
Making the Rounds: Exploring the Role of Circulating Tumor DNA (ctDNA) in Non-Small Cell Lung Cancer



Abstract
:1. Introduction
2. Pre-Analytical Conditions for ctDNA Assessment

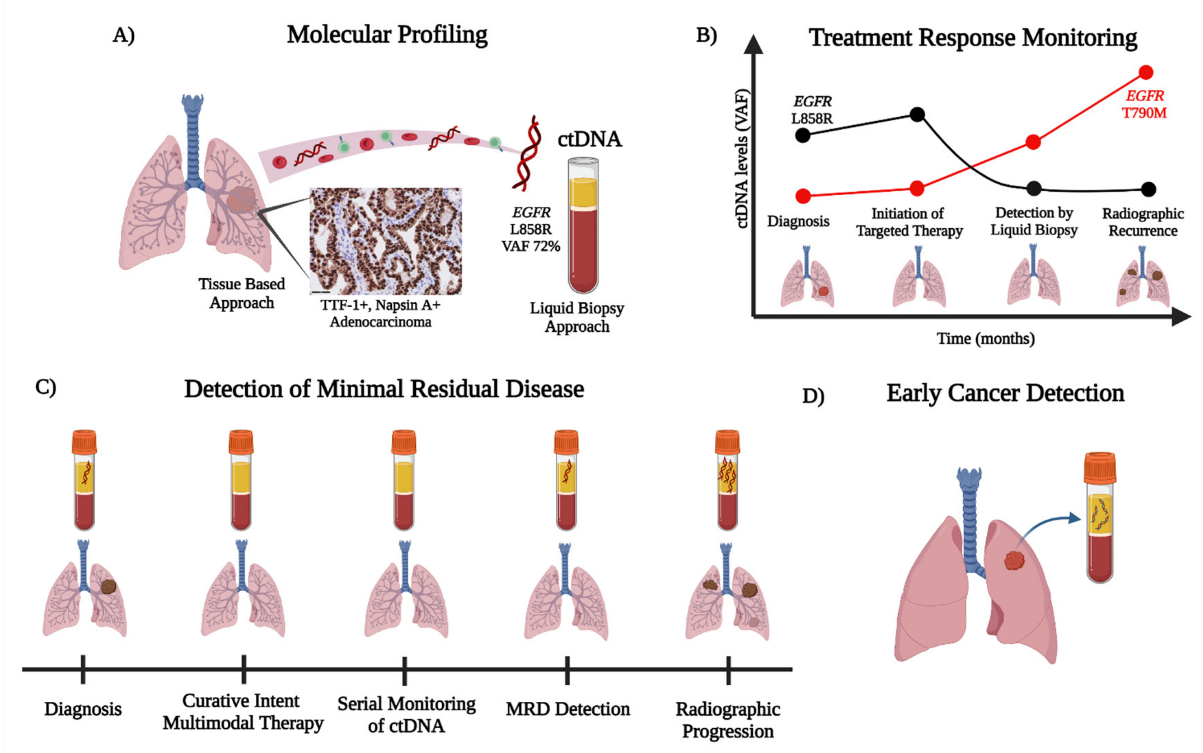{kind=link}
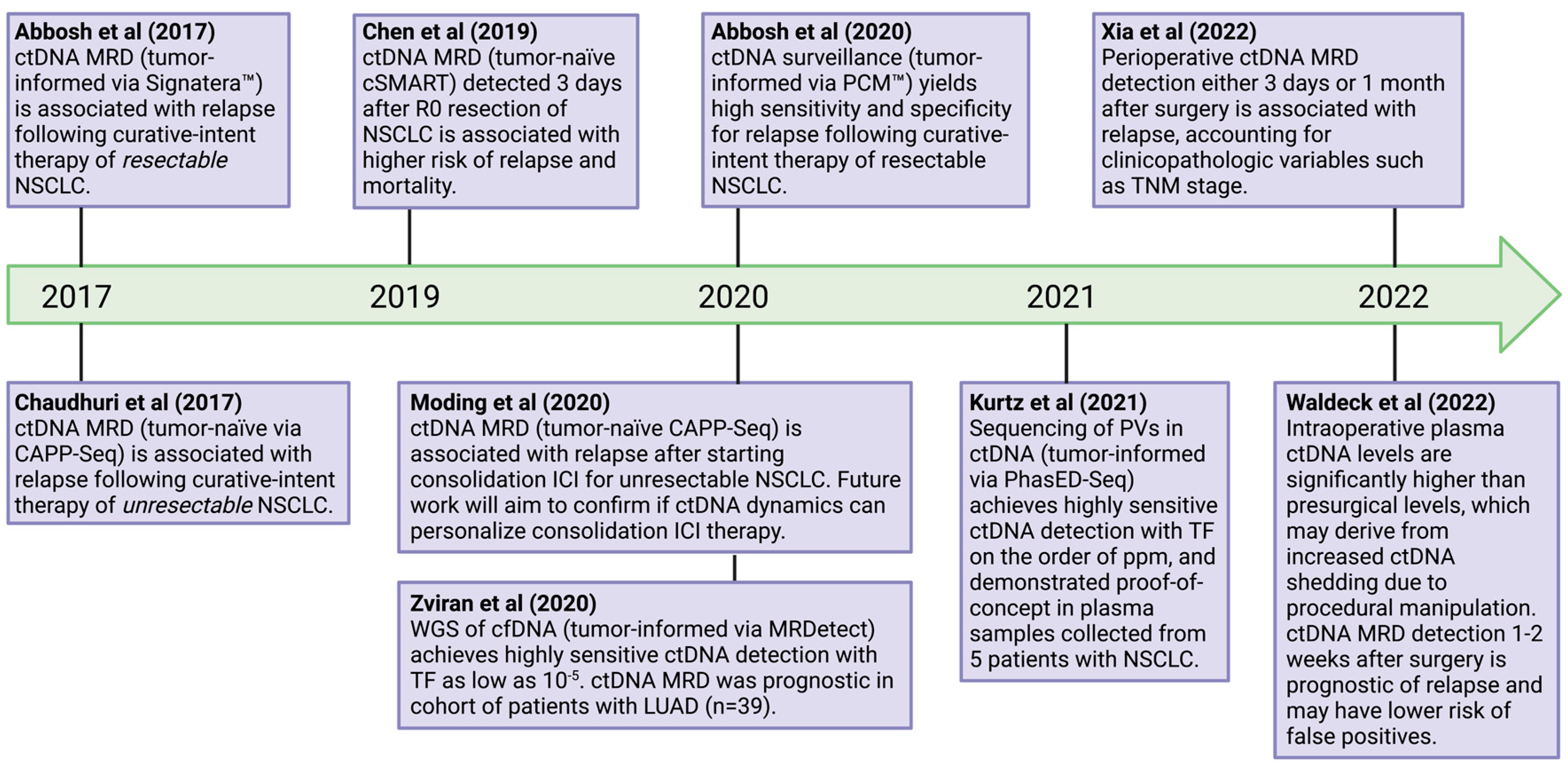{kind=link}
| Technology | Examples | Assay Personalization | Reported LOD (% of cfDNA) | Potential Clinical Uses | Limitations |
|---|---|---|---|---|---|
| AS-PCR | ARMS [32], Cobas EGFR mutation test v2 (Roche) [33] | Some Required | ~0.1–1 | Molecular Profiling | Detects only known mutations or a small number of variants concurrently; low sensitivity |
| dPCR | dPCR [34], ddPCR [35], BEAMing [36] | Some Required | ~0.01 | Molecular Profiling, Treatment Monitoring, ctDNA MRD | Detects only known mutations or a small number of variants |
| Multiplex PCR-based NGS | TAm-Seq [4], TAm-Seq Enhanced [37], Safe-SeqS [38], Natera® [39] | Some Required | ~0.01–2.0 | Molecular Profiling, Treatment Monitoring, ctDNA MRD | Detects only known mutations; less comprehensive than other NGS methods; incapable of detecting SCNAs and fusions without assay personalization |
| Hybrid captured-based NGS | CAPP-Seq [40,41], TEC-Seq [42], Guardant360® [25,43,44], FoundationOne® Liquid CDx [26,45] | Not Required | ~0.001–0.5 | Molecular Profiling, Treatment Monitoring, ctDNA MRD | Less comprehensive than WGS and WES |
| WES | WES [46] | Not Required | ~5 | Molecular Profiling, ctDNA MRD | Low sensitivity; high cost |
| WGS | WGS [47,48] | Not Required | ~10 | Molecular Profiling, ctDNA MRD | Low sensitivity; mostly limited to SCNA detection; high cost |
| cfDNA methylation | cfMeDIP-seq [49], cfMBD-seq [50], GRAIL Galleri™ [51] | Not Required | Variable | Early Lung Cancer Detection, ctDNA MRD | Variable/limited sensitivity for early-stage disease detection |
| Fragmentomics | DELFI [52] | Not Required | Variable | Early Lung Cancer Detection, ctDNA MRD | Variable/limited sensitivity for early-stage disease detection |
| Combined approaches | CAPP-Seq + GRP [53], CancerSEEK [54] | Variable | Variable | Early Lung Cancer Detection, ctDNA MRD | May require more time and resources |
3. Molecular Profiling
4. Treatment Response Monitoring
5. Minimal Residual Disease (MRD) Detection
6. ctDNA for Early Cancer Detection
7. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Leighl, N.B.; Page, R.D.; Raymond, V.M.; Daniel, D.B.; Divers, S.G.; Reckamp, K.L.; Villalona-Calero, M.A.; Dix, D.; Odegaard, J.I.; Lanman, R.B.; et al. Clinical Utility of Comprehensive Cell-free DNA Analysis to Identify Genomic Biomarkers in Patients with Newly Diagnosed Metastatic Non–small Cell Lung Cancer. Clin. Cancer Res. 2019, 25, 4691–4700. [Google Scholar] [CrossRef] [PubMed]
- Aggarwal, C.; Thompson, J.C.; Black, T.A.; Katz, S.I.; Fan, R.; Yee, S.S.; Chien, A.; Evans, T.L.; Bauml, J.M.; Alley, E.W.; et al. Clinical Implications of Plasma-Based Genotyping With the Delivery of Personalized Therapy in Metastatic Non–Small Cell Lung Cancer. JAMA Oncol. 2019, 5, 173–180. [Google Scholar] [CrossRef] [PubMed]
- Rolfo, C.; Mack, P.; Scagliotti, G.V.; Aggarwal, C.; Arcila, M.E.; Barlesi, F.; Bivona, T.; Diehn, M.; Dive, C.; Dziadziuszko, R.; et al. Liquid Biopsy for Advanced NSCLC: A Consensus Statement From the International Association for the Study of Lung Cancer. J. Thorac. Oncol. 2021, 16, 1647–1662. [Google Scholar] [CrossRef]
- Forshew, T.; Murtaza, M.; Parkinson, C.; Gale, D.; Tsui, D.W.Y.; Kaper, F.; Dawson, S.-J.; Piskorz, A.M.; Jimenez-Linan, M.; Bentley, D.; et al. Noninvasive Identification and Monitoring of Cancer Mutations by Targeted Deep Sequencing of Plasma DNA. Sci. Transl. Med. 2012, 4, 136ra68. [Google Scholar] [CrossRef]
- Miller, A.; Shah, R.; Pentsova, E.I.; Pourmaleki, M.; Briggs, S.; Distefano, N.; Zheng, Y.; Skakodub, A.; Mehta, S.A.; Campos, C.; et al. Tracking tumour evolution in glioma through liquid biopsies of cerebrospinal fluid. Nature 2019, 565, 654–658. [Google Scholar] [CrossRef]
- Crowley, E.; Di Nicolantonio, F.; Loupakis, F.; Bardelli, A. Liquid biopsy: Monitoring cancer-genetics in the blood. Nat. Rev. Clin. Oncol. 2013, 10, 472–484. [Google Scholar] [CrossRef] [PubMed]
- Diaz, L.A., Jr.; Bardelli, A. Liquid Biopsies: Genotyping Circulating Tumor DNA. J. Clin. Oncol. 2014, 32, 579–586. [Google Scholar] [CrossRef]
- Zhu, L.; Sun, H.-T.; Wang, S.; Huang, S.-L.; Zheng, Y.; Wang, C.-Q.; Hu, B.-Y.; Qin, W.; Zou, T.-T.; Fu, Y.; et al. Isolation and characterization of exosomes for cancer research. J. Hematol. Oncol. 2020, 13, 152. [Google Scholar] [CrossRef] [PubMed]
- Alix-Panabières, C.; Pantel, K. Liquid Biopsy: From Discovery to Clinical Application. Cancer Discov. 2021, 11, 858–873. [Google Scholar] [CrossRef] [PubMed]
- Nuzzo, P.V.; Berchuck, J.E.; Korthauer, K.; Spisak, S.; Nassar, A.H.; Alaiwi, S.A.; Chakravarthy, A.; Shen, S.Y.; Bakouny, Z.; Boccardo, F.; et al. Detection of renal cell carcinoma using plasma and urine cell-free DNA methylomes. Nat. Med. 2020, 26, 1041–1043. [Google Scholar] [CrossRef] [PubMed]
- Best, M.G.; Wesseling, P.; Wurdinger, T. Tumor-Educated Platelets as a Noninvasive Biomarker Source for Cancer Detection and Progression Monitoring. Cancer Res. 2018, 78, 3407–3412. [Google Scholar] [CrossRef] [PubMed]
- Mandel, P.; Metais, P. Nuclear Acids In Human Blood Plasma. Comptes Rendus Seances Soc. Biol. Ses Fil. 1948, 142, 241–243. [Google Scholar]
- Corcoran, R.B.; Chabner, B.A. Application of Cell-free DNA Analysis to Cancer Treatment. N. Engl. J. Med. 2018, 379, 1754–1765. [Google Scholar] [CrossRef] [PubMed]
- Jahr, S.; Hentze, H.; Englisch, S.; Hardt, D.; Fackelmayer, F.O.; Hesch, R.D.; Knippers, R. DNA fragments in the blood plasma of cancer patients: Quantitations and evidence for their origin from apoptotic and necrotic cells. Cancer Res. 2001, 61, 1659–1665. [Google Scholar] [PubMed]
- Wan, J.C.M.; Massie, C.; Garcia-Corbacho, J.; Mouliere, F.; Brenton, J.D.; Caldas, C.; Pacey, S.; Baird, R.; Rosenfeld, N. Liquid biopsies come of age: Towards implementation of circulating tumour DNA. Nat. Rev. Cancer 2017, 17, 223–238. [Google Scholar] [CrossRef] [PubMed]
- Pisetsky, D.S.; Fairhurst, A.-M. The origin of extracellular DNA during the clearance of dead and dying cells. Autoimmunity 2007, 40, 281–284. [Google Scholar] [CrossRef] [PubMed]
- El Messaoudi, S.; Rolet, F.; Mouliere, F.; Thierry, A.R. Circulating cell free DNA: Preanalytical considerations. Clin. Chim. Acta 2013, 424, 222–230. [Google Scholar] [CrossRef]
- Kustanovich, A.; Schwartz, R.; Peretz, T.; Grinshpun, A. Life and death of circulating cell-free DNA. Cancer Biol. Ther. 2019, 20, 1057–1067. [Google Scholar] [CrossRef]
- Thijssen, M.A.; Swinkels, D.W.; Ruers, T.J.M.; De Kok, J.B. Difference between free circulating plasma and serum DNA in patients with colorectal liver metastases. Anticancer Res. 2002, 22, 421–425. [Google Scholar]
- Umetani, N.; Hiramatsu, S.; Hoon, D.S. Higher Amount of Free Circulating DNA in Serum than in Plasma Is Not Mainly Caused by Contaminated Extraneous DNA during Separation. Ann. N. Y. Acad. Sci. 2006, 1075, 299–307. [Google Scholar] [CrossRef]
- Siravegna, G.; Marsoni, S.; Siena, S.; Bardelli, A. Integrating liquid biopsies into the management of cancer. Nat. Rev. Clin. Oncol. 2017, 14, 531–548. [Google Scholar] [CrossRef]
- Bettegowda, C.; Sausen, M.; Leary, R.J.; Kinde, I.; Wang, Y.; Agrawal, N.; Bartlett, B.R.; Wang, H.; Luber, B.; Alani, R.M.; et al. Detection of circulating tumor DNA in early- and late-stage human malignancies. Sci. Transl. Med. 2014, 6, 224. [Google Scholar] [CrossRef]
- Pellini, B.; Szymanski, J.; Chin, R.-I.; Jones, P.A.; Chaudhuri, A.A. Liquid Biopsies Using Circulating Tumor DNA in Non-Small Cell Lung Cancer. Thorac. Surg. Clin. 2020, 30, 165–177. [Google Scholar] [CrossRef] [PubMed]
- Lim, C.; Tsao, M.S.; Le, L.W.; Shepherd, F.A.; Feld, R.; Burkes, R.L.; Liu, G.; Kamel-Reid, S.; Hwang, D.; Tanguay, J.; et al. Biomarker testing and time to treatment decision in patients with advanced nonsmall-cell lung cancer. Ann. Oncol. 2015, 26, 1415–1421. [Google Scholar] [CrossRef]
- U.S. Food and Drug Administration. FDA Approves First Liquid Biopsy Next-Generation Sequencing Companion Diagnostic Test. 2020. Available online: https://www.fda.gov/news-events/press-announcements/fda-approves-first-liquid-biopsy-next-generation-sequencing-companion-diagnostic-test (accessed on 23 March 2022).
