
Design, Synthesis, Molecular Modeling and Anti-Hyperglycemic Evaluation of Quinazoline-Sulfonylurea Hybrids as Peroxisome Proliferator-Activated Receptor Gamma (PPARγ) and Sulfonylurea Receptor (SUR) Agonists

 ,
, 
Abstract
:1. Introduction
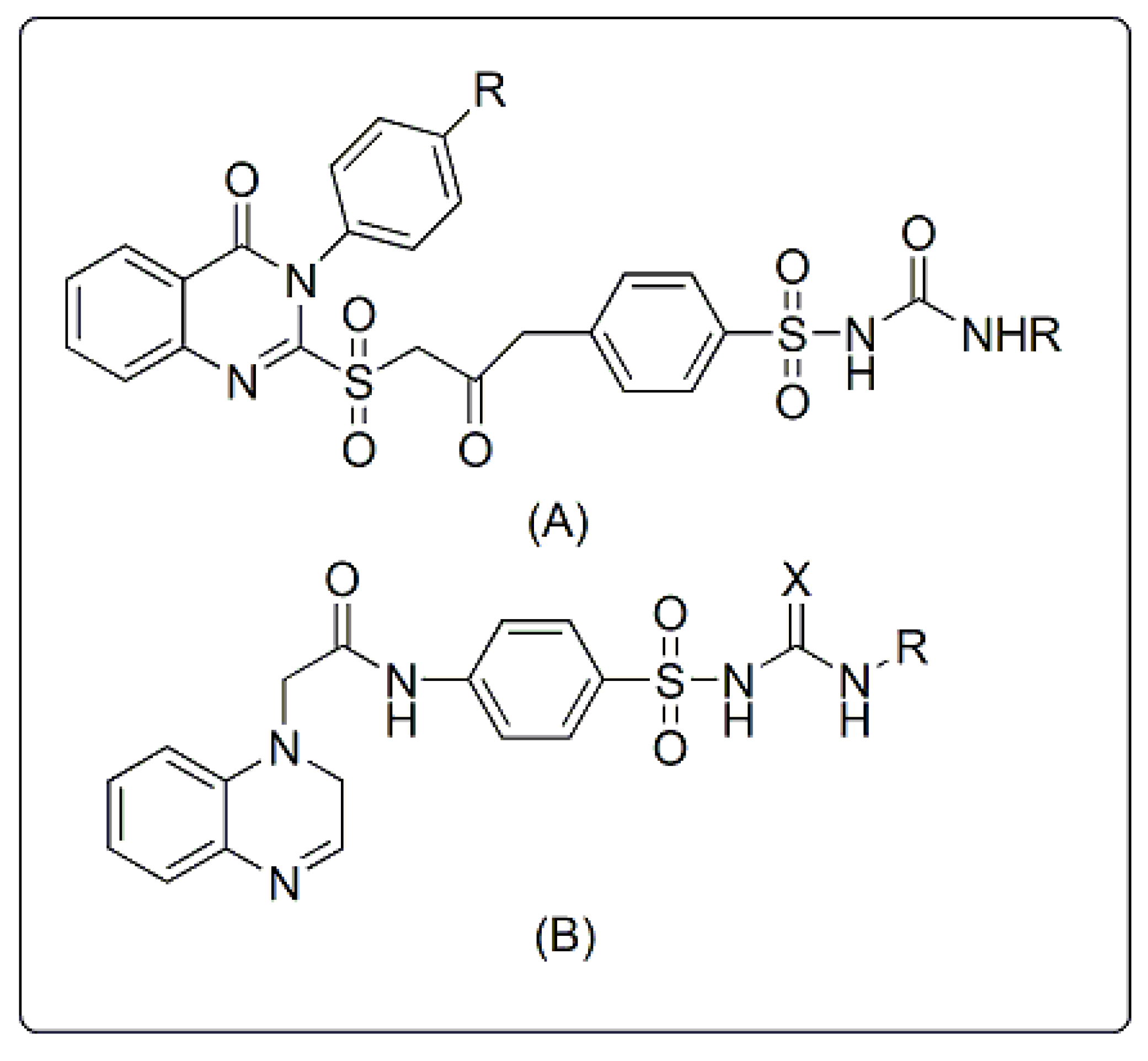
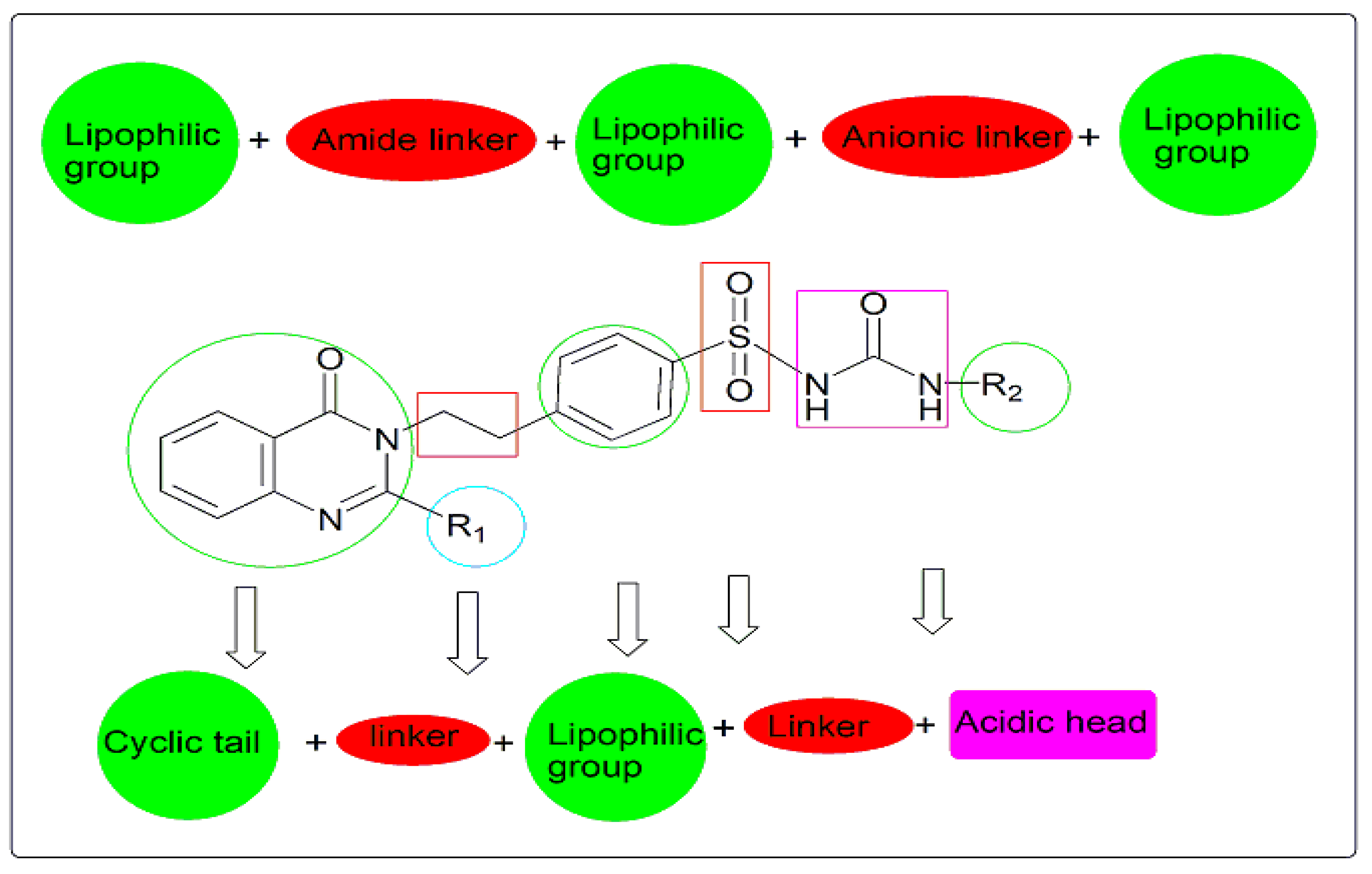
Rationale
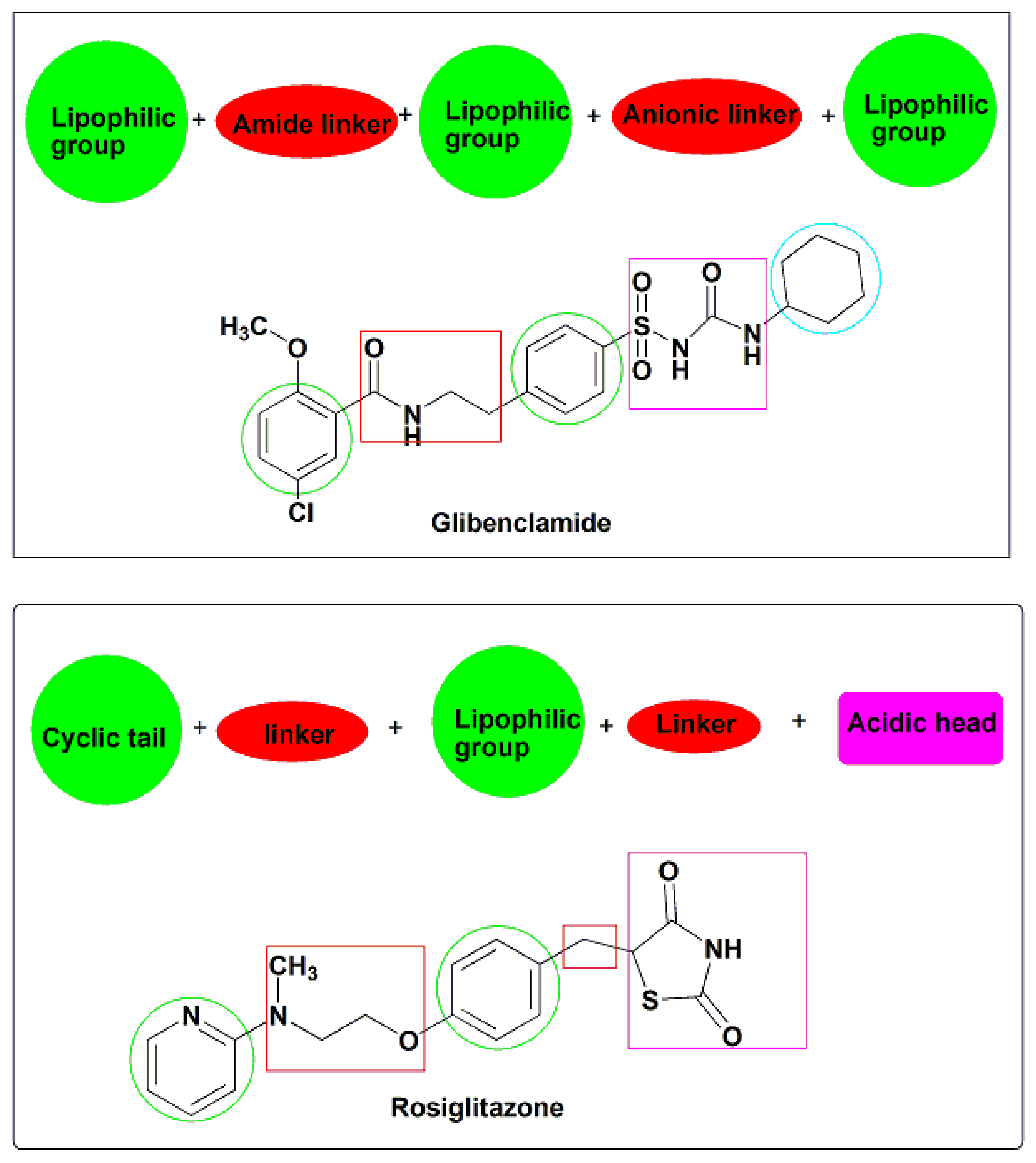
- Quinazolinone nucleus as a hydrophobic domain
- Aromatic sulfonylurea or sulphonamide group miming sulfonylurea derivatives
