
Consequences of Acute or Chronic Methylphenidate Exposure Using Ex Vivo Neurochemistry and In Vivo Electrophysiology in the Prefrontal Cortex and Striatum of Rats †


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
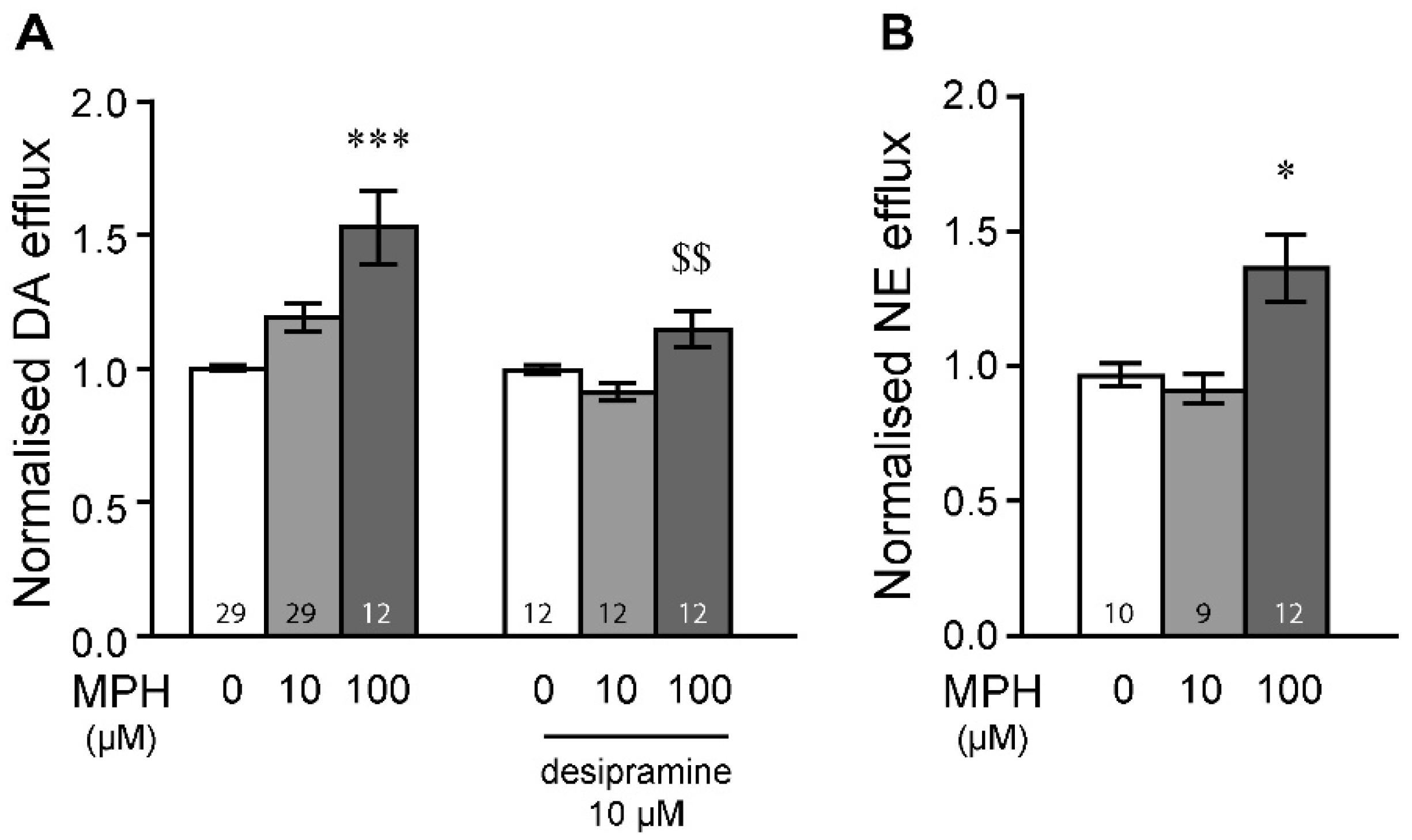
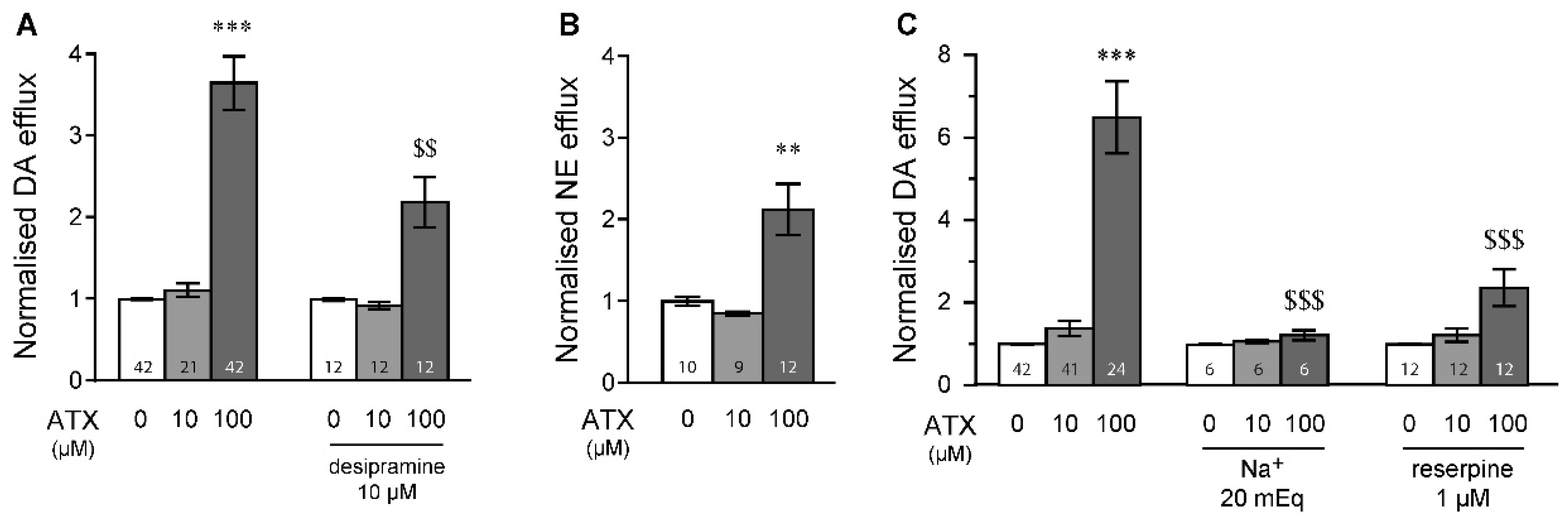
2.1. MPH Induces Dopamine and Norepinephrine Efflux in the Prefrontal Cortex
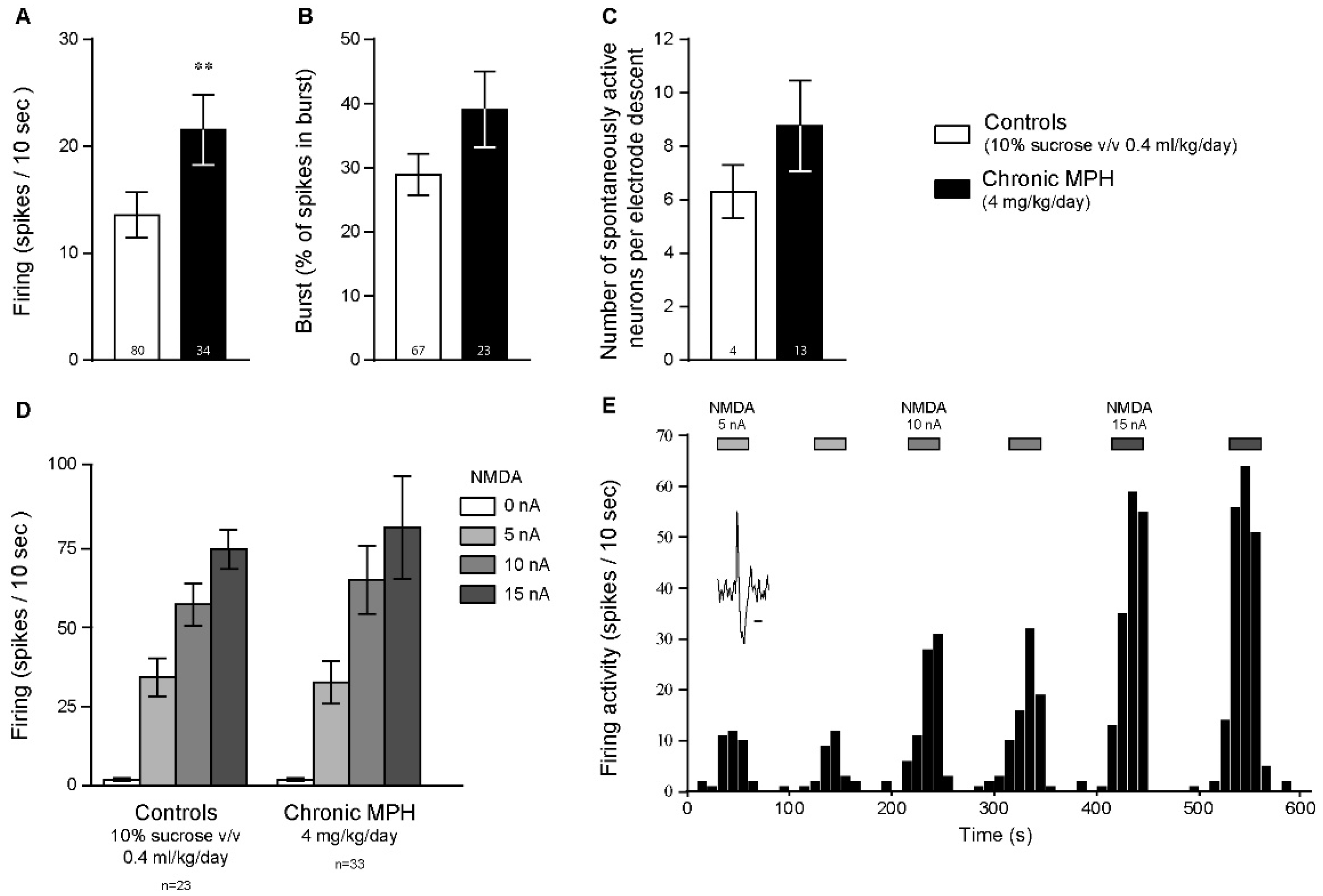
2.2. Chronic MPH Increases the Firing Activity of PFC Pyramidal Neurons without Altering Glutamatergic Neurotransmission
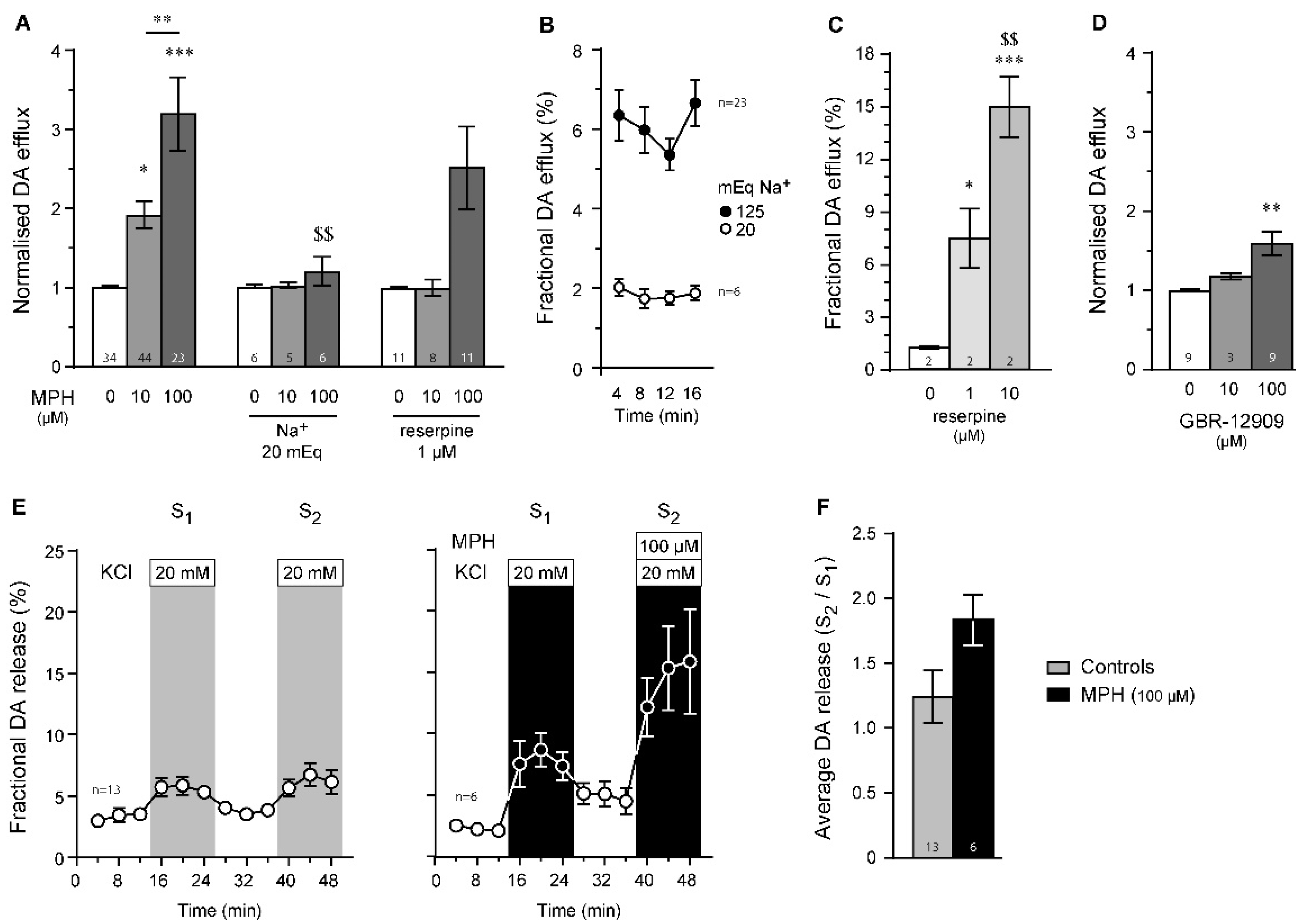
2.3. Dopamine Efflux, but Not Dopamine Release, Is Modulated by MPH in the Striatum
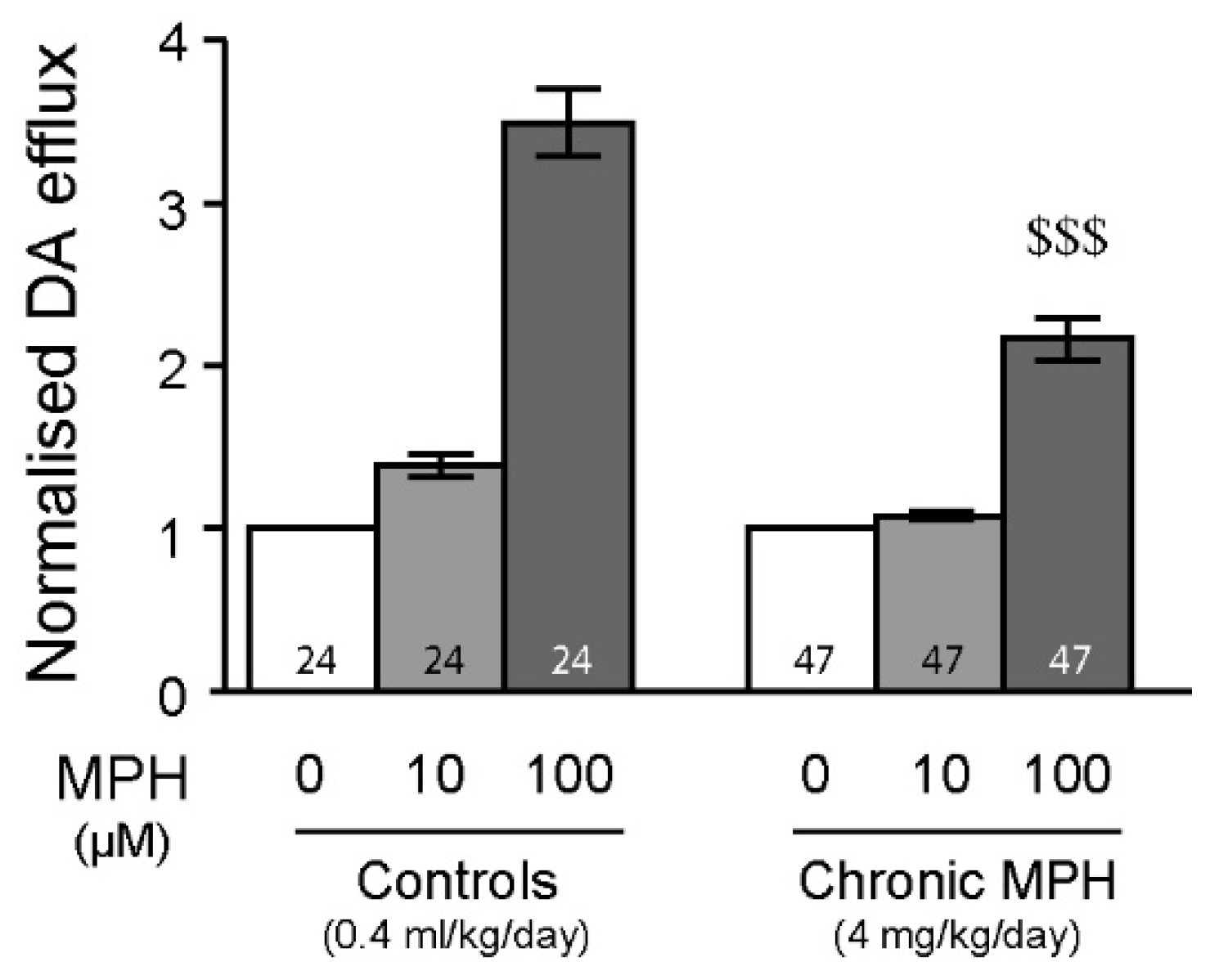
2.4. Tolerance to MPH and Dopamine in Chronically Treated Animals
3. Discussion
4. Materials and Methods
4.1. Animals and Drug Treatments
4.2. Ex Vivo Radiolabelled Neurotransmitter Efflux
4.3. In Vivo Extracellular Single-Unit Electrophysiology
4.4. Drugs and Reagents
4.5. Resampling
4.6. Data Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Swanson, J.; Baler, R.D.; Volkow, N.D. Understanding the Effects of Stimulant Medications on Cognition in Individuals with Attention-Deficit Hyperactivity Disorder: A Decade of Progress. Neuropsychopharmacology 2011, 36, 207–226. [Google Scholar] [CrossRef] [PubMed]
- De Sousa, A.; Kalra, G. Drug Therapy of Attention Deficit Hyperactivity Disorder: Current Trends. Mens Sana Monogr. 2012, 10, 45–69. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vitiello, B. Understanding the Risk of Using Medications for Attention Deficit Hyperactivity Disorder with Respect to Physical Growth and Cardiovascular Function. Child Adolesc. Psychiatr. Clin. N. Am. 2008, 17, 459–474. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heal, D.J.; Cheetham, S.C.; Smith, S.L. The Neuropharmacology of ADHD Drugs in Vivo: Insights on Efficacy and Safety. Neuropharmacology 2009, 57, 608–618. [Google Scholar] [CrossRef]
- Spiller, H.A.; Hays, H.L.; Aleguas, A. Overdose of Drugs for Attention-Deficit Hyperactivity Disorder: Clinical Presentation, Mechanisms of Toxicity, and Management. CNS Drugs 2013, 27, 531–543. [Google Scholar] [CrossRef] [PubMed]
- Kolar, D.; Keller, A.; Golfinopoulos, M.; Cumyn, L.; Syer, C.; Hechtman, L. Treatment of Adults with Attention-Deficit/Hyperactivity Disorder. Neuropsychiatr. Dis. Treat. 2008, 4, 389–403. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bushe, C.J.; Savill, N.C. Systematic Review of Atomoxetine Data in Childhood and Adolescent Attention-Deficit Hyperactivity Disorder 2009-2011: Focus on Clinical Efficacy and Safety. J. Psychopharmacol. 2014, 28, 204–211. [Google Scholar] [CrossRef] [PubMed]
- Bymaster, F.P.; Katner, J.S.; Nelson, D.L.; Hemrick-Luecke, S.K.; Threlkeld, P.G.; Heiligenstein, J.H.; Morin, S.M.; Gehlert, D.R.; Perry, K.W. Atomoxetine Increases Extracellular Levels of Norepinephrine and Dopamine in Prefrontal Cortex of Rat: A Potential Mechanism for Efficacy in Attention Deficit/Hyperactivity Disorder. Neuropsychopharmacology 2002, 27, 699–711. [Google Scholar] [CrossRef]
- Reith, M.E.A.; Wang, L.C.; Dutta, A.K. Pharmacological Profile of Radioligand Binding to the Norepinephrine Transporter: Instances of Poor Indication of Functional Activity. J. Neurosci. Methods 2005, 143, 87–94. [Google Scholar] [CrossRef] [PubMed]
- Erickson, J.D.; Schafer, M.K.; Bonner, T.I.; Eiden, L.E.; Weihe, E. Distinct Pharmacological Properties and Distribution in Neurons and Endocrine Cells of Two Isoforms of the Human Vesicular Monoamine Transporter. Proc. Natl. Acad. Sci. USA 1996, 93, 5166–5171. [Google Scholar] [CrossRef] [Green Version]
- Kalivas, P.W. Cocaine and Amphetamine-like Psychostimulants: Neurocircuitry and Glutamate Neuroplasticity. Dialogues Clin. Neurosci. 2007, 9, 389–397. [Google Scholar] [CrossRef] [PubMed]
- Dela Peña, I.; Gevorkiana, R.; Shi, W.-X. Psychostimulants Affect Dopamine Transmission through Both Dopamine Transporter-Dependent and Independent Mechanisms. Eur. J. Pharmacol. 2015, 764, 562–570. [Google Scholar] [CrossRef] [Green Version]
- Gamo, N.J.; Wang, M.; Arnsten, A.F.T. Methylphenidate and Atomoxetine Enhance Prefrontal Function through A2-Adrenergic and Dopamine D1 Receptors. J. Am. Acad. Child Adolesc. Psychiatry 2010, 49, 1011–1023. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kieling, C.; Goncalves, R.R.F.; Tannock, R.; Castellanos, F.X. Neurobiology of Attention Deficit Hyperactivity Disorder. Child Adolesc. Psychiatr. Clin. N. Am. 2008, 17, 285–307. [Google Scholar] [CrossRef] [PubMed]
- Koda, K.; Ago, Y.; Cong, Y.; Kita, Y.; Takuma, K.; Matsuda, T. Effects of Acute and Chronic Administration of Atomoxetine and Methylphenidate on Extracellular Levels of Noradrenaline, Dopamine and Serotonin in the Prefrontal Cortex and Striatum of Mice. J. Neurochem. 2010, 114, 259–270. [Google Scholar] [CrossRef] [PubMed]
- Floresco, S.B. Prefrontal Dopamine and Behavioral Flexibility: Shifting from an “Inverted-U” toward a Family of Functions. Front. Neurosci. 2013, 7, 62. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Devoto, P.; Flore, G. On the Origin of Cortical Dopamine: Is It a Co-Transmitter in Noradrenergic Neurons? Curr. Neuropharmacol. 2006, 4, 115–125. [Google Scholar] [CrossRef] [Green Version]
- Morón, J.A.; Brockington, A.; Wise, R.A.; Rocha, B.A.; Hope, B.T. Dopamine Uptake through the Norepinephrine Transporter in Brain Regions with Low Levels of the Dopamine Transporter: Evidence from Knock-out Mouse Lines. J. Neurosci. 2002, 22, 389–395. [Google Scholar] [CrossRef] [Green Version]
- Devoto, P.; Flore, G.; Pani, L.; Gessa, G.L. Evidence for Co-Release of Noradrenaline and Dopamine from Noradrenergic Neurons in the Cerebral Cortex. Mol. Psychiatry 2001, 6, 657–664. [Google Scholar] [CrossRef] [Green Version]
- Quansah, E.; Zetterström, T.S.C. Chronic Methylphenidate Preferentially Alters Catecholamine Protein Targets in the Parietal Cortex and Ventral Striatum. Neurochem. Int. 2019, 124, 193–199. [Google Scholar] [CrossRef] [PubMed]
- Quansah, E.; Sgamma, T.; Jaddoa, E.; Zetterström, T.S.C. Chronic Methylphenidate Regulates Genes and Proteins Mediating Neuroplasticity in the Juvenile Rat Brain. Neurosci. Lett. 2017, 654, 93–98. [Google Scholar] [CrossRef]
- Matsuda, W.; Furuta, T.; Nakamura, K.C.; Hioki, H.; Fujiyama, F.; Arai, R.; Kaneko, T. Single Nigrostriatal Dopaminergic Neurons Form Widely Spread and Highly Dense Axonal Arborizations in the Neostriatum. J. Neurosci. 2009, 29, 444–453. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lou, H.C.; Henriksen, L.; Bruhn, P.; Børner, H.; Nielsen, J.B. Striatal Dysfunction in Attention Deficit and Hyperkinetic Disorder. Arch. Neurol. 1989, 46, 48–52. [Google Scholar] [CrossRef] [PubMed]
- Fusar-Poli, P.; Rubia, K.; Rossi, G.; Sartori, G.; Balottin, U. Striatal Dopamine Transporter Alterations in ADHD: Pathophysiology or Adaptation to Psychostimulants? A Meta-Analysis. Am. J. Psychiatry 2012, 169, 264–272. [Google Scholar] [CrossRef]
- Krause, K.H.; Dresel, S.H.; Krause, J.; Kung, H.F.; Tatsch, K. Increased Striatal Dopamine Transporter in Adult Patients with Attention Deficit Hyperactivity Disorder: Effects of Methylphenidate as Measured by Single Photon Emission Computed Tomography. Neurosci. Lett. 2000, 285, 107–110. [Google Scholar] [CrossRef]
- Krause, J.; la Fougere, C.; Krause, K.-H.; Ackenheil, M.; Dresel, S.H. Influence of Striatal Dopamine Transporter Availability on the Response to Methylphenidate in Adult Patients with ADHD. Eur. Arch. Psychiatry Clin. Neurosci. 2005, 255, 428–431. [Google Scholar] [CrossRef]
- Mckenzie, A.; Meshkat, S.; Lui, L.M.W.; Ho, R.; Di Vincenzo, J.D.; Ceban, F.; Cao, B.; McIntyre, R.S. The Effects of Psychostimulants on Cognitive Functions in Individuals with Attention-Deficit Hyperactivity Disorder: A Systematic Review. J. Psychiatr. Res. 2022, 149, 252–259. [Google Scholar] [CrossRef]
- Hong, S.-B.; Harrison, B.J.; Fornito, A.; Sohn, C.-H.; Song, I.-C.; Kim, J.-W. Functional Dysconnectivity of Corticostriatal Circuitry and Differential Response to Methylphenidate in Youth with Attention-Deficit/Hyperactivity Disorder. J. Psychiatry Neurosci. JPN 2015, 40, 46–57. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Braz, B.Y.; Galiñanes, G.L.; Taravini, I.R.E.; Belforte, J.E.; Murer, M.G. Altered Corticostriatal Connectivity and Exploration/Exploitation Imbalance Emerge as Intermediate Phenotypes for a Neonatal Dopamine Dysfunction. Neuropsychopharmacology 2015, 40, 2576–2587. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alberquilla, S.; Gonzalez-Granillo, A.; Martín, E.D.; Moratalla, R. Dopamine Regulates Spine Density in Striatal Projection Neurons in a Concentration-Dependent Manner. Neurobiol. Dis. 2020, 134, 104666. [Google Scholar] [CrossRef] [PubMed]
- Di Miceli, M.; Gronier, B. Psychostimulants and Atomoxetine Alter the Electrophysiological Activity of Prefrontal Cortex Neurons, Interaction with Catecholamine and Glutamate NMDA Receptors. Psychopharmacology 2015, 232, 2191–2205. [Google Scholar] [CrossRef] [PubMed]
- Di Miceli, M.; Omoloye, A.; Gronier, B. Chronic Methylphenidate Treatment during Adolescence Has Long-Term Effects on Monoaminergic Function. J. Psychopharmacol. 2019, 33, 109–121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cvetkovic-Lopes, V.; Eggermann, E.; Uschakov, A.; Grivel, J.; Bayer, L.; Jones, B.E.; Serafin, M.; Mühlethaler, M. Rat Hypocretin/Orexin Neurons Are Maintained in a Depolarized State by TRPC Channels. PLoS ONE 2010, 5, e15673. [Google Scholar] [CrossRef]
- Wheeler, D.D.; Edwards, A.M.; Chapman, B.M.; Ondo, J.G. A Model of the Sodium Dependence of Dopamine Uptake in Rat Striatal Synaptosomes. Neurochem. Res. 1993, 18, 927–936. [Google Scholar] [CrossRef] [PubMed]
- Khvotchev, M.; Lonart, G.; Südhof, T.C. Role of Calcium in Neurotransmitter Release Evoked by Alpha-Latrotoxin or Hypertonic Sucrose. Neuroscience 2000, 101, 793–802. [Google Scholar] [CrossRef]
- Thanos, P.K.; Robison, L.S.; Steier, J.; Hwang, Y.F.; Cooper, T.; Swanson, J.M.; Komatsu, D.E.; Hadjiargyrou, M.; Volkow, N.D. A Pharmacokinetic Model of Oral Methylphenidate in the Rat and Effects on Behavior. Pharmacol. Biochem. Behav. 2015, 131, 143–153. [Google Scholar] [CrossRef] [Green Version]
- Berridge, C.W.; Devilbiss, D.M.; Andrzejewski, M.E.; Arnsten, A.F.T.; Kelley, A.E.; Schmeichel, B.; Hamilton, C.; Spencer, R.C. Methylphenidate Preferentially Increases Catecholamine Neurotransmission within the Prefrontal Cortex at Low Doses That Enhance Cognitive Function. Biol. Psychiatry 2006, 60, 1111–1120. [Google Scholar] [CrossRef]
- Wargin, W.; Patrick, K.; Kilts, C.; Gualtieri, C.T.; Ellington, K.; Mueller, R.A.; Kraemer, G.; Breese, G.R. Pharmacokinetics of Methylphenidate in Man, Rat and Monkey. J. Pharmacol. Exp. Ther. 1983, 226, 382–386. [Google Scholar]
- el Mansari, M.; Blier, P. In Vivo Electrophysiological Characterization of 5-HT Receptors in the Guinea Pig Head of Caudate Nucleus and Orbitofrontal Cortex. Neuropharmacology 1997, 36, 577–588. [Google Scholar] [CrossRef]
- Chuhma, N.; Tanaka, K.F.; Hen, R.; Rayport, S. Functional Connectome of the Striatal Medium Spiny Neuron. J. Neurosci. 2011, 31, 1183–1192. [Google Scholar] [CrossRef] [Green Version]
- Gertler, T.S.; Chan, C.S.; Surmeier, D.J. Dichotomous Anatomical Properties of Adult Striatal Medium Spiny Neurons. J. Neurosci. 2008, 28, 10814–10824. [Google Scholar] [CrossRef]
- Frederick, A.L.; Yano, H.; Trifilieff, P.; Vishwasrao, H.D.; Biezonski, D.; Mészáros, J.; Urizar, E.; Sibley, D.R.; Kellendonk, C.; Sonntag, K.C.; et al. Evidence against Dopamine D1/D2 Receptor Heteromers. Mol. Psychiatry 2015, 20, 1373–1385. [Google Scholar] [CrossRef] [Green Version]
- Soares-Cunha, C.; Coimbra, B.; Sousa, N.; Rodrigues, A.J. Reappraising Striatal D1- and D2-Neurons in Reward and Aversion. Neurosci. Biobehav. Rev. 2016, 68, 370–386. [Google Scholar] [CrossRef] [PubMed]
- Devoto, P.; Flore, G.; Pira, L.; Longu, G.; Gessa, G.L. Alpha2-Adrenoceptor Mediated Co-Release of Dopamine and Noradrenaline from Noradrenergic Neurons in the Cerebral Cortex. J. Neurochem. 2004, 88, 1003–1009. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Potter, W.P.; Partoens, P.; Schoups, A.; Llona, I.; Coen, E.P. Noradrenergic Neurons Release Both Noradrenaline and Neuropeptide Y from a Single Pool: The Large Dense Cored Vesicles. Synapse 1997, 25, 44–55. [Google Scholar] [CrossRef]
- Hendley, E.D.; Snyder, S.H.; Fauley, J.J.; LaPidus, J.B. Stereoselectivity of Catecholamine Uptake by Brain Synaptosomes: Studies with Ephedrine, Methylphenidate and Phenyl-2-Piperidyl Carbinol. J. Pharmacol. Exp. Ther. 1972, 183, 103–116. [Google Scholar] [PubMed]
- Han, D.D.; Gu, H.H. Comparison of the Monoamine Transporters from Human and Mouse in Their Sensitivities to Psychostimulant Drugs. BMC Pharmacol. 2006, 6, 6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gerasimov, M.R.; Franceschi, M.; Volkow, N.D.; Rice, O.; Schiffer, W.K.; Dewey, S.L. Synergistic Interactions between Nicotine and Cocaine or Methylphenidate Depend on the Dose of Dopamine Transporter Inhibitor. Synapse 2000, 38, 432–437. [Google Scholar] [CrossRef]
- Balcioglu, A.; Ren, J.-Q.; McCarthy, D.; Spencer, T.J.; Biederman, J.; Bhide, P.G. Plasma and Brain Concentrations of Oral Therapeutic Doses of Methylphenidate and Their Impact on Brain Monoamine Content in Mice. Neuropharmacology 2009, 57, 687–693. [Google Scholar] [CrossRef] [Green Version]
- Nomikos, G.G.; Damsma, G.; Wenkstern, D.; Fibiger, H.C. In Vivo Characterization of Locally Applied Dopamine Uptake Inhibitors by Striatal Microdialysis. Synapse 1990, 6, 106–112. [Google Scholar] [CrossRef]
- Schmeichel, B.E.; Berridge, C.W. Neurocircuitry Underlying the Preferential Sensitivity of Prefrontal Catecholamines to Low-Dose Psychostimulants. Neuropsychopharmacology 2013, 38, 1078–1084. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Swanson, L.W.; Hartman, B.K. The Central Adrenergic System. An Immunofluorescence Study of the Location of Cell Bodies and Their Efferent Connections in the Rat Utilizing Dopamine-Beta-Hydroxylase as a Marker. J. Comp. Neurol. 1975, 163, 467–505. [Google Scholar] [CrossRef] [PubMed]
- Berridge, C.W.; Stratford, T.L.; Foote, S.L.; Kelley, A.E. Distribution of Dopamine Beta-Hydroxylase-like Immunoreactive Fibers within the Shell Subregion of the Nucleus Accumbens. Synapse 1997, 27, 230–241. [Google Scholar] [CrossRef]
- Arai, A.; Tomiyama, M.; Kannari, K.; Kimura, T.; Suzuki, C.; Watanabe, M.; Kawarabayashi, T.; Shen, H.; Shoji, M. Reuptake of L-DOPA-Derived Extracellular DA in the Striatum of a Rodent Model of Parkinson’s Disease via Norepinephrine Transporter. Synapse 2008, 62, 632–635. [Google Scholar] [CrossRef]
- Chotibut, T.; Apple, D.M.; Jefferis, R.; Salvatore, M.F. Dopamine Transporter Loss in 6-OHDA Parkinson’s Model Is Unmet by Parallel Reduction in Dopamine Uptake. PLoS ONE 2012, 7, e52322. [Google Scholar] [CrossRef] [Green Version]
- Lauderdale, K.; Murphy, T.; Tung, T.; Davila, D.; Binder, D.K.; Fiacco, T.A. Osmotic Edema Rapidly Increases Neuronal Excitability through Activation of NMDA Receptor-Dependent Slow Inward Currents in Juvenile and Adult Hippocampus. ASN Neuro 2015, 7, 1759091415605115. [Google Scholar] [CrossRef]
- Roitman, M.F.; Patterson, T.A.; Sakai, R.R.; Bernstein, I.L.; Figlewicz, D.P. Sodium Depletion and Aldosterone Decrease Dopamine Transporter Activity in Nucleus Accumbens but Not Striatum. Am. J. Physiol. 1999, 276, R1339–R1345. [Google Scholar] [CrossRef]
- Claussen, C.M.; Chong, S.L.; Dafny, N. Nucleus Accumbens Neuronal Activity Correlates to the Animal’s Behavioral Response to Acute and Chronic Methylphenidate. Physiol. Behav. 2014, 129, 85–94. [Google Scholar] [CrossRef] [Green Version]
- Salek, R.L.; Claussen, C.M.; Pérez, A.; Dafny, N. Acute and Chronic Methylphenidate Alters Prefrontal Cortex Neuronal Activity Recorded from Freely Behaving Rats. Eur. J. Pharmacol. 2012, 679, 60–67. [Google Scholar] [CrossRef] [Green Version]
- Medina, A.C.; Reyes-Vasquez, C.; Kharas, N.; Dafny, N. Adolescent Rats Respond Differently to Methylphenidate as Compared to Adult Rats-Concomitant VTA Neuronal and Behavioral Recordings. Brain Res. Bull. 2022, 183, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Thanos, P.K.; Michaelides, M.; Benveniste, H.; Wang, G.J.; Volkow, N.D. Effects of Chronic Oral Methylphenidate on Cocaine Self-Administration and Striatal Dopamine D2 Receptors in Rodents. Pharmacol. Biochem. Behav. 2007, 87, 426–433. [Google Scholar] [CrossRef] [PubMed]
- Caprioli, D.; Jupp, B.; Hong, Y.T.; Sawiak, S.J.; Ferrari, V.; Wharton, L.; Williamson, D.J.; McNabb, C.; Berry, D.; Aigbirhio, F.I.; et al. Dissociable Rate-Dependent Effects of Oral Methylphenidate on Impulsivity and D2/3 Receptor Availability in the Striatum. J. Neurosci. 2015, 35, 3747–3755. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Richer, K.; Hamilton, J.; Delis, F.; Martin, C.; Fricke, D.; Yao, R.; Sajjad, M.; Blum, K.; Hadjiargyrou, M.; Komatsu, D.; et al. Chronic Treatment and Abstinence from Methylphenidate Exposure Dose-Dependently Changes Glucose Metabolism in the Rat Brain. Brain Res. 2022, 1780, 147799. [Google Scholar] [CrossRef] [PubMed]
- Foschiera, L.N.; Schmitz, F.; Wyse, A.T.S. Evidence of Methylphenidate Effect on Mitochondria, Redox Homeostasis, and Inflammatory Aspects: Insights from Animal Studies. Prog. Neuropsychopharmacol. Biol. Psychiatry 2022, 116, 110518. [Google Scholar] [CrossRef] [PubMed]
- Alam, N.; Ikram, R.; Naeem, S.; Khan, S.S.; Siddiqui, T.; Khatoon, H.; Kashif, S.S. Effect of Methylphenidate and Buspirone-Methylphenidate Co-Administration on Biochemical and Hematological Parameters in Rats: Implications for Safe and Confrontational Use. Pak. J. Pharm. Sci. 2021, 34, 2131–2139. [Google Scholar]
- Crowley, N.A.; Cody, P.A.; Davis, M.I.; Lovinger, D.M.; Mateo, Y. Chronic Methylphenidate Exposure during Adolescence Reduces Striatal Synaptic Responses to Ethanol. Eur. J. Neurosci. 2014, 39, 548–556. [Google Scholar] [CrossRef] [Green Version]
- Yang, P.B.; Swann, A.C.; Dafny, N. Chronic Administration of Methylphenidate Produces Neurophysiological and Behavioral Sensitization. Brain Res. 2007, 1145, 66–80. [Google Scholar] [CrossRef] [Green Version]
- Jones, Z.; Dafny, N. Acute and Chronic Dose Response Effect of Methylphenidate on Ventral Tegmental Area Neurons Correlated with Animal Behavior. J. Neural Transm. 2014, 121, 327–345. [Google Scholar] [CrossRef] [Green Version]
- Venkataraman, S.S.; Joseph, M.; Dafny, N. Concomitant Behavioral and Prefrontal Cortex Neuronal Responses Following Acute and Chronic Methylphenidate Exposure in Adolescent and Adult Rats. Brain Res. Bull. 2019, 144, 200–212. [Google Scholar] [CrossRef]
- King, N.; Floren, S.; Kharas, N.; Thomas, M.; Dafny, N. Glutaminergic Signaling in the Caudate Nucleus Is Required for Behavioral Sensitization to Methylphenidate. Pharmacol. Biochem. Behav. 2019, 184, 172737. [Google Scholar] [CrossRef]
- Venkataraman, S.S.; Claussen, C.M.; Kharas, N.; Dafny, N. The Prefrontal Cortex and the Caudate Nucleus Respond Conjointly to Methylphenidate (Ritalin). Concomitant Behavioral and Neuronal Recording Study. Brain Res. Bull. 2020, 157, 77–89. [Google Scholar] [CrossRef] [PubMed]
- Broussard, E.; Reyes-Vazquez, C.; Dafny, N. Methylphenidate Dose-Response Behavioral and Neurophysiological Study of the Ventral Tegmental Area and Nucleus Accumbens in Adolescent Rats. Eur. J. Neurosci. 2019, 50, 2635–2652. [Google Scholar] [CrossRef]
- Dos Santos Pereira, M.; Sathler, M.F.; Valli, T.; da, R.; Marques, R.S.; Ventura, A.L.M.; Peccinalli, N.R.; Fraga, M.C.; Manhães, A.C.; Kubrusly, R. Long Withdrawal of Methylphenidate Induces a Differential Response of the Dopaminergic System and Increases Sensitivity to Cocaine in the Prefrontal Cortex of Spontaneously Hypertensive Rats. PLoS ONE 2015, 10, e0141249. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Urban, K.R.; Waterhouse, B.D.; Gao, W.-J. Distinct Age-Dependent Effects of Methylphenidate on Developing and Adult Prefrontal Neurons. Biol. Psychiatry 2012, 72, 880–888. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zetterström, T.S.C.; Quansah, E.; Grootveld, M. Effects of Methylphenidate on the Dopamine Transporter and Beyond. In Current Topics in Behavioral Neurosciences; Springer: Berlin/Heidelberg, Germany, 2022. [Google Scholar] [CrossRef]
- Dipasquale, O.; Martins, D.; Sethi, A.; Veronese, M.; Hesse, S.; Rullmann, M.; Sabri, O.; Turkheimer, F.; Harrison, N.A.; Mehta, M.A.; et al. Unravelling the Effects of Methylphenidate on the Dopaminergic and Noradrenergic Functional Circuits. Neuropsychopharmacology 2020, 45, 1482–1489. [Google Scholar] [CrossRef]
- Krause, K.H.; Dresel, S.H.; Krause, J.; la Fougere, C.; Ackenheil, M. The Dopamine Transporter and Neuroimaging in Attention Deficit Hyperactivity Disorder. Neurosci. Biobehav. Rev. 2003, 27, 605–613. [Google Scholar] [CrossRef]
- Spencer, T.J.; Biederman, J.; Madras, B.K.; Faraone, S.V.; Dougherty, D.D.; Bonab, A.A.; Fischman, A.J. In Vivo Neuroreceptor Imaging in Attention-Deficit/Hyperactivity Disorder: A Focus on the Dopamine Transporter. Biol. Psychiatry 2005, 57, 1293–1300. [Google Scholar] [CrossRef]
- Volkow, N.D.; Fowler, J.S.; Wang, G.; Ding, Y.; Gatley, S.J. Mechanism of Action of Methylphenidate: Insights from PET Imaging Studies. J. Atten. Disord. 2002, 6 (Suppl. S1), S31–S43. [Google Scholar] [CrossRef]
- Hannestad, J.; Gallezot, J.-D.; Planeta-Wilson, B.; Lin, S.-F.; Williams, W.A.; van Dyck, C.H.; Malison, R.T.; Carson, R.E.; Ding, Y.-S. Clinically Relevant Doses of Methylphenidate Significantly Occupy Norepinephrine Transporters in Humans In Vivo. Biol. Psychiatry 2010, 68, 854–860. [Google Scholar] [CrossRef] [Green Version]
- Wang, G.-J.; Volkow, N.D.; Wigal, T.; Kollins, S.H.; Newcorn, J.H.; Telang, F.; Logan, J.; Jayne, M.; Wong, C.T.; Han, H.; et al. Long-Term Stimulant Treatment Affects Brain Dopamine Transporter Level in Patients with Attention Deficit Hyperactive Disorder. PLoS ONE 2013, 8, e63023. [Google Scholar] [CrossRef] [Green Version]
- Aster, H.-C.; Romanos, M.; Walitza, S.; Gerlach, M.; Mühlberger, A.; Rizzo, A.; Andreatta, M.; Hasenauer, N.; Hartrampf, P.E.; Nerlich, K.; et al. Responsivity of the Striatal Dopamine System to Methylphenidate—A within-Subject I-123-β-CIT-SPECT Study in Male Children and Adolescents with Attention-Deficit/Hyperactivity Disorder. Front. Psychiatry 2022, 13, 804730. [Google Scholar] [CrossRef] [PubMed]
- Dresel, S.; Krause, J.; Krause, K.H.; LaFougere, C.; Brinkbäumer, K.; Kung, H.F.; Hahn, K.; Tatsch, K. Attention Deficit Hyperactivity Disorder: Binding of [99mTc]TRODAT-1 to the Dopamine Transporter before and after Methylphenidate Treatment. Eur. J. Nucl. Med. 2000, 27, 1518–1524. [Google Scholar] [CrossRef] [PubMed]
- Szobot, C.M.; Shih, M.C.; Schaefer, T.; Júnior, N.; Hoexter, M.Q.; Fu, Y.K.; Pechansky, F.; Bressan, R.A.; Rohde, L.A.P. Methylphenidate DAT Binding in Adolescents with Attention-Deficit/ Hyperactivity Disorder Comorbid with Substance Use Disorder—A Single Photon Emission Computed Tomography with [Tc(99m)]TRODAT-1 Study. NeuroImage 2008, 40, 1195–1201. [Google Scholar] [CrossRef] [PubMed]
- Akay, A.P.; Kaya, G.Ç.; Kose, S.; Yazıcıoğlu, Ç.E.; Erkuran, H.Ö.; Güney, S.A.; Oğuz, K.; Keskin, D.; Baykara, B.; Emiroğlu, N.İ.; et al. Genetic Imaging Study with [Tc-99m] TRODAT-1 SPECT in Adolescents with ADHD Using OROS-Methylphenidate. Prog. Neuropsychopharmacol. Biol. Psychiatry 2018, 86, 294–300. [Google Scholar] [CrossRef]
- Volkow, N.D.; Wang, G.-J.; Tomasi, D.; Kollins, S.H.; Wigal, T.L.; Newcorn, J.H.; Telang, F.W.; Fowler, J.S.; Logan, J.; Wong, C.T.; et al. Methylphenidate-Elicited Dopamine Increases in Ventral Striatum Are Associated with Long-Term Symptom Improvement in Adults with Attention Deficit Hyperactivity Disorder. J. Neurosci. 2012, 32, 841–849. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, F.-M.; Liang, Y.; Salas, R.; Zhang, L.; De Biasi, M.; Dani, J.A. Corelease of Dopamine and Serotonin from Striatal Dopamine Terminals. Neuron 2005, 46, 65–74. [Google Scholar] [CrossRef] [Green Version]
- Navailles, S.; Bioulac, B.; Gross, C.; De Deurwaerdère, P. Serotonergic Neurons Mediate Ectopic Release of Dopamine Induced by L-DOPA in a Rat Model of Parkinson’s Disease. Neurobiol. Dis. 2010, 38, 136–143. [Google Scholar] [CrossRef]
- Larsen, M.B.; Sonders, M.S.; Mortensen, O.V.; Larson, G.A.; Zahniser, N.R.; Amara, S.G. Dopamine Transport by the Serotonin Transporter: A Mechanistically Distinct Mode of Substrate Translocation. J. Neurosci. 2011, 31, 6605–6615. [Google Scholar] [CrossRef] [Green Version]