- U.S. Food and Drug Administration. FDA Approves Liquid Biopsy NGS Companion Diagnostic Test for Multiple Cancers and Biomarkers. 2020. Available online: https://www.fda.gov/drugs/resources-information-approved-drugs/fda-approves-liquid-biopsy-ngs-companion-diagnostic-test-multiple-cancers-and-biomarkers (accessed on 23 March 2022).
- Marrugo-Ramírez, J.; Mir, M.; Samitier, J. Blood-Based Cancer Biomarkers in Liquid Biopsy: A Promising Non-Invasive Alternative to Tissue Biopsy. Int. J. Mol. Sci. 2018, 19, 2877. [Google Scholar] [CrossRef]
- Alborelli, I.; Generali, D.; Jermann, P.; Cappelletti, M.R.; Ferrero, G.; Scaggiante, B.; Bortul, M.; Zanconati, F.; Nicolet, S.; Haegele, J.; et al. Cell-free DNA analysis in healthy individuals by next-generation sequencing: A proof of concept and technical validation study. Cell Death Dis. 2019, 10, 534. [Google Scholar] [CrossRef]
- Danesi, R.; Lo, Y.; Oellerich, M.; Beck, J.; Galbiati, S.; Del Re, M.; Lianidou, E.; Neumaier, M.; van Schaik, R. What do we need to obtain high quality circulating tumor DNA (ctDNA) for routine diagnostic test in oncology?—Considerations on pre-analytical aspects by the IFCC workgroup cfDNA. Clin. Chim. Acta 2021, 520, 168–171. [Google Scholar] [CrossRef]
- Nikolaev, S.; Lemmens, L.; Koessler, T.; Blouin, J.-L.; Nouspikel, T. Circulating tumoral DNA: Preanalytical validation and quality control in a diagnostic laboratory. Anal. Biochem. 2018, 542, 34–39. [Google Scholar] [CrossRef]
- Parpart-Li, S.; Bartlett, B.; Popoli, M.; Adleff, V.; Tucker, L.; Steinberg, R.; Georgiadis, A.; Phallen, J.; Brahmer, J.R.; Azad, N.A.; et al. The Effect of Preservative and Temperature on the Analysis of Circulating Tumor DNA. Clin. Cancer Res. 2017, 23, 2471–2477. [Google Scholar] [CrossRef]
- Newton, C.R.; Graham, A.; Heptinstall, L.E.; Powell, S.J.; Summers, C.; Kalsheker, N.; Smith, J.C.; Markham, A.F. Analysis of any point mutation in DNA. The amplification refractory mutation system (ARMS). Nucleic Acids Res. 1989, 17, 2503–2516. [Google Scholar] [CrossRef] [PubMed]
- Malapelle, U.; Sirera, R.; Jantus-Lewintre, E.; Reclusa, P.; Calabuig, S.; Blasco, A.; Pisapia, P.; Rolfo, C.; Camps, C. Profile of the Roche cobas® EGFR mutation test v2 for non-small cell lung cancer. Expert Rev. Mol. Diagn. 2017, 17, 209–215. [Google Scholar] [CrossRef] [PubMed]
- Vogelstein, B.; Kinzler, K.W. Digital PCR. Proc. Natl. Acad. Sci. USA 1999, 96, 9236–9241. [Google Scholar] [CrossRef] [PubMed]
- Hindson, B.J.; Ness, K.D.; Masquelier, D.A.; Belgrader, P.; Heredia, N.J.; Makarewicz, A.J.; Bright, I.J.; Lucero, M.Y.; Hiddessen, A.L.; Legler, T.C.; et al. High-throughput droplet digital PCR system for absolute quantitation of DNA copy number. Anal Chem. 2011, 83, 8604–8610. [Google Scholar] [CrossRef]
- Diehl, F.; Li, M.; He, Y.; Kinzler, K.W.; Vogelstein, B.; Dressman, D. BEAMing: Single-molecule PCR on microparticles in water-in-oil emulsions. Nat Methods 2006, 3, 551–559. [Google Scholar] [CrossRef]
- Gale, D.; Plagnol, V.; Lawson, A.; Pugh, M.; Smalley, S.; Howarth, K.; Madi, M.; Durham, B.; Kumanduri, V.; Lo, K.; et al. Abstract 3639: Analytical performance and validation of an enhanced TAm-Seq circulating tumor DNA sequencing assay. Cancer Res. 2016, 76, 3639. Available online: https://aacrjournals.org/cancerres/article/76/14_Supplement/3639/611041/Abstract-3639-Analytical-performance-and (accessed on 16 April 2022). [CrossRef]
- Kinde, I.; Wu, J.; Papadopoulos, N.; Kinzler, K.W.; Vogelstein, B. Detection and quantification of rare mutations with massively parallel sequencing. Proc. Natl. Acad. Sci. USA 2011, 108, 9530–9535. [Google Scholar] [CrossRef]
- Abbosh, C.; Birkbak, N.J.; Wilson, G.A.; Jamal-Hanjani, M.; Constantin, T.; Salari, R.; Le Quesne, J.; Moore, D.A.; Veeriah, S.; Rosenthal, R.; et al. Phylogenetic ctDNA analysis depicts early-stage lung cancer evolution. Nature 2017, 545, 446–451. [Google Scholar] [CrossRef]
- Newman, A.M.; Bratman, S.V.; To, J.; Wynne, J.F.; Eclov, N.C.W.; Modlin, L.A.; Liu, C.L.; Neal, J.W.; Wakelee, H.A.; Merritt, R.E.; et al. An ultrasensitive method for quantitating circulating tumor DNA with broad patient coverage. Nat. Med. 2014, 20, 548–554. [Google Scholar] [CrossRef]
- Newman, A.M.; Lovejoy, A.F.; Klass, D.M.; Kurtz, D.M.; Chabon, J.J.; Scherer, F.; Stehr, H.; Liu, C.L.; Bratman, S.V.; Say, C.; et al. Integrated digital error suppression for improved detection of circulating tumor DNA. Nat. Biotechnol. 2016, 34, 547–555. [Google Scholar] [CrossRef] [PubMed]
- Phallen, J.; Sausen, M.; Adleff, V.; Leal, A.; Hruban, C.; White, J.; Anagnostou, V.; Fiksel, J.; Cristiano, S.; Papp, E.; et al. Direct detection of early-stage cancers using circulating tumor DNA. Sci. Transl. Med. 2017, 9, 1252–1255. [Google Scholar] [CrossRef]
- Lanman, R.B.; Mortimer, S.A.; Zill, O.A.; Sebisanovic, D.; Lopez, R.; Blau, S.; Collisson, E.A.; Divers, S.G.; Hoon, D.; Kopetz, S.; et al. Analytical and Clinical Validation of a Digital Sequencing Panel for Quantitative, Highly Accurate Evaluation of Cell-Free Circulating Tumor DNA. PLoS ONE 2015, 10, e0140712. [Google Scholar] [CrossRef]
- Odegaard, J.I.; Vincent, J.J.; Mortimer, S.; Vowles, J.V.; Ulrich, B.C.; Banks, K.C.; Fairclough, S.R.; Zill, O.A.; Sikora, M.; Mokhtari, R.; et al. Validation of a Plasma-Based Comprehensive Cancer Genotyping Assay Utilizing Orthogonal Tissue- and Plasma-Based Methodologies. Clin. Cancer Res. 2018, 24, 3539–3549. [Google Scholar] [CrossRef]
- Lee, J.K.; Hazar-Rethinam, M.; Decker, B.; Gjoerup, O.; Madison, R.W.; Lieber, D.S.; Chung, J.H.; Schrock, A.B.; Creeden, J.; Venstrom, J.M.; et al. The Pan-Tumor Landscape of Targetable Kinase Fusions in Circulating Tumor DNA. Clin. Cancer Res. 2022, 28, 728–737. [Google Scholar] [CrossRef] [PubMed]
- Murtaza, M.; Dawson, S.-J.; Tsui, D.W.Y.; Gale, D.; Forshew, T.; Piskorz, A.M.; Parkinson, C.; Chin, S.-F.; Kingsbury, Z.; Wong, A.S.C.; et al. Non-invasive analysis of acquired resistance to cancer therapy by sequencing of plasma DNA. Nature 2013, 497, 108–112. [Google Scholar] [CrossRef]
- Chan, K.A.; Jiang, P.; Zheng, Y.W.; Liao, G.J.; Sun, H.; Wong, J.; Siu, S.S.N.; Chan, W.C.; Chan, S.L.; Chan, A.T.; et al. Cancer Genome Scanning in Plasma: Detection of Tumor-Associated Copy Number Aberrations, Single-Nucleotide Variants, and Tumoral Heterogeneity by Massively Parallel Sequencing. Clin. Chem. 2013, 59, 211–224. [Google Scholar] [CrossRef]
- Welch, J.S. Use of Whole-Genome Sequencing to Diagnose a Cryptic Fusion Oncogene. JAMA 2011, 305, 1577–1584. [Google Scholar] [CrossRef]
- Shen, S.Y.; Burgener, J.M.; Bratman, S.V.; De Carvalho, D.D. Preparation of cfMeDIP-seq libraries for methylome profiling of plasma cell-free DNA. Nat. Protoc. 2019, 14, 2749–2780. [Google Scholar] [CrossRef]
- Huang, J.; Soupir, A.C.; Schlick, B.D.; Teng, M.; Sahin, I.H.; Permuth, J.B.; Siegel, E.M.; Manley, B.J.; Pellini, B.; Wang, L. Cancer Detection and Classification by CpG Island Hypermethylation Signatures in Plasma Cell-Free DNA. Cancers 2021, 13, 5611. [Google Scholar] [CrossRef]
- Klein, E.; Richards, D.; Cohn, A.; Tummala, M.; Lapham, R.; Cosgrove, D.; Chung, G.; Clement, J.; Gao, J.; Hunkapiller, N.; et al. Clinical validation of a targeted methylation-based multi-cancer early detection test using an independent validation set. Ann. Oncol. 2021, 32, 1167–1177. [Google Scholar] [CrossRef]
- Mathios, D.; Johansen, J.S.; Cristiano, S.; Medina, J.E.; Phallen, J.; Larsen, K.R.; Bruhm, D.C.; Niknafs, N.; Ferreira, L.; Adleff, V.; et al. Detection and characterization of lung cancer using cell-free DNA fragmentomes. Nat. Commun. 2021, 12, 5060. [Google Scholar] [CrossRef]
- Przybyl, J.; Chabon, J.J.; Spans, L.; Ganjoo, K.N.; Vennam, S.; Newman, A.M.; Forgó, E.; Varma, S.; Zhu, S.; Debiec-Rychter, M.; et al. Combination Approach for Detecting Different Types of Alterations in Circulating Tumor DNA in Leiomyosarcoma. Clin. Cancer Res. 2018, 24, 2688–2699. [Google Scholar] [CrossRef]
- Cohen, J.D.; Li, L.; Wang, Y.; Thoburn, C.; Afsari, B.; Danilova, L.; Douville, C.; Javed, A.A.; Wong, F.; Mattox, A.; et al. Detection and localization of surgically resectable cancers with a multi-analyte blood test. Science 2018, 359, 926–930. [Google Scholar] [CrossRef]
- Chin, R.-I.; Chen, K.; Usmani, A.; Chua, C.; Harris, P.K.; Binkley, M.S.; Azad, T.D.; Dudley, J.C.; Chaudhuri, A.A. Detection of Solid Tumor Molecular Residual Disease (MRD) Using Circulating Tumor DNA (ctDNA). Mol. Diagn. Ther. 2019, 23, 311–331. [Google Scholar] [CrossRef]
- Herbst, R.S.; Morgensztern, D.; Boshoff, C. The biology and management of non-small cell lung cancer. Nature 2018, 553, 446–454. [Google Scholar] [CrossRef]
- Shields, M.D.; Marin-Acevedo, J.A.; Pellini, B. Immunotherapy for Advanced Non–Small Cell Lung Cancer: A Decade of Progress. Am. Soc. Clin. Oncol. Educ. Book 2021, 41, e105–e127. [Google Scholar] [CrossRef]
- Schiller, J.H.; Harrington, D.; Belani, C.P.; Langer, C.; Sandler, A.; Krook, J.; Zhu, J.; Johnson, D.H. Comparison of Four Chemotherapy Regimens for Advanced Non–Small-Cell Lung Cancer. N. Engl. J. Med. 2002, 346, 92–98. [Google Scholar] [CrossRef]
- Yamamoto, N.; Nambu, Y.; Fujimoto, T.; Koshiji, M. A Landmark Point Analysis with Cytotoxic Agents for Advanced NSCLC. J. Thorac. Oncol. 2009, 4, 697–701. [Google Scholar] [CrossRef]
- Zhou, C.; Wu, Y.-L.; Chen, G.; Feng, J.; Liu, X.-Q.; Wang, C.; Zhang, S.; Wang, J.; Zhou, S.; Ren, S.; et al. Erlotinib versus chemotherapy as first-line treatment for patients with advanced EGFR mutation-positive non-small-cell lung cancer (OPTIMAL, CTONG-0802): A multicentre, open-label, randomised, phase 3 study. Lancet Oncol. 2011, 12, 735–742. [Google Scholar] [CrossRef]
- Mok, T.S.; Wu, Y.-L.; Ahn, M.-J.; Garassino, M.C.; Kim, H.R.; Ramalingam, S.S.; Shepherd, F.A.; He, Y.; Akamatsu, H.; Theelen, W.S.; et al. Osimertinib or Platinum–Pemetrexed in EGFR T790M–Positive Lung Cancer. N. Engl. J. Med. 2017, 376, 629–640. [Google Scholar] [CrossRef]
- Shaw, A.T.; Kim, D.-W.; Nakagawa, K.; Seto, T.; Crinó, L.; Ahn, M.-J.; De Pas, T.; Besse, B.; Solomon, B.J.; Blackhall, F.; et al. Crizotinib versus Chemotherapy in AdvancedALK-Positive Lung Cancer. N. Engl. J. Med. 2013, 368, 2385–2394. [Google Scholar] [CrossRef]
- Mok, T.S.; Wu, Y.L.; Yu, C.J.; Zhou, C.; Chen, Y.M.; Zhang, L.; Ignacio, J.; Liao, M.; Srimuninnimit, V.; Boyer, M.J.; et al. Randomized, placebo-controlled, phase II study of sequential erlotinib and chemotherapy as first-line treatment for advanced non-small-cell lung cancer. J. Clin. Oncol. 2009, 27, 5080–5087. [Google Scholar] [CrossRef]
- Hong, D.S.; Fakih, M.G.; Strickler, J.H.; Desai, J.; Durm, G.A.; Shapiro, G.I.; Falchook, G.S.; Price, T.J.; Sacher, A.; Denlinger, C.S.; et al. KRASG12C Inhibition with Sotorasib in Advanced Solid Tumors. N. Engl. J. Med. 2020, 383, 1207–1217. [Google Scholar] [CrossRef]
- Shaw, A.T.; Solomon, B.J. Crizotinib in ROS1-rearranged non-small-cell lung cancer. N. Engl. J. Med. 2015, 372, 683–684. [Google Scholar] [CrossRef]
- Brown, N.A.; Aisner, D.L.; Oxnard, G.R. Precision Medicine in Non–Small Cell Lung Cancer: Current Standards in Pathology and Biomarker Interpretation. Am. Soc. Clin. Oncol. Educ. Book 2018, 38, 708–715. [Google Scholar] [CrossRef] [PubMed]
- Schwartzberg, L.; Kim, E.S.; Liu, D.; Schrag, D. Precision Oncology: Who, How, What, When, and When Not? Am. Soc. Clin. Oncol. Educ. Book 2017, 160–169. [Google Scholar] [CrossRef] [PubMed]
- Bratman, S.; Newman, A.; Alizadeh, A.A.; Diehn, M. Potential clinical utility of ultrasensitive circulating tumor DNA detection with CAPP-Seq. Expert Rev. Mol. Diagn. 2015, 15, 715–719. [Google Scholar] [CrossRef]
- Elazezy, M.; Joosse, S.A. Techniques of using circulating tumor DNA as a liquid biopsy component in cancer management. Comput. Struct. Biotechnol. J. 2018, 16, 370–378. [Google Scholar] [CrossRef]
- Huggett, J.F.; Whale, A. Digital PCR as a Novel Technology and Its Potential Implications for Molecular Diagnostics. Clin. Chem. 2013, 59, 1691–1693. [Google Scholar] [CrossRef] [PubMed]
- Oxnard, G.R.; Paweletz, C.P.; Kuang, Y.; Mach, S.L.; O’Connell, A.; Messineo, M.M.; Luke, J.J.; Butaney, M.; Kirschmeier, P.; Jackman, D.M.; et al. Noninvasive Detection of Response and Resistance in EGFR-Mutant Lung Cancer Using Quantitative Next-Generation Genotyping of Cell-Free Plasma DNA. Clin. Cancer Res. 2014, 20, 1698–1705. [Google Scholar] [CrossRef] [PubMed]
- Leary, R.J.; Sausen, M.; Kinde, I.; Papadopoulos, N.; Carpten, J.D.; Craig, D.; O’Shaughnessy, J.; Kinzler, K.W.; Parmigiani, G.; Vogelstein, B.; et al. Detection of Chromosomal Alterations in the Circulation of Cancer Patients with Whole-Genome Sequencing. Sci. Transl. Med. 2012, 4, 162ra154. [Google Scholar] [CrossRef] [PubMed]
- Narayan, A.; Carriero, N.J.; Gettinger, S.N.; Kluytenaar, J.; Kozak, K.R.; Yock, T.I.; Muscato, N.E.; Ugarelli, P.; Decker, R.H.; Patel, A.A. Ultrasensitive Measurement of Hotspot Mutations in Tumor DNA in Blood Using Error-Suppressed Multiplexed Deep Sequencing. Cancer Res. 2012, 72, 3492–3498. [Google Scholar] [CrossRef] [PubMed]
- Paweletz, C.P.; Sacher, A.G.; Raymond, C.K.; Alden, R.S.; O’Connell, A.; Mach, S.L.; Kuang, Y.; Gandhi, L.; Kirschmeier, P.; English, J.M.; et al. Bias-Corrected Targeted Next-Generation Sequencing for Rapid, Multiplexed Detection of Actionable Alterations in Cell-Free DNA from Advanced Lung Cancer Patients. Clin. Cancer Res. 2016, 22, 915–922. [Google Scholar] [CrossRef]
- Chabon, J.J.; Simmons, A.D.; Lovejoy, A.F.; Esfahani, M.S.; Newman, A.M.; Haringsma, H.J.; Kurtz, D.M.; Stehr, H.; Scherer, F.; Karlovich, C.A.; et al. Circulating tumour DNA profiling reveals heterogeneity of EGFR inhibitor resistance mechanisms in lung cancer patients. Nat. Commun. 2016, 7, 11815. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.J.; Ritterhouse, L.L.; Ali, S.M.; Bailey, M.; Schrock, A.B.; Gainor, J.F.; Ferris, L.A.; Mino-Kenudson, M.; Miller, V.A.; Iafrate, A.J.; et al. ROS1 Fusions Rarely Overlap with Other Oncogenic Drivers in Non–Small Cell Lung Cancer. J. Thorac. Oncol. 2017, 12, 872–877. [Google Scholar] [CrossRef]
- Sholl, L.M.; Sun, H.; Butaney, M.; Zhang, C.; Lee, C.; Jänne, P.A.; Rodig, S.J. ROS1 Immunohistochemistry for Detection of ROS1-Rearranged Lung Adenocarcinomas. Am. J. Surg. Pathol. 2013, 37, 1441–1449. [Google Scholar] [CrossRef]
- Zheng, Z.; Liebers, M.; Zhelyazkova, B.; Cao, Y.; Panditi, D.; Lynch, K.D.; Chen, J.; Robinson, H.E.; Shim, H.S.; Chmielecki, J.; et al. Anchored multiplex PCR for targeted next-generation sequencing. Nat. Med. 2014, 20, 1479–1484. [Google Scholar] [CrossRef] [PubMed]
- Dagogo-Jack, I.; Rooney, M.; Nagy, R.J.; Lin, J.J.; Chin, E.; Ferris, L.A.; Ackil, J.; Lennerz, J.K.; Lanman, R.B.; Gainor, J.F.; et al. Molecular Analysis of Plasma From Patients With ROS1-Positive NSCLC. J. Thorac. Oncol. 2019, 14, 816–824. [Google Scholar] [CrossRef] [PubMed]
- FoundationOne®Liquid CDx. 2022. Available online: https://www.foundationmedicine.com/test/foundationone-liquid-cdx (accessed on 12 June 2022).
- Li, M.; Hou, X.; Zheng, L.; Ma, Y.; Li, D.; Lv, Y.; Chen, J.; Zheng, W.; Shao, Y.; Mou, Y.; et al. Utilizing phenotypic characteristics of metastatic brain tumors to improve the probability of detecting circulating tumor DNA from cerebrospinal fluid in non-small-cell lung cancer patients: Development and validation of a prediction model in a prospective cohort study. ESMO Open 2022, 7, 100305. [Google Scholar]
- Sudhindra, A.; Ochoa, R.; Santos, E.S. Biomarkers, prediction, and prognosis in non-small-cell lung cancer: A platform for personalized treatment. Clin. Lung Cancer 2011, 12, 360–368. [Google Scholar] [CrossRef]
- Wei, L.; Wu, W.; Han, L.; Yu, W.; Du, Y. A quantitative analysis of the potential biomarkers of non-small cell lung cancer by circulating cell-free DNA. Oncol. Lett. 2018, 16, 4353–4360. [Google Scholar]
- Normanno, N.; Rachiglio, A.M.; Roma, C.; Fenizia, F.; Esposito, C.; Pasquale, R.; La Porta, M.L.; Iannaccone, A.; Micheli, F.; Santangelo, M.; et al. Molecular diagnostics and personalized medicine in oncology: Challenges and opportunities. J. Cell. Biochem. 2012, 114, 514–524. [Google Scholar] [CrossRef]
- Dagogo-Jack, I.; Fabrizio, D.; Lennerz, J.; Schrock, A.B.; Young, L.; Mino-Kenudson, M.; Digumarthy, S.R.; Heist, R.S.; Ali, S.M.; Miller, V.A.; et al. Circulating Tumor DNA Identifies EGFR Coamplification as a Mechanism of Resistance to Crizotinib in a Patient with Advanced MET-Amplified Lung Adenocarcinoma. J. Thorac. Oncol. 2017, 12, e155–e157. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.-L.; Lee, J.S.; Thongprasert, S.; Yu, C.-J.; Zhang, L.; Ladrera, G.; Srimuninnimit, V.; Sriuranpong, V.; Sandoval-Tan, J.; Zhu, Y.; et al. Intercalated combination of chemotherapy and erlotinib for patients with advanced stage non-small-cell lung cancer (FASTACT-2): A randomised, double-blind trial. Lancet Oncol. 2013, 14, 777–786. [Google Scholar] [CrossRef]
- Mok, T.; Wu, Y.-L.; Lee, J.S.; Yu, C.-J.; Sriuranpong, V.; Sandoval-Tan, J.; Ladrera, G.; Thongprasert, S.; Srimuninnimit, V.; Liao, M.; et al. Detection and Dynamic Changes of EGFR Mutations from Circulating Tumor DNA as a Predictor of Survival Outcomes in NSCLC Patients Treated with First-line Intercalated Erlotinib and Chemotherapy. Clin. Cancer Res. 2015, 21, 3196–3203. [Google Scholar] [CrossRef] [PubMed]
- Phallen, J.; Leal, A.; Woodward, B.D.; Forde, P.M.; Naidoo, J.; Marrone, K.A.; Brahmer, J.R.; Fiksel, J.; Medina, J.E.; Cristiano, S.; et al. Early Noninvasive Detection of Response to Targeted Therapy in Non–Small Cell Lung Cancer. Cancer Res. 2019, 79, 1204–1213. [Google Scholar] [CrossRef] [PubMed]
- Serrano, C.; Leal, A.; Kuang, Y.; Morgan, J.A.; Barysauskas, C.M.; Phallen, J.; Triplett, O.; Mariño-Enríquez, A.; Wagner, A.J.; Demetri, G.D.; et al. Phase I Study of Rapid Alternation of Sunitinib and Regorafenib for the Treatment of Tyrosine Kinase Inhibitor Refractory Gastrointestinal Stromal Tumors. Clin. Cancer Res. 2019, 25, 7287–7293. [Google Scholar] [CrossRef]
- Garrido, P.; Paz-Ares, L.; Majem, M.; Morán, T.; Trigo, J.M.; Bosch-Barrera, J.; Garcίa-Campelo, R.; González-Larriba, J.L.; Sánchez-Torres, J.M.; Isla, D.; et al. LungBEAM: A prospective multicenter study to monitor stage IV NSCLC patients with EGFR mutations using BEAMing technology. Cancer Med. 2021, 10, 5878–5888. [Google Scholar] [CrossRef] [PubMed]
- Peters, S.; Camidge, D.R.; Shaw, A.T.; Gadgeel, S.; Ahn, J.S.; Kim, D.W.; Ou, S.H.I.; Pérol, M.; Dziadziuszko, R.; Rosell, R.; et al. Alectinib versus Crizotinib in Untreated ALK-Positive Non–Small-Cell Lung Cancer. N. Engl. J. Med. 2017, 377, 829–838. [Google Scholar] [CrossRef] [PubMed]
- Dziadziuszko, R.; Peters, S.; Mok, T.; Camidge, D.R.; Gadgeel, S.M.; Ou, S.-H.I.; Konopa, K.; Noé, J.; Nowicka, M.; Bordogna, W.; et al. Circulating Cell-free DNA as a Prognostic Biomarker in Patients with Advanced ALK+ Non–small Cell Lung Cancer in the Global Phase III ALEX Trial. Clin. Cancer Res. 2022, 28, 1800–1808. [Google Scholar] [CrossRef]
- Shien, K.; Papadimitrakopoulou, V.A.; Wistuba, I.I. Predictive biomarkers of response to PD-1/PD-L1 immune checkpoint inhibitors in non–small cell lung cancer. Lung Cancer 2016, 99, 79–87. [Google Scholar] [CrossRef] [PubMed]
- Havel, J.J.; Chowell, D.; Chan, T.A. The evolving landscape of biomarkers for checkpoint inhibitor immunotherapy. Nat. Cancer 2019, 19, 133–150. [Google Scholar] [CrossRef] [PubMed]
- Camidge, D.R.; Doebele, R.C.; Kerr, K.M. Comparing and contrasting predictive biomarkers for immunotherapy and targeted therapy of NSCLC. Nat. Rev. Clin. Oncol. 2019, 16, 341–355. [Google Scholar] [CrossRef]
- Shukuya, T.; Carbone, D.P. Predictive Markers for the Efficacy of Anti–PD-1/PD-L1 Antibodies in Lung Cancer. J. Thorac. Oncol. 2016, 11, 976–988. [Google Scholar] [CrossRef] [PubMed]
- McGranahan, N.; Furness, A.J.S.; Rosenthal, R.; Ramskov, S.; Lyngaa, R.; Saini, S.K.; Jamal-Hanjani, M.; Wilson, G.A.; Birkbak, N.J.; Hiley, C.T.; et al. Clonal neoantigens elicit T cell immunoreactivity and sensitivity to immune checkpoint blockade. Science 2016, 351, 1463–1469. [Google Scholar] [CrossRef] [PubMed]
- Ricciuti, B.; Jones, G.; Severgnini, M.; Alessi, J.V.; Recondo, G.; Lawrence, M.; Forshew, T.; Lydon, C.; Nishino, M.; Cheng, M.; et al. Early plasma circulating tumor DNA (ctDNA) changes predict response to first-line pembrolizumab-based therapy in non-small cell lung cancer (NSCLC). J. Immunother. Cancer 2021, 9, e001504. [Google Scholar] [CrossRef] [PubMed]
- Le, D.T.; Uram, J.N.; Wang, H.; Bartlett, B.R.; Kemberling, H.; Eyring, A.D.; Skora, A.D.; Luber, B.S.; Azad, N.S.; Laheru, D.; et al. PD-1 Blockade in Tumors with Mismatch-Repair Deficiency. N. Engl. J. Med. 2015, 372, 2509–2520. [Google Scholar] [CrossRef] [PubMed]
- Fang, W.; Ma, Y.; Yin, J.C.; Hong, S.; Zhou, H.; Wang, A.; Wang, F.; Bao, H.; Wu, X.; Yang, Y.; et al. Comprehensive Genomic Profiling Identifies Novel Genetic Predictors of Response to Anti–PD-(L)1 Therapies in Non–Small Cell Lung Cancer. Clin. Cancer Res. 2019, 25, 5015–5026. [Google Scholar] [CrossRef] [PubMed]
- Pu, X.; Wu, L.; Su, D.; Mao, W.; Fang, B. Immunotherapy for non-small cell lung cancers: Biomarkers for predicting responses and strategies to overcome resistance. BMC Cancer 2018, 18, 1082. [Google Scholar] [CrossRef] [PubMed]
- Cabel, L.; Riva, F.; Servois, V.; Livartowski, A.; Daniel, C.; Rampanou, A.; Lantz, O.; Romano, E.; Milder, M.; Buecher, B.; et al. Circulating tumor DNA changes for early monitoring of anti-PD1 immunotherapy: A proof-of-concept study. Ann. Oncol. 2017, 28, 1996–2001. [Google Scholar] [CrossRef] [PubMed]
- Wolchok, J.D.; Hoos, A.; O’Day, S.; Weber, J.S.; Hamid, O.; Lebbé, C.; Maio, M.; Binder, M.; Bohnsack, O.; Nichol, G.; et al. Guidelines for the Evaluation of Immune Therapy Activity in Solid Tumors: Immune-Related Response Criteria. Clin. Cancer Res. 2009, 15, 7412–7420. [Google Scholar] [CrossRef] [PubMed]
