; Agarwal, A. Heme Oxygenase-1 in Kidney Health and Disease. Antioxid. Redox Signal. 2016, 25, 165–183. [Google Scholar] [CrossRef] [Green Version]
- Maines, M.D. The Heme Oxygenase System: A Regulator of Second Messenger Gases. Annu. Rev. Pharmacol. Toxicol. 1997, 37, 517–554. [Google Scholar] [CrossRef]
- Stocker, R. Induction of Haem Oxygenase as a Defence against Oxidative Stress. Free. Radic. Res. Commun. 1990, 9, 101–112. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, H.; Takahashi, T.; Suzuki, T.; Yamasaki, A.; Fujiwara, T.; Odaka, Y.; Hirakawa, M.; Fujita, H.; Akagi, R. Protective effect of heme oxygenase induction in ischemic acute renal failure. Crit. Care Med. 2000, 28, 809–817. [Google Scholar] [CrossRef] [PubMed]
- Blydt-Hansen, T.D.; Katori, M.; Lassman, C.; Ke, B.; Coito, A.J.; Iyer, S.; Buelow, R.; Ettenger, R.; Busuttil, R.W.; Kupiec-Weglinski, J.W. Gene transfer-induced local heme oxygenase-1 overexpression protects rat kidney transplants from ischemia/reperfusion injury. J. Am. Soc. Nephrol. 2003, 14, 745–754. [Google Scholar] [CrossRef] [Green Version]
- Bolisetty, S.; Traylor, A.M.; Kim, J.; Joseph, R.; Ricart, K.; Landar, A.; Agarwal, A. Heme Oxygenase-1 Inhibits Renal Tubular Macroautophagy in Acute Kidney Injury. J. Am. Soc. Nephrol. 2010, 21, 1702–1712. [Google Scholar] [CrossRef] [Green Version]
- Shiraishi, F.; Curtis, L.M.; Truong, L.; Poss, K.; Visner, G.A.; Madsen, K.; Nick, H.S.; Agarwal, A. Heme oxygenase-1 gene ablation or expression modulates cisplatin-induced renal tubular apoptosis. Am. J. Physiol. Physiol. 2000, 278, F726–F736. [Google Scholar] [CrossRef] [Green Version]
- Tracz, M.J.; Juncos, J.P.; Grande, J.P.; Croatt, A.J.; Ackerman, A.W.; Rajagopalan, G.; Knutson, K.L.; Badley, A.D.; Griffin, M.D.; Alam, J.; et al. Renal hemodynamic, inflammatory, and apoptotic responses to lipopolysaccharide in HO-1−/− mice. Am. J. Pathol. 2007, 170, 1820–1830. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pósa, A.; Szabó, R.; Csonka, A.; Veszelka, M.; Berkó, A.M.; Baráth, Z.; Ménesi, R.; Pávó, I.; Gyongyosi, M.; László, F.; et al. Endogenous Estrogen-Mediated Heme Oxygenase Regulation in Experimental Menopause. Oxid. Med. Cell. Longev. 2015, 2015, 429713. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.M.; Cheng, P.-Y.; Hong, S.-F.; Chen, S.-Y.; Lam, K.-K.; Sheu, J.-R.; Yen, M.-H. Oxidative stress induces vascular heme oxygenase-1 expression in ovariectomized rats. Free Radic. Biol. Med. 2005, 39, 108–117. [Google Scholar] [CrossRef]
- Tian, J.; Zheng, Y.; Zhang, J.; Yang, C.; Wang, J.; Zhang, Z. Expression of heme oxygenase-1 in rat prostate and effects of androgen and estrogen on it. Sichuan Da Xue Xue Bao. Yi Xue Ban = J. Sichuan Univ. Med Sci. Ed. 2003, 34, 234–237. [Google Scholar]
- Baruscotti, I.; Barchiesi, F.; Jackson, E.K.; Imthurn, B.; Stiller, R.; Kim, J.-H.; Schaufelberger, S.; Rosselli, M.; Hughes, C.C.W.; Dubey, K. Estradiol stimulates capillary formation by human endothelial progenitor cells: Role of estrogen receptor-α/β, heme oxygenase 1, and tyrosine kinase. Hypertension 2010, 56, 397–404. [Google Scholar] [CrossRef] [PubMed]
- Sul, O.; Hyun, H.; Rajasekaran, M.; Suh, J.; Choi, H. Estrogen enhances browning in adipose tissue by M2 macrophage polarization via heme oxygenase-1. J. Cell. Physiol. 2020, 236, 1875–1888. [Google Scholar] [CrossRef]
- Barta, T.; Tosaki, A.; Haines, D.; Balla, G.; Lekli, I.; Tosaki, A. Endothelin-1-induced hypertrophic alterations and heme oxygenase-1 expression in cardiomyoblasts are counteracted by beta estradiol: In vitro and in vivo studies. Naunyn-Schmiedebergs Arch. Pharmacol. 2018, 391, 371–383. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hsu, J.-T.; Yeh, H.-C.; Chen, T.-H.; Kuo, C.-J.; Lin, C.-J.; Chiang, K.-C.; Yeh, T.-S.; Hwang, T.-L.; Chaudry, I.I. Role of Akt/HO-1 pathway in estrogen-mediated attenuation of trauma-hemorrhage-induced lung injury. J. Surg. Res. 2013, 182, 319–325. [Google Scholar] [CrossRef] [PubMed]
- Nath, M.; Agarwal, A. New insights into the role of heme oxygenase-1 in acute kidney injury. Kidney Res. Clin. Pract. 2020, 39, 387–401. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Sun, X.; Lin, M.S.; Ferrario, C.M.; Van Remmen, H.; Groban, L. G protein-coupled estrogen receptor (GPER) deficiency induces cardiac remodeling through oxidative stress. Transl. Res. 2018, 199, 39–51. [Google Scholar] [CrossRef]
- De Francesco, E.M.; Rocca, C.; Scavello, F.; Amelio, D.; Pasqua, T.; Rigiracciolo, D.C.; Scarpelli, A.; Avino, S.; Cirillo, F.; Amodio, N.; et al. Protective Role of GPER Agonist G-1 on Cardiotoxicity Induced by Doxorubicin. J. Cell. Physiol. 2017, 232, 1640–1649. [Google Scholar] [CrossRef]
- Liu, S.; Le May, C.; Wong, W.P.; Ward, R.D.; Clegg, D.J.; Marcelli, M.; Korach, K.S.; Mauvais-Jarvis, F. Importance of Extranuclear Estrogen Receptor-α and Membrane G Protein–Coupled Estrogen Receptor in Pancreatic Islet Survival. Diabetes 2009, 58, 2292–2302. [Google Scholar] [CrossRef] [Green Version]
- Grott, M.; Karakaya, S.; Mayer, F.; Baertling, F.; Beyer, C.; Kipp, M.; Kopp, H.-G. Progesterone and estrogen prevent cisplatin-induced apoptosis of lung cancer cells. Anticancer Res. 2013, 33, 791–800. [Google Scholar]
- Wang, X.; Xu, Z.; Sun, J.; Lv, H.; Wang, Y.; Ni, Y.; Chen, S.; Hu, C.; Wang, L.; Chen, W.; et al. Cisplatin resistance in gastric cancer cells is involved with GPR30-mediated epithelial-mesenchymal transition. J. Cell. Mol. Med. 2020, 24, 3625–3633. [Google Scholar] [CrossRef]
- Zhang, F.; Peng, L.; Huang, Y.; Lin, X.; Zhou, L.; Chen, J. Chronic BDE-47 Exposure Aggravates Malignant Phenotypes and Chemoresistance by Activating ERK Through ERalpha and GPR30 in Endometrial Carcinoma. Front. Oncol. 2019, 9, 1079. [Google Scholar] [CrossRef]
- Cheng, L.; Poulsen, S.B.; Wu, Q.; Esteva-Font, C.; Olesen, E.T.B.; Peng, L.; Olde, B.; Leeb-Lundberg, L.M.F.; Pisitkun, T.; Rieg, T.; et al. Rapid Aldosterone-Mediated Signaling in the DCT Increases Activity of the Thiazide-Sensitive NaCl Cotransporter. J. Am. Soc. Nephrol. 2019, 30, 1454–1470. [Google Scholar] [CrossRef]
- Gros, R.; Ding, Q.; Liu, B.; Chorazyczewski, J.; Feldman, R.D. Aldosterone mediates its rapid effects in vascular endothelial cells through GPER activation. Am. J. Physiol. Physiol. 2013, 304, C532–C540. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferreira, N.S.; Cau, S.; Silva, M.; Manzato, C.P.; Mestriner, F.L.A.C.; Matsumoto, T.; Carneiro, F.; Tostes, R.C. Diabetes impairs the vascular effects of aldosterone mediated by G protein-coupled estrogen receptor activation. Front. Pharmacol. 2015, 6, 34. [Google Scholar] [CrossRef] [Green Version]
- Rigiracciolo, D.C.; Scarpelli, A.; Lappano, R.; Pisano, A.; Santolla, M.F.; Avino, S.; De Marco, P.; Bussolati, B.; Maggiolini, M.; De Francesco, E.M. GPER is involved in the stimulatory effects of aldosterone in breast cancer cells and breast tumor-derived endothelial cells. Oncotarget 2015, 7, 94–111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caroccia, B.; Seccia, T.M.; Piazza, M.; Prisco, S.; Zanin, S.; Iacobone, M.; Lenzini, L.; Pallafacchina, G.; Domening, O.; Poglitsch, M.; et al. Aldosterone Stimulates Its Biosynthesis Via a Novel GPER-Mediated Mechanism. J. Clin. Endocrinol. Metab. 2019, 104, 6316–6324. [Google Scholar] [CrossRef] [PubMed]
- Rossi, G.P.; Caroccia, B.; Seccia, T.M. Role of estrogen receptors in modulating aldosterone biosynthesis and blood pressure. Steroids 2019, 152, 108486. [Google Scholar] [CrossRef]
- Tomsa, A.M.; Alexa, A.L.; Junie, M.L.; Rachisan, A.L.; Ciumarnean, L. Oxidative stress as a potential target in acute kidney injury. PeerJ 2019, 7, e8046. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.J.; Yoon, Y.M.; Lee, J.H.; Lee, S.H. Protective Role of Fucoidan on Cisplatin-mediated ER Stress in Renal Proximal Tubule Epithelial Cells. Anticancer Res. 2019, 39, 5515–5524. [Google Scholar] [CrossRef]
- Mapuskar, K.A.; Steinbach, E.J.; Zaher, A.; Riley, D.P.; Beardsley, R.A.; Keene, J.L.; Holmlund, J.T.; Anderson, C.M.; Zepeda-Orozco, D.; Buatti, J.M.; et al. Mitochondrial Superoxide Dismutase in Cisplatin-Induced Kidney Injury. Antioxidants 2021, 10, 1329. [Google Scholar] [CrossRef]
- Soni, H.; Kaminski, D.; Gangaraju, R.; Adebiyi, A. Cisplatin-induced oxidative stress stimulates renal Fas ligand shedding. Ren. Fail. 2018, 40, 314–322. [Google Scholar] [CrossRef] [Green Version]
- Li, W.-L.; Xiang, W.; Ping, Y. Activation of novel estrogen receptor GPER results in inhibition of cardiocyte apoptosis and cardioprotection. Mol. Med. Rep. 2015, 12, 2425–2430. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meyer, M.R.; Rosemann, T.; Barton, M.; Prossnitz, E.R. GPER Mediates Functional Endothelial Aging in Renal Arteries. Pharmacology 2017, 100, 188–193. [Google Scholar] [CrossRef] [PubMed]
- Huang, M.; Huang, M.; Li, X.; Liu, S.; Fu, L.; Jiang, X.; Yang, M. Bisphenol A induces apoptosis through GPER-dependent activation of the ROS/Ca2+-ASK1-JNK pathway in human granulosa cell line KGN. Ecotoxicol. Environ. Saf. 2021, 208, 111429. [Google Scholar] [CrossRef] [PubMed]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gohar, E.Y.; Almutlaq, R.N.; Fan, C.; Balkawade, R.S.; Butt, M.K.; Curtis, L.M. Does G Protein-Coupled Estrogen Receptor 1 Contribute to Cisplatin-Induced Acute Kidney Injury in Male Mice? Int. J. Mol. Sci. 2022, 23, 8284. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23158284
Gohar EY, Almutlaq RN, Fan C, Balkawade RS, Butt MK, Curtis LM. Does G Protein-Coupled Estrogen Receptor 1 Contribute to Cisplatin-Induced Acute Kidney Injury in Male Mice? International Journal of Molecular Sciences. 2022; 23(15):8284. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23158284
Chicago/Turabian StyleGohar, Eman Y., Rawan N. Almutlaq, Chunlan Fan, Rohan S. Balkawade, Maryam K. Butt, and Lisa M. Curtis. 2022. "Does G Protein-Coupled Estrogen Receptor 1 Contribute to Cisplatin-Induced Acute Kidney Injury in Male Mice?" International Journal of Molecular Sciences 23, no. 15: 8284. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23158284