- Different attached hydrophobic groups with different electronic environment to study and compare the difference in their biological activities.
2. Results and Discussion
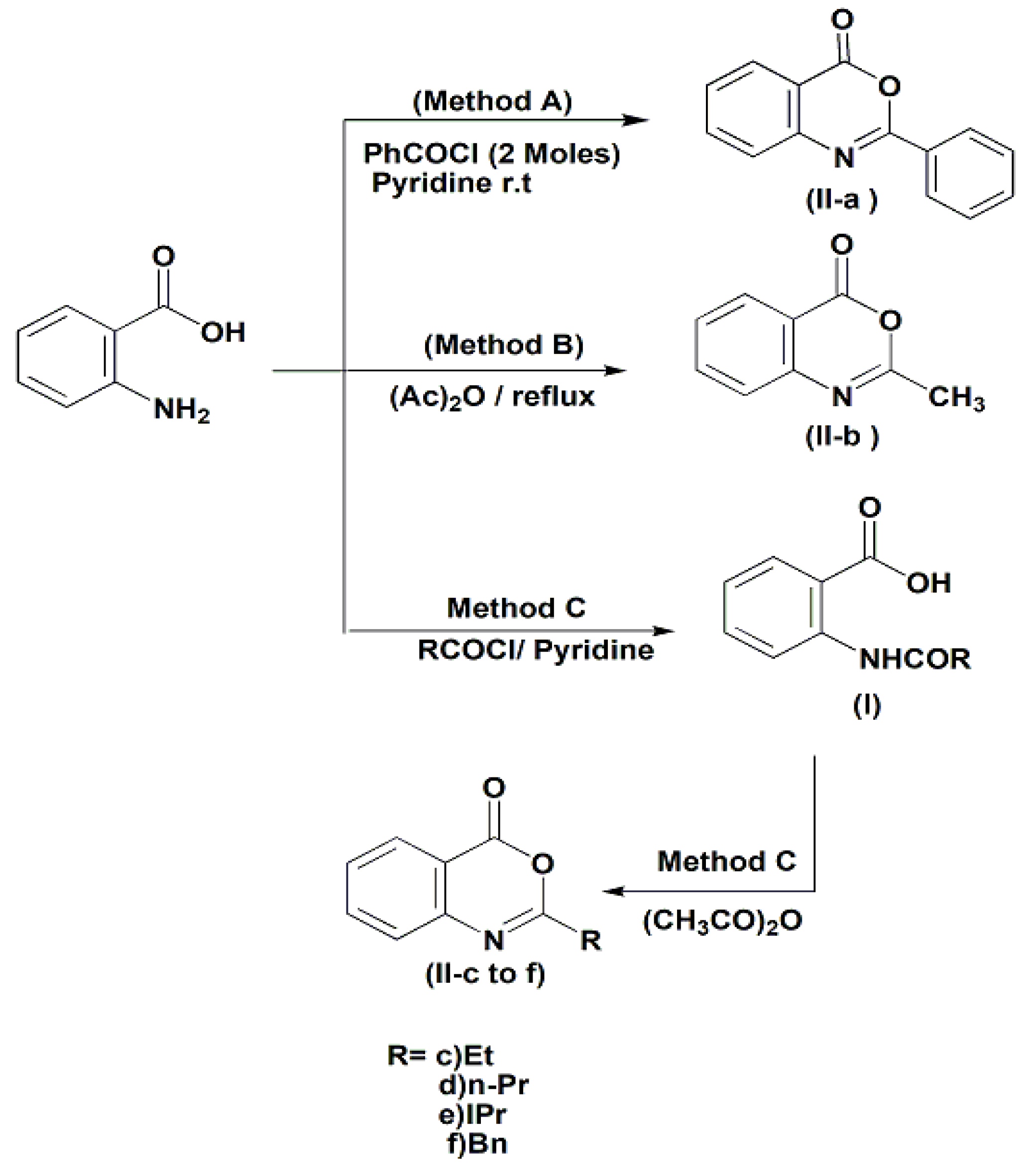
2.1. Chemistry
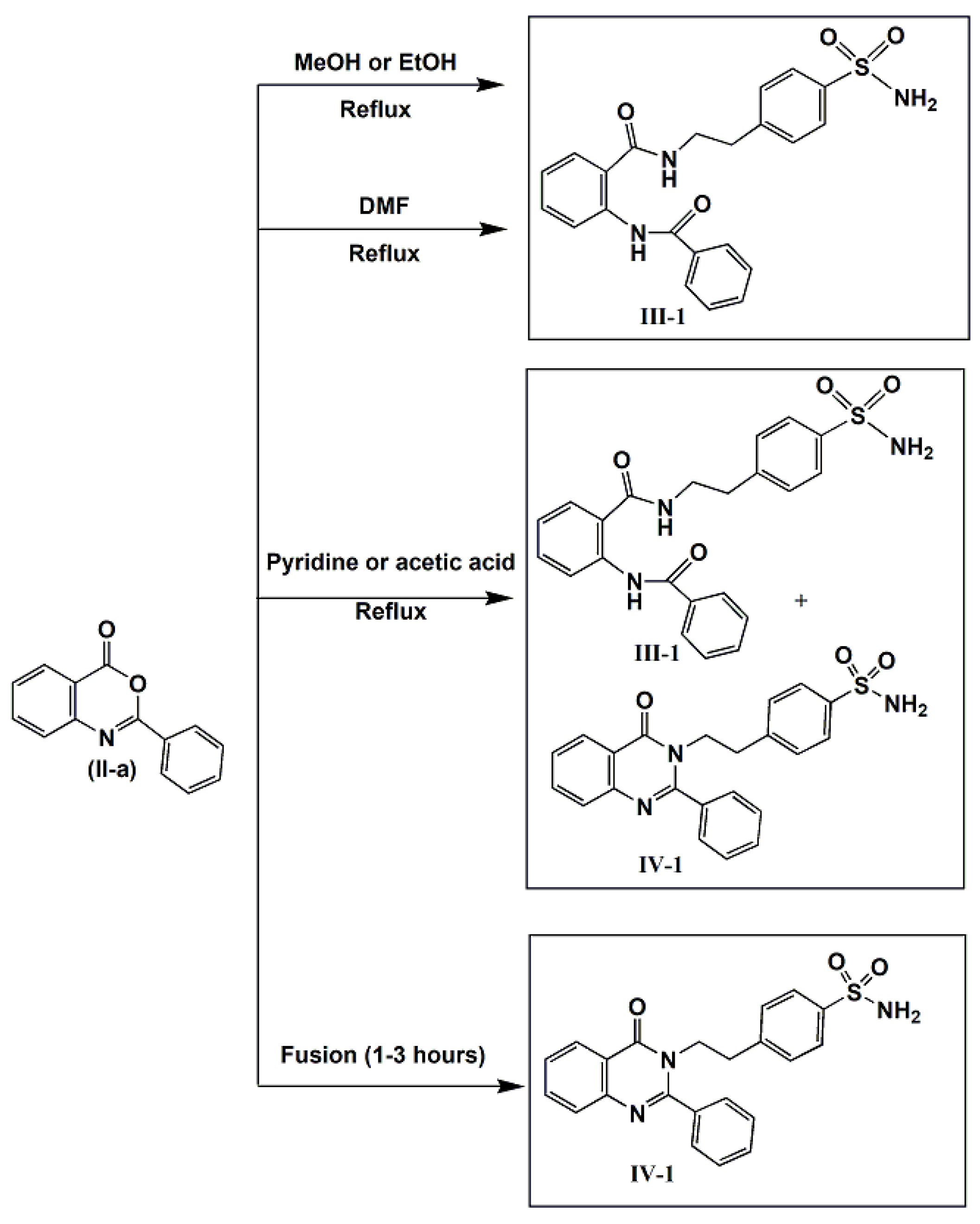
- (1)
- Overnight reflux in methanol or ethanol or DMF afforded the corresponding open structure 2-benzamido-N-(p-aminosulfonyl]phenylethyl)-benzamide III-1 with trace amount of the corresponding cyclic structure IV-1.
- (2)
- Refluxing in dry pyridine for 4 h afforded a mixture of both the open and cyclic intermediates III-1 and IV-1, respectively, where the cyclic intermediate was the major.
- (3)
- Overnight reflux in glacial acetic acid afforded the cyclic IV-1 as a major product.
- (4)
- Fusion for two hours at appropriate temperature gave the target cyclic intermediate IV-1 in best yields in comparison with other cyclization procedures.
2.2. Biology
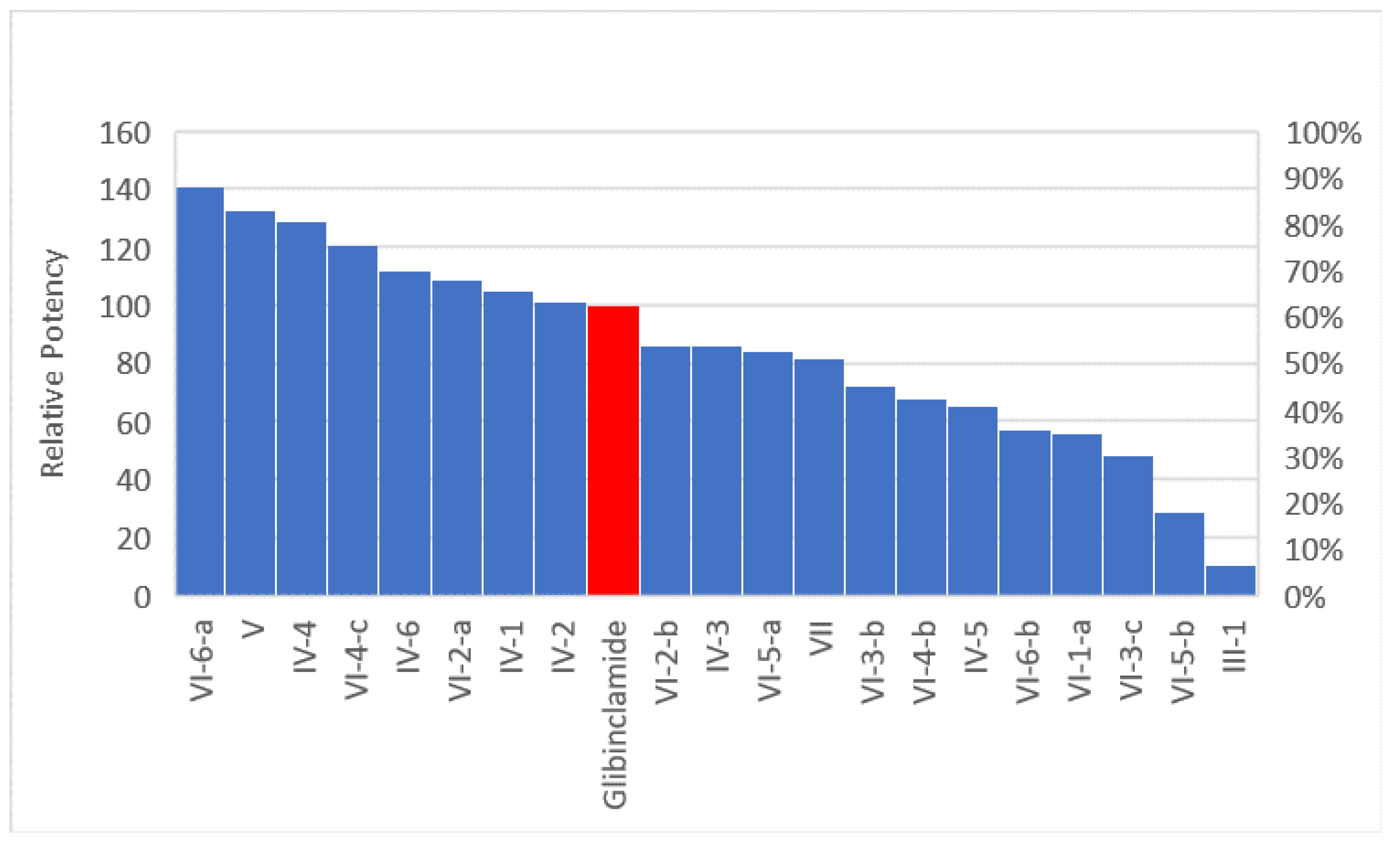
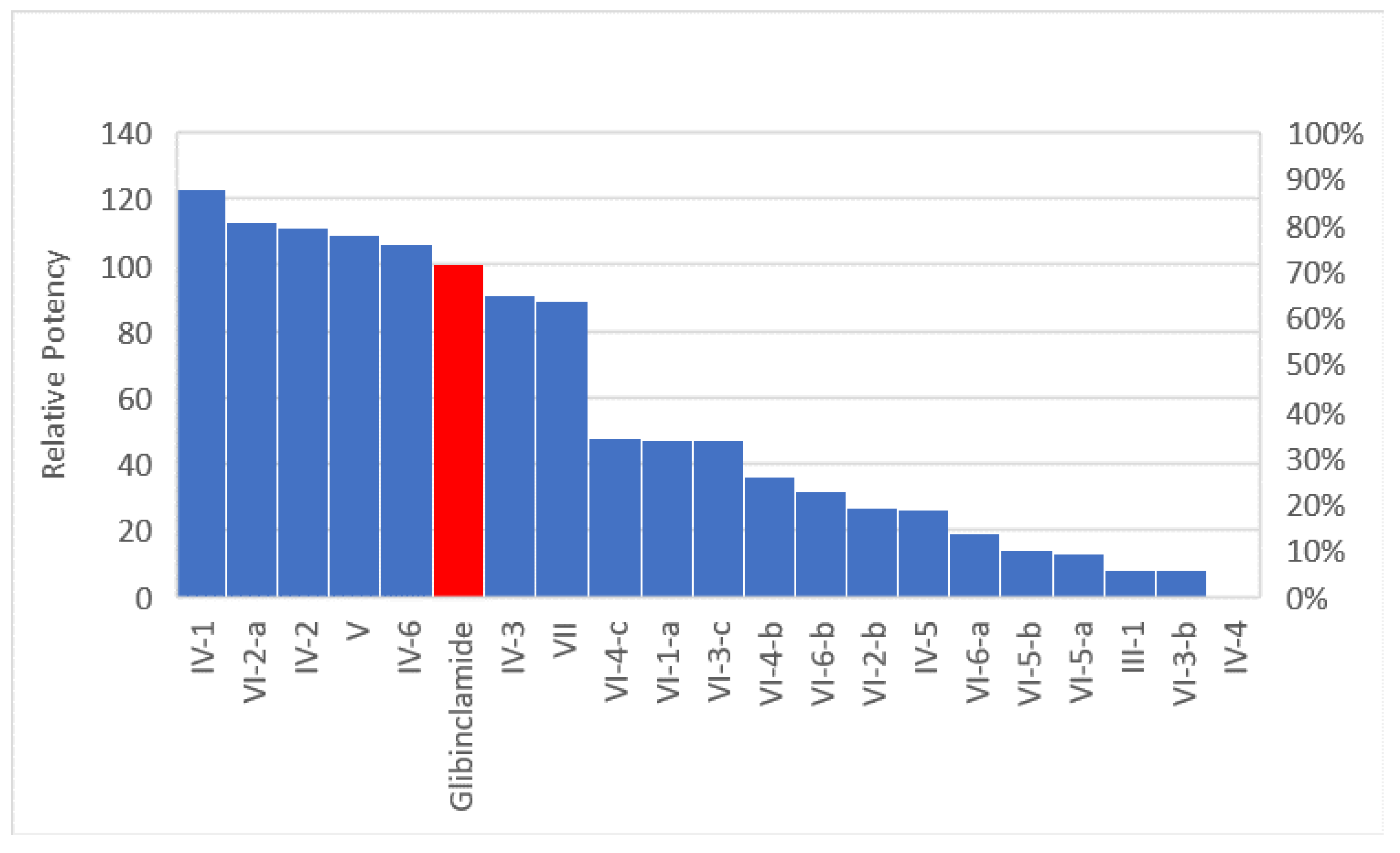
2.2.1. In Vivo Antihyperglycemic Screening
Effect of Glibenclamide and the Tested Compounds in a Daily Dose of 2 mg/kg for 6 Days on Blood Glucose Levels of Diabetic Rats
Effect of Discontinuation of Drugs for 6 Days on the Rebound Elevation of Blood Glucose of Diabetic Rats
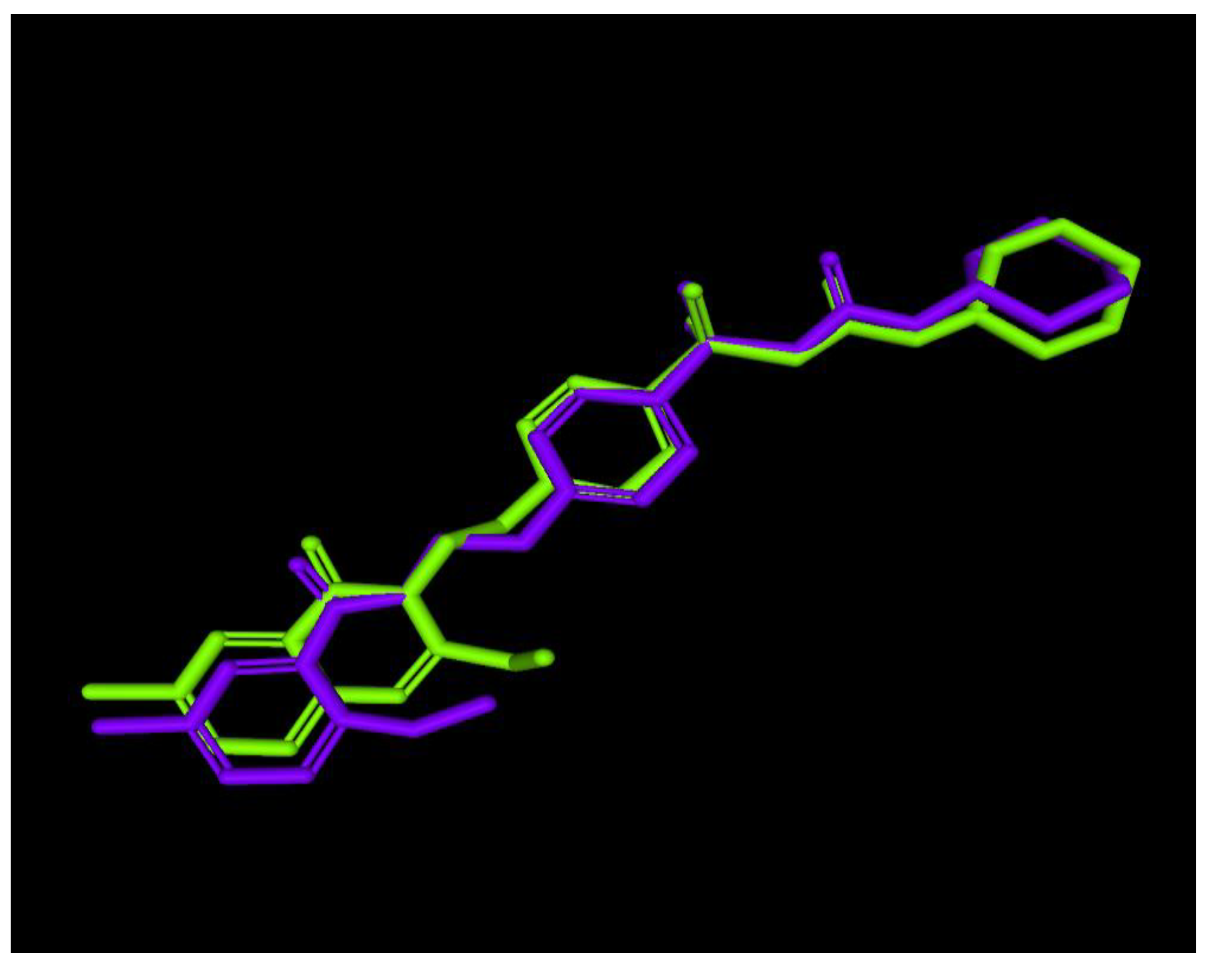
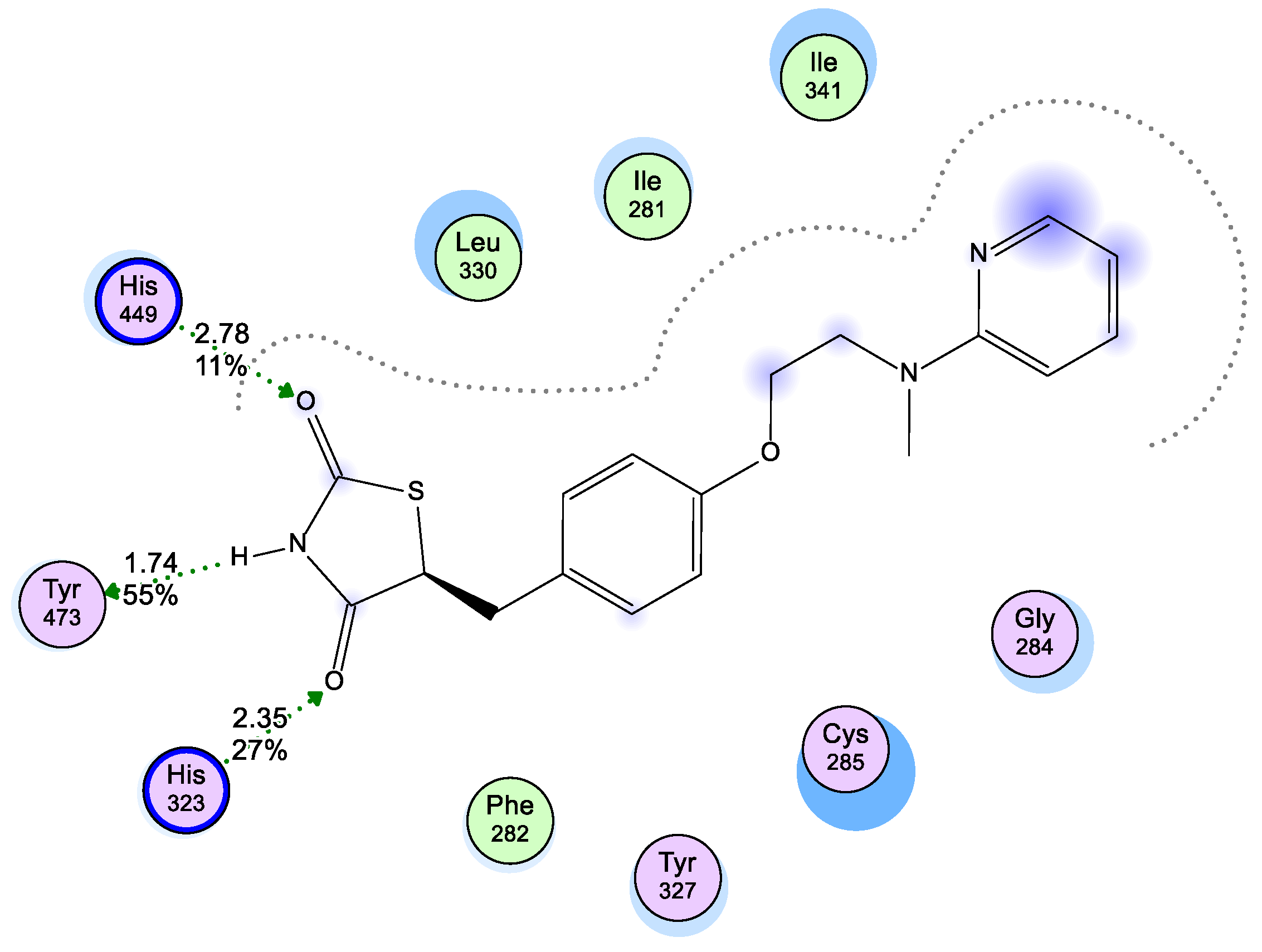
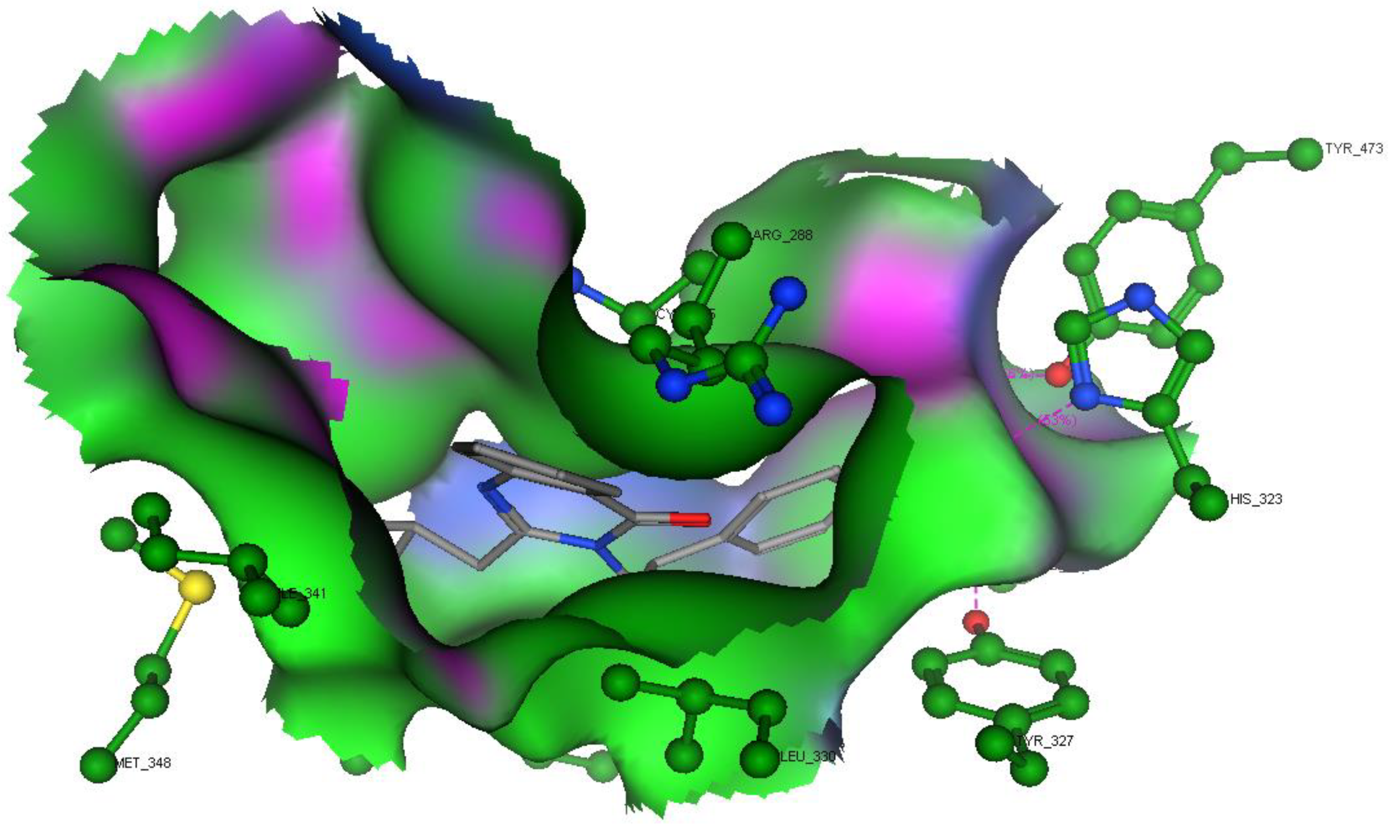
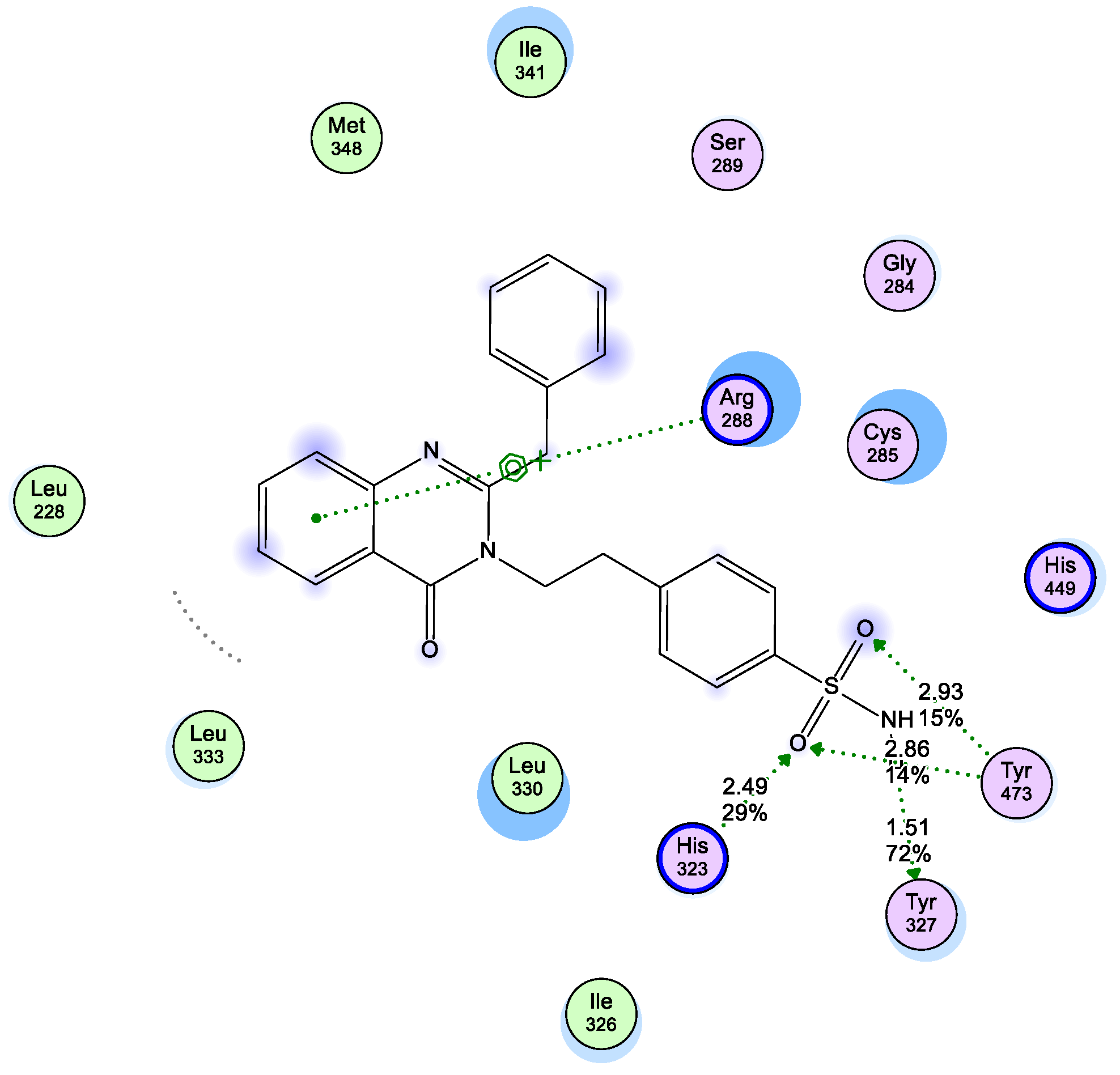
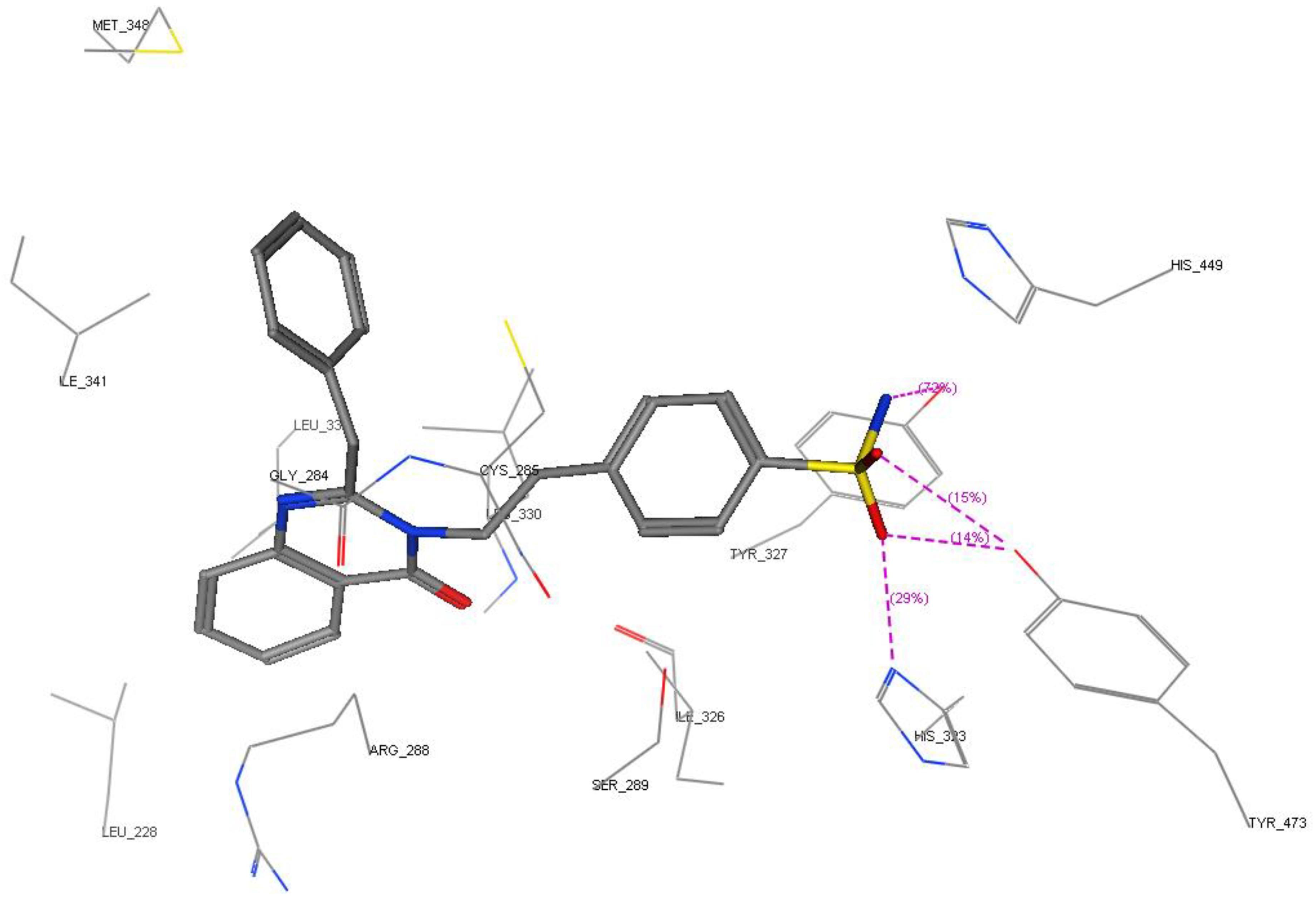
2.3. Molecular Modeling
2.3.1. Modeling Studies
2.3.2. In Silico ADMET Studies
Evaluation of Lipophilicity, Solubility, Drug Likeness, GIT Absorption and Toxicity Risk
2.4. Structural Activity Relationship
- In group IV, the propyl analog (IV-4) was the most potent among all six derivatives of the quinazolinone ring analogs followed by the benzyl (IV-6) and then the methyl. The least active derivative among all was the isopropyl derivative (IV-5).
- Comparing compound (IV-1) (in which two methylene spacers are present between the quinazolinone ring and the phenyl sulfonamide group) and compound V which no spacers has unexpectedly shown that the two ethylene bridge was not essential for activity.
- There was big difference between the open structure (III-1) and its cyclic congener (IV-1), which showed more than ten folds the activity of the corresponding open structure.
- Compounds having benzyl, phenyl, n-propyl and methyl groups at position 2 of quinazolinone nucleus were the most potent among all groups.
- Compounds (IV-1), (VI-2-a), (IV-2), (V), (IV-6) which have phenyl, methyl and benzyl groups at the 2-position of the quinazolinone nucleus have shown prolonged actions while compounds (IV-4) and (IV-5) (having the n-propyl or isopropyl groups at 2-position of the quinazolinone nucleus) showed short duration of action.
- These compounds are so promising that this work could be performed by modifying the 5-position of the quinazolinone nucleus and attaching other groups to the sulfonamide group.
3. Materials and Methods
3.1. Chemistry
3.1.1. N-Acylnthranilic Acids (I)
3.1.2. 4H-3,1-Benzoxazin-4-Ones (II)
3.1.3. o-Benzamido-N-(p-aminosulfonyl]phenylethyl)-benzamide (III-1)
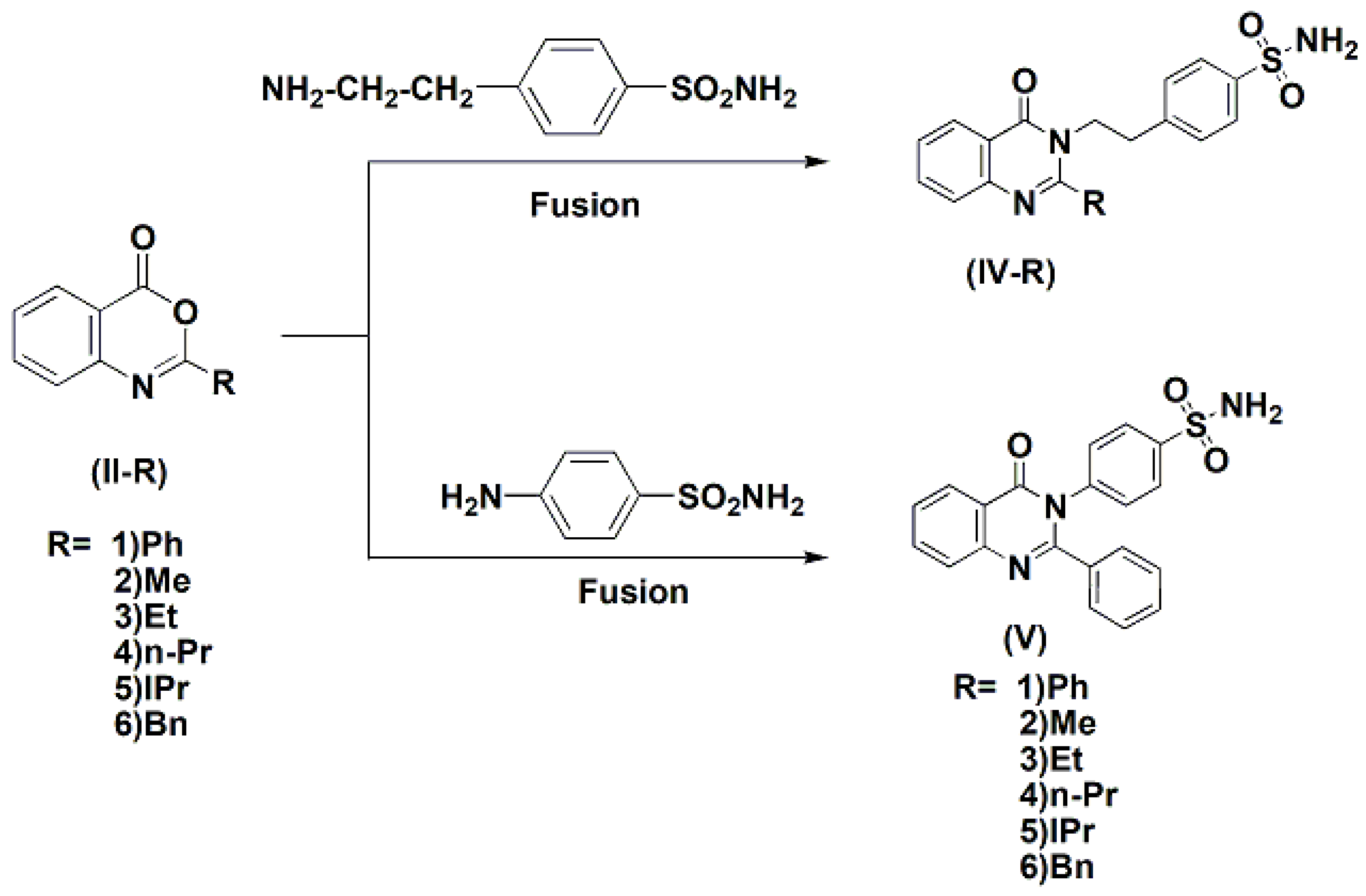
3.1.4. General Procedures for Synthesis of 3-(p-Sulfamoylaralkyl)-4-(3H)-quinazolinones (IV-1–6)
2-Phenyl-3-(p-aminosulfonyl]phenylethyl)-2-phenyl-4(3H)-quinazo-linone (IV-1)
2-Methyl-3-(p-aminosulfonyl]phenylethyl)-4(3H)-quinazolinone (IV-2)
2-Ethyl-3-(p-aminosulfonyl]phenylethyl)-4(3H)-quinazolinone (IV-3)
2-n-Propyl-3-(p-aminosulfonyl]phenylethyl)-4(3H)-quinazolinone (IV-4)
2-iso-Propyl-3-(p-aminosulfonyl]phenylethyl)-4(3H)-quinazolinone (IV-5)
2-iso-Propyl-3-(p-aminosulfonyl]phenylethyl)-4(3H)-quinazolinone (IV-6)
2-Phenyl-3-(p-sulfonyl]phenyl)-4(3H)-quinazolinone (V)
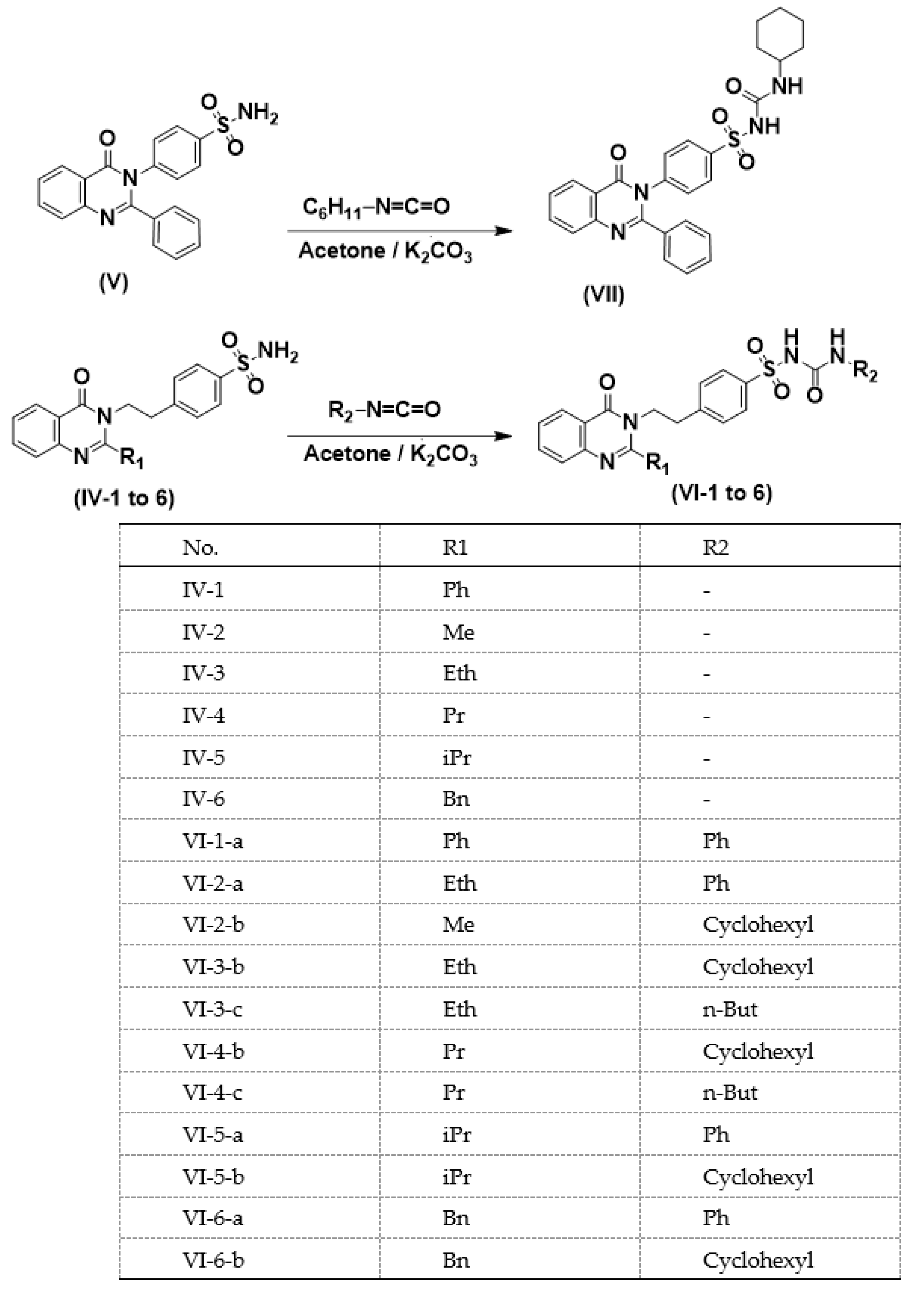
3.1.5. General Procedures for Synthesis of Sulfonyl Urea Derivatives of 3-(p-Sulfamoylaralkyl)-4-(3H)-quinazolinones (VI-1–6)
2-Phenyl-3-[4-[[[(phenylamino)carbonyl]aminosulfonyl]phenylethyl]-4(3H)-quinazolinone (VI-1-a)
2-Phenyl-3-[4-[[[(cyclohexylamino)carbonyl]aminosulfonyl]phenyl]-4(3H)-quinazolinone (VI-1-b)
2-Phenyl-3-[4-[[[(n-butylamino)carbonyl]aminosulfonyl]phenylethyl]-4(3H)-quinazolinone (VI-1-c)
2-Methyl-3-[4-[[[(phenylamino)carbonyl]aminosulfonyl]phenylethyl]-4(3H)-quinazolinone (VI-2-a)
2-Methyl-3-[4-[[[(n-butylamino)carbonyl]aminosulfonyl]phenylethyl]-4(3H)-quinazolinone (VI-2-b)
2-Ethyl-3-[4-[[[(phenylamino)carbonyl]aminosulfonyl]phenylethyl]-4(3H)-quinazolinone (VI-3-a)
2-Ethyl-3-[4-[[[(cyclohexylamino)carbonyl]aminosulfonyl]phenylethyl]-4(3H)-quinazolinone (VI-3-b)
2-Ethyl-3-[4-[[[(n-butylamino)carbonyl]aminosulfonyl]phenylethyl]-4(3H)-quinazolinone (VI-3-c)
2-n-Propyl-3-[4-[[[(phenylamino)carbonyl]aminosulfonyl]phenylethyl]-4(3H)-quinazolinone (VI-4-a)
2-n-Propyl-3-[4-[[[(cyclohexylamino)carbonyl]aminosulfonyl]phenylethyl]-4(3H)-quinazolinone (VI-4-b)
2-n-Propyl-3-[4-[[[(n-butylamino)carbonyl]aminosulfonyl]phenylethyl]-4(3H)-quinazolinone (VI-4-c)
2-iso-pr-3-[4-[[[(Cyclohexylamino)carbonyl]aminosulfonyl]phenylethyl]-4(3H)-quinazolinone (VI-5-a)
2-iso-Propyl-3-[4-[[[(n-butylamino)carbonyl]aminosulfonyl]phenylethyl]-4(3H)-quinazolinone (VI-5-b)
2-Benzyl-3-[4-[[[(phenylamino)carbonyl]aminosulfonyl]phenylethyl]-4(3H)-quinazolinone (VI-6-a)
2-Benzyl-3-[4-[[[(cyclohexylamino)carbonyl]aminosulfonyl]phenylethyl]-4(3H)-quinazolinone (VI-6-b)
3.1.6. 2-Phenyl-3-[4-[[[(cyclohexylamino)carbonyl]aminosulfonyl]phenyl]-4(3H)-quinazolinone (VII)
3.2. Antidiabetic Activity
3.3. Molecular Modeling
3.3.1. Docking Studies
3.3.2. ADMET Studies
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Krentz, A.J.; Ferner, R.E.; Bailey, C.J. Comparative tolerability profiles of oral antidiabetic agents. Drug Saf. 1994, 11, 223–241. [Google Scholar] [CrossRef] [PubMed]
- Diabetes, D.O.F. Diagnosis and classification of diabetes mellitus. Diabetes Care 2013, 36, 67–74. [Google Scholar]
- Schmidt, A.M. Highlighting Diabetes Mellitus. Arterioscler. Thromb. Vasc. Biol. 2018, 38, e1–e8. [Google Scholar] [CrossRef] [PubMed]
- Keefer, L.M.; Piron, M.A.; Meyts, D.P. Human insulin prepared by recombinant DNA techniques and native human insulin interact identically with insulin receptors. Proc. Natl. Acad. Sci. USA 1981, 78, 1391–1405. [Google Scholar] [CrossRef]
- Kobayashi, M.; Sasaoka, T.; Sugibayashi, M.; Iwanishi, M.; Shigeta, Y. Receptor binding and biologic activity of biosynthetic human insulin and mini-proinsulin produced by recombinant gene technology. Diabetes Res. Clin. Pract. 1989, 7, 25–38. [Google Scholar] [CrossRef]
- Owens, D.R.; Zinman, B.; Bolli, G. Alternative routes of insulin delivery. Diabet. Med. 2003, 20, 886–898. [Google Scholar] [CrossRef]
- Weiss, S.R.; Cheng, S.L.; Kourides, I.A.; Gelfand, R.A.; Landschulz, W.H. Inhaled Insulin Phase II Study Group. Inhaled insulin provides improved glycemic control in patients with type 2 diabetes mellitus inadequately controlled with oral agents: A randomized controlled trial. Arch. Intern. Med. 2003, 163, 2277–2282. [Google Scholar] [CrossRef]
- Bayes, M.; Rabasseda, X.; Prous, J.R. Gateways to clinical trials. Methods Find Exp. Clin. Pharmacol. 2003, 25, 565–597. [Google Scholar]
- Appel, S.J.; Wright, M.A. Teach your patient to administer: Inhaled insulin. Nursing 2007, 37, 49–50. [Google Scholar] [CrossRef]
- Wright, M.A.; Appel, S.J. Inhaled insulin: Breathing new life into diabetes therapy. Nursing 2007, 37, 46–48. [Google Scholar] [CrossRef]
- Magkos, F.; Hjorth, M.F.; Astrup, A. Diet and exercise in the prevention and treatment of type 2 diabetes mellitus. Nat. Rev. Endocrinol. 2020, 16, 545–555. [Google Scholar] [CrossRef] [PubMed]
- Marshall, V.; Wilton, L.; Shakir, S. Safety profile of repaglinide as used in general practice in England: Results of a prescription-event monitoring study. Acta Diabetol. 2006, 43, 6–14. [Google Scholar] [CrossRef] [PubMed]
- Bodmer, M.; Meier, C.; Krähenbühl, S.; Jick, S.S.; Meier, R.C. Metformin, sulfonylureas, or other antidiabetes drugs and the risk of lactic acidosis or hypoglycemi. Diabetes Care 2008, 31, 2086–2091. [Google Scholar] [CrossRef] [PubMed]
- Groop, L.C. Sulfonylureas in NIDDM. Diabetes Care 1992, 15, 737–754. [Google Scholar] [CrossRef]
- Fukuen, S.; Iwaki, M.; Yasui, A.; Makishima, M.; