
Inhibition of Vps34 and p110δ PI3K Impairs Migration, Invasion and Three-Dimensional Spheroid Growth in Breast Cancer Cells


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Analysis of Vps34 and p110 δ Gene Expression
2.2. Effect of IC87114 and Vps34-IN1 on MCF-7 Cell Migration and Proliferation
2.3. Effect of p110δ and Vps34 Inhibitors on MDA-MB-231 Cell Migration, Invasion, and Proliferation
2.4. IC87114 and Vsp34-IN1 Impair the Growth of MCF-7- and MDA-MB-231-Derived Spheroids
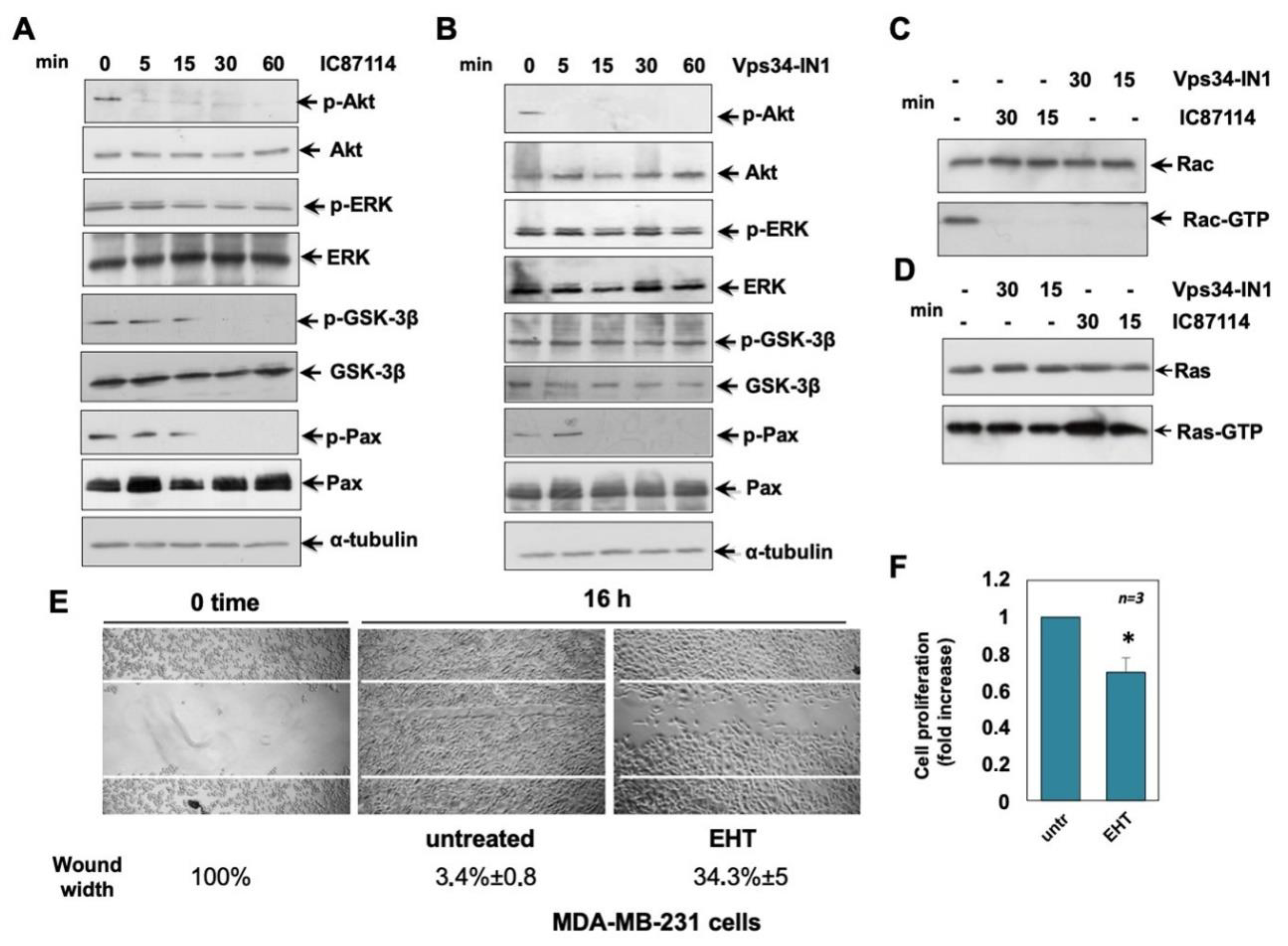
2.5. Effects of IC87114 and Vsp34-IN1 on MCF-7 Cell Signalling Pathways
2.6. Effects of IC87114 and Vsp34-IN1 on MDA-MB-231 Cell Signalling Pathways
3. Discussion
4. Materials and Methods
4.1. Chemicals, Reagents, and Constructs
4.2. Cell Culture
4.3. DNA Synthesis and WST-1 Assay
4.4. Wound Scratch and Boyden’s Chambers Migration Assays
4.5. D Culture
4.6. Protein Lysates, Rac and Ras Assays, and Western Blot
4.7. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: Globocan estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Di Donato, M.; Giovannelli, P.; Cernera, G.; Di Santi, A.; Marino, I.; Bilancio, A.; Galasso, G.; Auricchio, F.; Migliaccio, A.; Castoria, G. Non-genomic androgen action regulates proliferative/migratory signaling in stromal cells. Front. Endocrinol. 2014, 5, 225. [Google Scholar] [CrossRef] [PubMed]
- Giovannelli, P.; Di Donato, M.; Galasso, G.; Di Zazzo, E.; Medici, N.; Bilancio, A.; Migliaccio, A.; Castoria, G. Breast cancer stem cells: The role of sex steroid receptors. World J. Stem Cells 2019, 11, 594–603. [Google Scholar] [CrossRef] [PubMed]
- Migliaccio, A.; Castoria, G.; Bilancio, A.; Giovannelli, P.; Di Donato, M.; Auricchio, F. Non-genomic Action of Steroid Hormones: More Questions than Answers. In Advances in Rapid Sex-Steroid Action: New Challenges and New Chances in Breast and Prostate Cancers; Castoria, G., Migliaccio, A., Eds.; Springer: New York, NY, USA, 2012; pp. 1–15. [Google Scholar]
- Belachew, E.B.; Sewasew, D.T. Molecular Mechanisms of Endocrine Resistance in Estrogen-Positive Breast Cancer. Front. Endocrinol. 2021, 12, 599586. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Becerra, R.; Santos, N.; Diaz, L.; Camacho, J. Mechanisms of resistance to endocrine therapy in breast cancer: Focus on signaling pathways, miRNAs and genetically based resistance. Int. J. Mol. Sci. 2012, 14, 108–145. [Google Scholar] [CrossRef]
- Gururaj, A.E.; Rayala, S.K.; Vadlamudi, R.K.; Kumar, R. Novel mechanisms of resistance to endocrine therapy: Genomic and nongenomic considerations. Clin. Cancer Res. 2006, 12, 1001s–1007s. [Google Scholar] [CrossRef]
- Ring, A.; Dowsett, M. Mechanisms of tamoxifen resistance. Endocr. Relat. Cancer 2004, 11, 643–658. [Google Scholar] [CrossRef] [PubMed]
- Castoria, G.; Migliaccio, A.; D’Amato, L.; Di Stasio, R.; Ciociola, A.; Lombardi, M.; Bilancio, A.; Di Domenico, M.; de Falco, A.; Auricchio, F. Integrating signals between cAMP and MAPK pathways in breast cancer. Front. Biosci. 2008, 13, 1318–1327. [Google Scholar] [CrossRef] [PubMed]
- Bilancio, A.; Migliaccio, A. Phosphoinositide 3-kinase assay in breast cancer cell extracts. Methods Mol. Biol. 2014, 1204, 145–153. [Google Scholar] [CrossRef]
- Miricescu, D.; Totan, A.; Stanescu, S., II.; Badoiu, S.C.; Stefani, C.; Greabu, M. PI3K/AKT/mTOR Signaling Pathway in Breast Cancer: From Molecular Landscape to Clinical Aspects. Int. J. Mol. Sci. 2020, 22, 173. [Google Scholar] [CrossRef] [PubMed]
- Lehmann, B.D.; Bauer, J.A.; Chen, X.; Sanders, M.E.; Chakravarthy, A.B.; Shyr, Y.; Pietenpol, J.A. Identification of human triple-negative breast cancer subtypes and preclinical models for selection of targeted therapies. J. Clin. Investig. 2011, 121, 2750–2767. [Google Scholar] [CrossRef] [PubMed]
- Giovannelli, P.; Di Donato, M.; Galasso, G.; Di Zazzo, E.; Bilancio, A.; Migliaccio, A. The Androgen Receptor in Breast Cancer. Front. Endocrinol. 2018, 9, 492. [Google Scholar] [CrossRef] [PubMed]
- Marra, A.; Trapani, D.; Viale, G.; Criscitiello, C.; Curigliano, G. Practical classification of triple-negative breast cancer: Intratumoral heterogeneity, mechanisms of drug resistance, and novel therapies. NPJ Breast Cancer 2020, 6, 54. [Google Scholar] [CrossRef] [PubMed]
- Uprety, D.; Adjei, A.A. KRAS: From undruggable to a druggable Cancer Target. Cancer Treat. Rev. 2020, 89, 102070. [Google Scholar] [CrossRef]
- Zimmer, A.S.; Gillard, M.; Lipkowitz, S.; Lee, J.M. Update on PARP Inhibitors in Breast Cancer. Curr. Treat. Options Oncol. 2018, 19, 21. [Google Scholar] [CrossRef] [PubMed]
- Doebele, R.C.; Drilon, A.; Paz-Ares, L.; Siena, S.; Shaw, A.T.; Farago, A.F.; Blakely, C.M.; Seto, T.; Cho, B.C.; Tosi, D.; et al. Entrectinib in patients with advanced or metastatic NTRK fusion-positive solid tumours: Integrated analysis of three phase 1-2 trials. Lancet Oncol. 2020, 21, 271–282. [Google Scholar] [CrossRef]
- Loibl, S.; Poortmans, P.; Morrow, M.; Denkert, C.; Curigliano, G. Breast cancer. Lancet 2021, 397, 1750–1769. [Google Scholar] [CrossRef]
- Bilanges, B.; Posor, Y.; Vanhaesebroeck, B. PI3K isoforms in cell signalling and vesicle trafficking. Nat. Rev. Mol. Cell Biol. 2019, 20, 515–534. [Google Scholar] [CrossRef]