- Hoos, A.; Wolchok, J.D.; Humphrey, R.W.; Hodi, F.S. CCR 20th Anniversary Commentary: Immune-Related Response Criteria—Capturing Clinical Activity in Immuno-Oncology. Clin. Cancer Res. 2015, 21, 4989–4991. [Google Scholar] [CrossRef] [PubMed]
- Goldberg, S.B.; Narayan, A.; Kole, A.J.; Decker, R.H.; Teysir, J.; Carriero, N.J.; Lee, A.; Nemati, R.; Nath, S.K.; Mane, S.M.; et al. Early Assessment of Lung Cancer Immunotherapy Response via Circulating Tumor DNA. Clin. Cancer Res. 2018, 24, 1872–1880. [Google Scholar] [CrossRef]
- Raja, R.; Kuziora, M.; Brohawn, P.Z.; Higgs, B.W.; Gupta, A.; Dennis, P.A.; Ranade, K. Early Reduction in ctDNA Predicts Survival in Patients with Lung and Bladder Cancer Treated with Durvalumab. Clin. Cancer Res. 2018, 24, 6212–6222. [Google Scholar] [CrossRef] [PubMed]
- Garassino, M.C.; Cho, B.-C.; Kim, J.-H.; Mazières, J.; Vansteenkiste, J.; Lena, H.; Jaime, J.C.; Gray, J.E.; Powderly, J.; Chouaid, C.; et al. Durvalumab as third-line or later treatment for advanced non-small-cell lung cancer (ATLANTIC): An open-label, single-arm, phase 2 study. Lancet Oncol. 2018, 19, 521–536. [Google Scholar] [CrossRef]
- Zhang, Q.; Luo, J.; Wu, S.; Si, H.; Gao, C.; Xu, W.; Abdullah, S.E.; Higgs, B.W.; Dennis, P.A.; van der Heijden, M.S.; et al. Prognostic and Predictive Impact of Circulating Tumor DNA in Patients with Advanced Cancers Treated with Immune Checkpoint Blockade. Cancer Discov. 2020, 10, 1842–1853. [Google Scholar] [CrossRef] [PubMed]
- Anagnostou, V.; Forde, P.M.; White, J.R.; Niknafs, N.; Hruban, C.; Naidoo, J.; Marrone, K.; Sivakumar, I.A.; Bruhm, D.C.; Rosner, S.; et al. Dynamics of Tumor and Immune Responses during Immune Checkpoint Blockade in Non–Small Cell Lung Cancer. Cancer Res. 2019, 79, 1214–1225. [Google Scholar] [CrossRef] [PubMed]
- Thompson, J.C.; Carpenter, E.L.; Silva, B.A.; Rosenstein, J.; Chien, A.L.; Quinn, K.; Espenschied, C.R.; Mak, A.; Kiedrowski, L.A.; Lefterova, M.; et al. Serial Monitoring of Circulating Tumor DNA by Next-Generation Gene Sequencing as a Biomarker of Response and Survival in Patients With Advanced NSCLC Receiving Pembrolizumab-Based Therapy. JCO Precis. Oncol. 2021, 5, 510–524. [Google Scholar] [CrossRef] [PubMed]
- Nabet, B.Y.; Esfahani, M.S.; Moding, E.J.; Hamilton, E.G.; Chabon, J.J.; Rizvi, H.; Steen, C.B.; Chaudhuri, A.A.; Liu, C.L.; Hui, A.B.; et al. Noninvasive Early Identification of Therapeutic Benefit from Immune Checkpoint Inhibition. Cell 2020, 183, 363–376.e13. [Google Scholar] [CrossRef] [PubMed]
- Pellini, B.; Chaudhuri, A.A. Circulating Tumor DNA Minimal Residual Disease Detection of Non–Small-Cell Lung Cancer Treated With Curative Intent. J. Clin. Oncol. 2022, 40, 567–575. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.; Shields, M.D.; Chauhan, P.S.; Ramirez, R.J.; Harris, P.K.; Reimers, M.A.; Zevallos, J.P.; Davis, A.A.; Pellini, B.; Chaudhuri, A.A. Commercial ctDNA Assays for Minimal Residual Disease Detection of Solid Tumors. Mol. Diagn. Ther. 2021, 25, 757–774. [Google Scholar] [CrossRef] [PubMed]
- Tie, J.; Cohen, J.D.; Lahouel, K.; Lo, S.N.; Wang, Y.; Kosmider, S.; Wong, R.; Shapiro, J.; Lee, M.; Harris, S.; et al. Circulating Tumor DNA Analysis Guiding Adjuvant Therapy in Stage II Colon Cancer. N. Engl. J. Med. 2022, 386, 2261–2272. [Google Scholar] [CrossRef] [PubMed]
- Abbosh, C.; Frankell, A.; Garnett, A.; Harrison, T.; Weichert, M.; Licon, A.; Veeriah, S.; Daber, B.; Moreau, M.; Chesh, A.; et al. Abstract CT023: Phylogenetic tracking and minimal residual disease detection using ctDNA in early-stage NSCLC: A lung TRACERx study. Cancer Res. 2020, 80, CT023. [Google Scholar] [CrossRef]
- Chaudhuri, A.A.; Chabon, J.J.; Lovejoy, A.F.; Newman, A.M.; Stehr, H.; Azad, T.D.; Khodadoust, M.S.; Esfahani, M.S.; Liu, C.L.; Zhou, L.; et al. Early Detection of Molecular Residual Disease in Localized Lung Cancer by Circulating Tumor DNA Profiling. Cancer Discov. 2017, 7, 1394–1403. [Google Scholar] [CrossRef]
- Moding, E.J.; Liu, Y.; Nabet, B.Y.; Chabon, J.J.; Chaudhuri, A.A.; Hui, A.B.; Bonilla, R.F.; Ko, R.B.; Yoo, C.H.; Gojenola, L.; et al. Circulating tumor DNA dynamics predict benefit from consolidation immunotherapy in locally advanced non-small-cell lung cancer. Nat. Cancer 2020, 1, 176–183. [Google Scholar] [CrossRef]
- Chen, K.; Zhao, H.; Shi, Y.; Yang, F.; Wang, L.T.; Kang, G.; Nie, Y.; Wang, J. Perioperative Dynamic Changes in Circulating Tumor DNA in Patients with Lung Cancer (DYNAMIC). Clin. Cancer Res. 2019, 25, 7058–7067. [Google Scholar] [CrossRef]
- Zviran, A.; Schulman, R.C.; Shah, M.; Hill, S.T.K.; Deochand, S.; Khamnei, C.C.; Maloney, D.; Patel, K.; Liao, W.; Widman, A.J.; et al. Genome-wide cell-free DNA mutational integration enables ultra-sensitive cancer monitoring. Nat. Med. 2020, 26, 1114–1124. [Google Scholar] [CrossRef]
- Kurtz, D.M.; Chabon, J.J.; Sworder, B.; Keh, L.C.T.; Soo, J.; Alig, S.; Schultz, A.; Garofalo, A.; Hamilton, E.G.; Chen, B.; et al. Leveraging phased variants for personalized minimal residual disease detection in localized non-small cell lung cancer. J. Clin. Oncol. 2021, 39, 8518. [Google Scholar] [CrossRef]
- Xia, L.; Mei, J.; Kang, R.; Deng, S.; Chen, Y.; Yang, Y.; Feng, G.; Deng, Y.; Gan, F.; Lin, Y.; et al. Perioperative ctDNA-Based Molecular Residual Disease Detection for Non-Small Cell Lung Cancer: A Prospective Multicenter Cohort Study (LUNGCA-1). Clin. Cancer Res. 2022, 28, 3308–3317. [Google Scholar] [CrossRef]
- Waldeck, S.; Mitschke, J.; Wiesemann, S.; Rassner, M.; Andrieux, G.; Deuter, M.; Mutter, J.; Lüchtenborg, A.; Kottmann, D.; Titze, L.; et al. Early assessment of circulating tumor DNA after curative-intent resection predicts tumor recurrence in early-stage and locally advanced non-small-cell lung cancer. Mol. Oncol. 2021, 16, 527–537. [Google Scholar] [CrossRef] [PubMed]
- Kurtz, D.M.; Soo, J.; Keh, L.C.T.; Alig, S.; Chabon, J.J.; Sworder, B.J.; Schultz, A.; Jin, M.C.; Scherer, F.; Garofalo, A.; et al. Enhanced detection of minimal residual disease by targeted sequencing of phased variants in circulating tumor DNA. Nat. Biotechnol. 2021, 39, 1537–1547. [Google Scholar] [CrossRef]
- Schmitt, M.W.; Kennedy, S.R.; Salk, J.J.; Fox, E.J.; Hiatt, J.B.; Loeb, L.A. Detection of ultra-rare mutations by next-generation sequencing. Proc. Natl. Acad. Sci. USA 2012, 109, 14508–14513. [Google Scholar] [CrossRef]
- Pellini, B.; Pejovic, N.; Feng, W.; Earland, N.; Harris, P.K.; Usmani, A.; Szymanski, J.J.; Qaium, F.; Mudd, J.; Petty, M.; et al. ctDNA MRD Detection and Personalized Oncogenomic Analysis in Oligometastatic Colorectal Cancer From Plasma and Urine. JCO Precis. Oncol. 2021, 5, 378–388. [Google Scholar] [CrossRef] [PubMed]
- Phase III Study to Determine the Efficacy of Durvalumab in Combination with Chemotherapy in Completely Resected Stage II-III Non-small Cell Lung Cancer (NSCLC) (MERMAID-1). Available online: https://clinicaltrials.gov/ct2/show/NCT04385368?term=NCT04385368&draw=2&rank=1 (accessed on 12 June 2022).
- Phase III Study to Determine Efficacy of Durvalumab in Stage II-III Non-small Cell Lung Cancer (NSCLC) After Curative Intent Therapy. (MERMAID-2). Available online: https://clinicaltrials.gov/ct2/show/NCT04642469?term=NCT04642469&draw=2&rank=1 (accessed on 12 June 2022).
- Adjuvant Durvalumab for Early Stage NSCLC Patients with ctDNA Minimal Residual Disease. Available online: https://clinicaltrials.gov/ct2/show/NCT04585477?term=NCT04585477&draw=2&rank=1 (accessed on 12 June 2022).
- Personalized Escalation of Consolidation Treatment Following Chemoradiotherapy and Immunotherapy in Stage III NSCLC in Stage III NSCLC. Available online: https://clinicaltrials.gov/ct2/show/NCT04585490?term=NCT04585490&draw=2&rank=1 (accessed on 12 June 2022).
- National Lung Screening Trial Research Team; Aberle, D.R.; Adams, A.M.; Berg, C.D.; Black, W.C.; Clapp, J.D.; Fagerstrom, R.M.; Gareen, I.F.; Gatsonis, C.; Marcus, P.M.; et al. Reduced Lung-Cancer Mortality with Low-Dose Computed Tomographic Screening. N. Engl. J. Med. 2011, 365, 395–409. [Google Scholar] [PubMed]
- Chabon, J.J.; Hamilton, E.G.; Kurtz, D.M.; Esfahani, M.S.; Moding, E.J.; Stehr, H.; Schroers-Martin, J.; Nabet, B.Y.; Chen, B.; Chaudhuri, A.A.; et al. Integrating genomic features for non-invasive early lung cancer detection. Nature 2020, 580, 245–251. [Google Scholar] [CrossRef] [PubMed]
- Abbosh, C.; Birkbak, N.; Swanton, C. Early stage NSCLC—Challenges to implementing ctDNA-based screening and MRD detection. Nat. Rev. Clin. Oncol. 2018, 15, 577–586. [Google Scholar] [CrossRef]
- Hong, Y.; Kim, W.J. DNA methylation markers in lung cancer. Curr. Genom. 2021, 21, 79–87. [Google Scholar] [CrossRef]
- Qi, J.; Hong, B.; Tao, R.; Sun, R.; Zhang, H.; Zhang, X.; Ji, J.; Wang, S.; Liu, Y.; Deng, Q.; et al. Prediction model for malignant pulmonary nodules based on cfMeDIP-seq and machine learning. Cancer Sci. 2021, 112, 3918–3923. [Google Scholar] [CrossRef]
- Liu, M.C.; Oxnard, G.R.; Klein, E.A.; Swanton, C.; Seiden, M.V. Sensitive and specific multi-cancer detection and localization using methylation signatures in cell-free DNA. Ann. Oncol. 2020, 31, 745–759. [Google Scholar] [CrossRef]
- The SUMMIT Study: A Cancer Screening Study (SUMMIT). Available online: https://clinicaltrials.gov/ct2/show/NCT03934866?term=NCT03934866&draw=2&rank=1 (accessed on 12 June 2022).
- Johns Hopkins Researchers Design New Blood Test That Uses DNA ‘Packaging’ Patterns to Detect Multiple Cancer Types. 2019. Available online: https://www.hopkinsmedicine.org/news/newsroom/news-releases/johns-hopkins-researchers-design-new-blood-test-that-uses-dna-packaging-patterns-to-detect-multiple-cancer-types (accessed on 1 August 2022).
- DNA Evaluation of Fragments for Early Interception—Lung Cancer Training Study (DELFI-L101 Study) (DELFI-L101). Available online: https://clinicaltrials.gov/ct2/show/NCT04825834?term=NCT04825834&draw=2&rank=1 (accessed on 12 June 2022).


| Trial (NCT #) | Primary Treatment | Stage | Estimated Enrollment | Recruitment Status | ctDNA Timepoint | ctDNA (+) Intervention | ctDNA (−) Intervention | Phase | Primary Endpoint | Type of Assay |
|---|---|---|---|---|---|---|---|---|---|---|
| SCION (NCT04944173) | SBRT+ 4 cycles of durvalumab | T1-2 N0 M0 | 94 | Not yet recruiting | After SBRT + 4 cycles durvalumab | Additional 8 cycles of durvalumab | No further treatment | II | ORR at 18 months | Avenio |
| NCT04585490 | CRT | III | 48 | Recruiting | After CRT | Four cycles of platinum doublet chemo + durvalumab (1500 mg IV every 21 days, for 1 year) | SoC durvalumab (10 mg/kg every 2 weeks, or equivalent, for 1 year) | III | Change in ctDNA level following chemo | Avenio |
| NCT04585477 | Surgery or definitive SBRT | I–III | 80 | Recruiting | After surgery or SBRT | Twelve cycles of durvalumab | SoC and no treatment | II | Decrease in ctDNA level | Avenio |
| MERMAID-1 (NCT04385368) | Resection of primary NSCLC | II–III | 332 | Active, not recruiting | After surgery | Durvalumab + SoC chemo vs. placebo + SoC chemo | N/A | III | DFS in MRD+ analysis set | ArcherDx |
| MERMAID-2 (NCT04642469) | Surgery +/− neoadjuvant or adjuvant Tx | II–III | 284 | Active, not recruiting | After surgery | Durvalumab vs. placebo | N/A | III | DFS in the PD-L1 TC ≥ 1% analysis set | ArcherDx |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shields, M.D.; Chen, K.; Dutcher, G.; Patel, I.; Pellini, B. Making the Rounds: Exploring the Role of Circulating Tumor DNA (ctDNA) in Non-Small Cell Lung Cancer. Int. J. Mol. Sci. 2022, 23, 9006. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23169006
Shields MD, Chen K, Dutcher G, Patel I, Pellini B. Making the Rounds: Exploring the Role of Circulating Tumor DNA (ctDNA) in Non-Small Cell Lung Cancer. International Journal of Molecular Sciences. 2022; 23(16):9006. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23169006
Chicago/Turabian StyleShields, Misty Dawn, Kevin Chen, Giselle Dutcher, Ishika Patel, and Bruna Pellini. 2022. "Making the Rounds: Exploring the Role of Circulating Tumor DNA (ctDNA) in Non-Small Cell Lung Cancer" International Journal of Molecular Sciences 23, no. 16: 9006. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23169006