- Easton, N.; Steward, C.; Marshall, F.; Fone, K.; Marsden, C. Effects of Amphetamine Isomers, Methylphenidate and Atomoxetine on Synaptosomal and Synaptic Vesicle Accumulation and Release of Dopamine and Noradrenaline in Vitro in the Rat Brain. Neuropharmacology 2007, 52, 405–414. [Google Scholar] [CrossRef]
- Wallace, L.J. Effects of Amphetamine on Subcellular Distribution of Dopamine and DOPAC. Synapse 2012, 66, 592–607. [Google Scholar] [CrossRef]
- Kuczenski, R.; Segal, D.S. Stimulant Actions in Rodents: Implications for Attention-Deficit/Hyperactivity Disorder Treatment and Potential Substance Abuse. Biol. Psychiatry 2005, 57, 1391–1396. [Google Scholar] [CrossRef]
- Schiffer, W.K.; Volkow, N.D.; Fowler, J.S.; Alexoff, D.L.; Logan, J.; Dewey, S.L. Therapeutic Doses of Amphetamine or Methylphenidate Differentially Increase Synaptic and Extracellular Dopamine. Synapse 2006, 59, 243–251. [Google Scholar] [CrossRef]
- Atcha, Z.; Rourke, C.; Neo, A.H.; Goh, C.W.; Lim, J.S.; Aw, C.-C.; Browne, E.R.; Pemberton, D.J. Alternative Method of Oral Dosing for Rats. J. Am. Assoc. Lab. Anim. Sci. 2010, 49, 335–343. [Google Scholar] [PubMed]
- Paxinos, G.; Watson, C. The Rat Brain in Stereotaxic Coordinates, 6th ed.; Academic Press: San Diego, CA, USA, 2007. [Google Scholar]
- Gronier, B. In Vivo Electrophysiological Effects of Methylphenidate in the Prefrontal Cortex: Involvement of Dopamine D1 and Alpha 2 Adrenergic Receptors. Eur. Neuropsychopharmacol. 2011, 21, 192–204. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Liu, J.; Gui, Z.H.; Ali, U.; Fan, L.L.; Hou, C.; Wang, T.; Chen, L.; Li, Q. A2-Adrenoceptor Regulates the Spontaneous and the GABA/Glutamate Modulated Firing Activity of the Rat Medial Prefrontal Cortex Pyramidal Neurons. Neuroscience 2011, 182, 193–202. [Google Scholar] [CrossRef] [PubMed]
- Mallet, N.; Ballion, B.; Moine, C.L.; Gonon, F. Cortical Inputs and GABA Interneurons Imbalance Projection Neurons in the Striatum of Parkinsonian Rats. J. Neurosci. 2006, 26, 3875–3884. [Google Scholar] [CrossRef] [Green Version]
- Wild, C.J.; Elliott, T.; Sporle, A. On Democratizing Data Science: Some INZights Into Empowering the Many. Harv. Data Sci. Rev. 2021, 3. [Google Scholar] [CrossRef]
- Elliott, T.; Soh, Y.H.; Barnett, D.; Anastasiadis, S. INZightPlots: Graphical Tools for Exploring Data with “INZight” 2022. Available online: https://inzight.nz (accessed on 1 April 2022).
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2013. [Google Scholar]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Di Miceli, M.; Derf, A.; Gronier, B. Consequences of Acute or Chronic Methylphenidate Exposure Using Ex Vivo Neurochemistry and In Vivo Electrophysiology in the Prefrontal Cortex and Striatum of Rats. Int. J. Mol. Sci. 2022, 23, 8588. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23158588
Di Miceli M, Derf A, Gronier B. Consequences of Acute or Chronic Methylphenidate Exposure Using Ex Vivo Neurochemistry and In Vivo Electrophysiology in the Prefrontal Cortex and Striatum of Rats. International Journal of Molecular Sciences. 2022; 23(15):8588. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23158588
Chicago/Turabian StyleDi Miceli, Mathieu, Asma Derf, and Benjamin Gronier. 2022. "Consequences of Acute or Chronic Methylphenidate Exposure Using Ex Vivo Neurochemistry and In Vivo Electrophysiology in the Prefrontal Cortex and Striatum of Rats" International Journal of Molecular Sciences 23, no. 15: 8588. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23158588