- Vanhaesebroeck, B.; Burke, J.E.; Madsen, R.R. Precision Targeting of Mutant PI3Kα in Cancer by Selective Degradation. Cancer Discov. 2022, 12, 20–22. [Google Scholar] [CrossRef]
- Vanhaesebroeck, B.; Perry, M.W.D.; Brown, J.R.; André, F.; Okkenhaug, K. PI3K inhibitors are finally coming of age. Nat. Rev. Drug Discov. 2021, 20, 741–769. [Google Scholar] [CrossRef] [PubMed]
- Bilancio, A.; Okkenhaug, K.; Camps, M.; Emery, J.L.; Ruckle, T.; Rommel, C.; Vanhaesebroeck, B. Key role of the p110delta isoform of PI3K in B-cell antigen and IL-4 receptor signaling: Comparative analysis of genetic and pharmacologic interference with p110delta function in B cells. Blood 2006, 107, 642–650. [Google Scholar] [CrossRef] [PubMed]
- Bilancio, A.; Rinaldi, B.; Oliviero, M.A.; Donniacuo, M.; Monti, M.G.; Boscaino, A.; Marino, I.; Friedman, L.; Rossi, F.; Vanhaesebroeck, B.; et al. Inhibition of p110δ PI3K prevents inflammatory response and restenosis after artery injury. Biosci. Rep. 2017, 37, BSR20171112. [Google Scholar] [CrossRef] [PubMed]
- Bilanges, B.; Vanhaesebroeck, B. Cinderella finds her shoe: The first Vps34 inhibitor uncovers a new PI3K-AGC protein kinase connection. Biochem. J. 2014, 464, e7–e10. [Google Scholar] [CrossRef]
- Hu, D.X.; Patel, S.; Chen, H.; Wang, S.; Staben, S.T.; Dimitrova, Y.N.; Wallweber, H.A.; Lee, J.Y.; Chan, G.K.Y.; Sneeringer, C.J.; et al. Structure-Based Design of Potent, Selective, and Orally Bioavailable VPS34 Kinase Inhibitors. J. Med. Chem. 2021. [Google Scholar] [CrossRef] [PubMed]
- Vanhaesebroeck, B.; Leevers, S.J.; Ahmadi, K.; Timms, J.; Katso, R.; Driscoll, P.C.; Woscholski, R.; Parker, P.J.; Waterfield, M.D. Synthesis and function of 3-phosphorylated inositol lipids. Annu. Rev. Biochem. 2001, 70, 535–602. [Google Scholar] [CrossRef] [PubMed]
- Papakonstanti, E.A.; Zwaenepoel, O.; Bilancio, A.; Burns, E.; Nock, G.E.; Houseman, B.; Shokat, K.; Ridley, A.J.; Vanhaesebroeck, B. Distinct roles of class IA PI3K isoforms in primary and immortalised macrophages. J. Cell Sci. 2008, 121, 4124–4133. [Google Scholar] [CrossRef] [PubMed]
- Shutes, A.; Onesto, C.; Picard, V.; Leblond, B.; Schweighoffer, F.; Der, C.J. Specificity and mechanism of action of EHT 1864, a novel small molecule inhibitor of Rac family small GTPases. J. Biol. Chem. 2007, 282, 35666–35678. [Google Scholar] [CrossRef] [PubMed]
- Castoria, G.; Lombardi, M.; Barone, M.V.; Bilancio, A.; Di Domenico, M.; Bottero, D.; Vitale, F.; Migliaccio, A.; Auricchio, F. Androgen-stimulated DNA synthesis and cytoskeletal changes in fibroblasts by a nontranscriptional receptor action. J. Cell. Biol. 2003, 161, 547–556. [Google Scholar] [CrossRef]
- Dong, C.; Wu, J.; Chen, Y.; Nie, J.; Chen, C. Activation of PI3K/AKT/mTOR Pathway Causes Drug Resistance in Breast Cancer. Front. Pharmacol. 2021, 12, 628690. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.J.; Loh, K.; Yap, Y.S. PI3K/Akt/mTOR inhibitors in breast cancer. Cancer Biol. Med. 2015, 12, 342–354. [Google Scholar] [CrossRef] [PubMed]
- Fares, J.; Fares, M.Y.; Khachfe, H.H.; Salhab, H.A.; Fares, Y. Molecular principles of metastasis: A hallmark of cancer revisited. Signal Transduct. Target. 2020, 5, 28. [Google Scholar] [CrossRef] [PubMed]
- Nunnery, S.E.; Mayer, I.A. Targeting the PI3K/AKT/mTOR Pathway in Hormone-Positive Breast Cancer. Drugs 2020, 80, 1685–1697. [Google Scholar] [CrossRef]
- Peng, Y.; Wang, Y.; Zhou, C.; Mei, W.; Zeng, C. PI3K/Akt/mTOR Pathway and Its Role in Cancer Therapeutics: Are We Making Headway? Front. Oncol. 2022, 12, 819128. [Google Scholar] [CrossRef] [PubMed]
- Skolariki, A.; D’Costa, J.; Little, M.; Lord, S. Role of PI3K/Akt/mTOR pathway in mediating endocrine resistance: Concept to clinic. Explor. Target. Anti Tumor Ther. 2022, 3, 172–199. [Google Scholar] [CrossRef]
- Ali, K.; Bilancio, A.; Thomas, M.; Pearce, W.; Gilfillan, A.M.; Tkaczyk, C.; Kuehn, N.; Gray, A.; Giddings, J.; Peskett, E.; et al. Essential role for the p110delta phosphoinositide 3-kinase in the allergic response. Nature 2004, 431, 1007–1011. [Google Scholar] [CrossRef] [PubMed]
- Okkenhaug, K.; Bilancio, A.; Farjot, G.; Priddle, H.; Sancho, S.; Peskett, E.; Pearce, W.; Meek, S.E.; Salpekar, A.; Waterfield, M.D.; et al. Impaired B and T cell antigen receptor signaling in p110delta PI 3-kinase mutant mice. Science 2002, 297, 1031–1034. [Google Scholar] [CrossRef]
- González-Cao, M.; Rodón, J.; Karachaliou, N.; Sánchez, J.; Santarpia, M.; Viteri, S.; Pilotto, S.; Teixidó, C.; Riso, A.; Rosell, R. Other targeted drugs in melanoma. Ann. Transl. Med. 2015, 3, 266. [Google Scholar] [CrossRef] [PubMed]
- Sawyer, C.; Sturge, J.; Bennett, D.C.; O’Hare, M.J.; Allen, W.E.; Bain, J.; Jones, G.E.; Vanhaesebroeck, B. Regulation of breast cancer cell chemotaxis by the phosphoinositide 3-kinase p110delta. Cancer Res. 2003, 63, 1667–1675. [Google Scholar] [PubMed]
- Goulielmaki, E.; Bermudez-Brito, M.; Andreou, M.; Tzenaki, N.; Tzardi, M.; de Bree, E.; Tsentelierou, E.; Makrigiannakis, A.; Papakonstanti, E.A. Pharmacological inactivation of the PI3K p110δ prevents breast tumour progression by targeting cancer cells and macrophages. Cell Death Dis. 2018, 9, 678. [Google Scholar] [CrossRef] [PubMed]
- Ali, K.; Soond, D.R.; Pineiro, R.; Hagemann, T.; Pearce, W.; Lim, E.L.; Bouabe, H.; Scudamore, C.L.; Hancox, T.; Maecker, H.; et al. Inactivation of PI(3)K p110δ breaks regulatory T-cell-mediated immune tolerance to cancer. Nature 2014, 510, 407–411. [Google Scholar] [CrossRef]
- Lauder, S.N.; Vanhaesebroeck, B.; Gallimore, A. Sequential targeting of PI3Kδ and LAG3 as an effective anti-cancer approach. Br. J. Cancer 2021, 125, 467–469. [Google Scholar] [CrossRef]
- Xenou, L.; Papakonstanti, E.A. p110δ PI3K as a therapeutic target of solid tumours. Clin. Sci. 2020, 134, 1377–1397. [Google Scholar] [CrossRef]
- Lim, J.; Murthy, A. Targeting Autophagy to Treat Cancer: Challenges and Opportunities. Front. Pharm. 2020, 11, 590344. [Google Scholar] [CrossRef] [PubMed]
- Bilanges, B.; Alliouachene, S.; Pearce, W.; Morelli, D.; Szabadkai, G.; Chung, Y.L.; Chicanne, G.; Valet, C.; Hill, J.M.; Voshol, P.J.; et al. Vps34 PI 3-kinase inactivation enhances insulin sensitivity through reprogramming of mitochondrial metabolism. Nat. Commun. 2017, 8, 1804. [Google Scholar] [CrossRef]
- Liang, J.; Xu, Z.X.; Ding, Z.; Lu, Y.; Yu, Q.; Werle, K.D.; Zhou, G.; Park, Y.Y.; Peng, G.; Gambello, M.J.; et al. Myristoylation confers noncanonical AMPK functions in autophagy selectivity and mitochondrial surveillance. Nat. Commun. 2015, 6, 7926. [Google Scholar] [CrossRef] [PubMed]
- Dyczynski, M.; Yu, Y.; Otrocka, M.; Parpal, S.; Braga, T.; Henley, A.B.; Zazzi, H.; Lerner, M.; Wennerberg, K.; Viklund, J.; et al. Targeting autophagy by small molecule inhibitors of vacuolar protein sorting 34 (Vps34) improves the sensitivity of breast cancer cells to Sunitinib. Cancer Lett. 2018, 435, 32–43. [Google Scholar] [CrossRef]
- Elgebaly, A.; Menshawy, A.; El Ashal, G.; Osama, O.; Ghanem, E.; Omar, A.; Negida, A. Sunitinib alone or in combination with chemotherapy for the treatment of advanced breast cancer: A systematic review and meta-analysis. Breast Dis. 2016, 36, 91–101. [Google Scholar] [CrossRef]
- Noman, M.Z.; Parpal, S.; Van Moer, K.; Xiao, M.; Yu, Y.; Viklund, J.; De Milito, A.; Hasmim, M.; Andersson, M.; Amaravadi, R.K.; et al. Inhibition of Vps34 reprograms cold into hot inflamed tumors and improves anti-PD-1/PD-L1 immunotherapy. Sci. Adv. 2020, 6, eaax7881. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.; Bao, Y.; Liu, H.; Kou, X.; Zhang, Z.; Sun, F.; Qian, Z.; Lin, Z.; Li, X.; Liu, X.; et al. VPS34 stimulation of p62 phosphorylation for cancer progression. Oncogene 2017, 36, 6850–6862. [Google Scholar] [CrossRef] [PubMed]
- Meunier, G.; Birsen, R.; Cazelles, C.; Belhadj, M.; Cantero-Aguilar, L.; Kosmider, O.; Fontenay, M.; Azar, N.; Mayeux, P.; Chapuis, N.; et al. Antileukemic activity of the VPS34-IN1 inhibitor in acute myeloid leukemia. Oncogenesis 2020, 9, 94. [Google Scholar] [CrossRef]
- Di Donato, M.; Galasso, G.; Giovannelli, P.; Sinisi, A.A.; Migliaccio, A.; Castoria, G. Targeting the Nerve Growth Factor Signaling Impairs the Proliferative and Migratory Phenotype of Triple-Negative Breast Cancer Cells. Front. Cell Dev. Biol. 2021, 9, 676568. [Google Scholar] [CrossRef] [PubMed]
- Di Donato, M.; Bilancio, A.; D’Amato, L.; Claudiani, P.; Oliviero, M.A.; Barone, M.V.; Auricchio, A.; Appella, E.; Migliaccio, A.; Auricchio, F.; et al. Cross-talk between androgen receptor/filamin A and TrkA regulates neurite outgrowth in PC12 cells. Mol. Biol. Cell 2015, 26, 2858–2872. [Google Scholar] [CrossRef] [PubMed]
- Giovannelli, P.; Di Donato, M.; Auricchio, F.; Castoria, G.; Migliaccio, A. Androgens Induce Invasiveness of Triple Negative Breast Cancer Cells Through AR/Src/PI3-K Complex Assembly. Sci. Rep. 2019, 9, 4490. [Google Scholar] [CrossRef] [PubMed]
- Di Donato, M.; Cernera, G.; Migliaccio, A.; Castoria, G. Nerve growth factor induces proliferation and aggressiveness in prostate cancer cells. Cancers 2019, 11, 784. [Google Scholar] [CrossRef] [PubMed]
- Bilancio, A.; Bontempo, P.; Di Donato, M.; Conte, M.; Giovannelli, P.; Altucci, L.; Migliaccio, A.; Castoria, G. Bisphenol A induces cell cycle arrest in primary and prostate cancer cells through EGFR/ERK/p53 signaling pathway activation. Oncotarget 2017, 8, 115620–115631. [Google Scholar] [CrossRef]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Di Donato, M.; Giovannelli, P.; Migliaccio, A.; Bilancio, A. Inhibition of Vps34 and p110δ PI3K Impairs Migration, Invasion and Three-Dimensional Spheroid Growth in Breast Cancer Cells. Int. J. Mol. Sci. 2022, 23, 9008. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23169008
Di Donato M, Giovannelli P, Migliaccio A, Bilancio A. Inhibition of Vps34 and p110δ PI3K Impairs Migration, Invasion and Three-Dimensional Spheroid Growth in Breast Cancer Cells. International Journal of Molecular Sciences. 2022; 23(16):9008. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23169008
Chicago/Turabian StyleDi Donato, Marzia, Pia Giovannelli, Antimo Migliaccio, and Antonio Bilancio. 2022. "Inhibition of Vps34 and p110δ PI3K Impairs Migration, Invasion and Three-Dimensional Spheroid Growth in Breast Cancer Cells" International Journal of Molecular Sciences 23, no. 16: 9008. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23169008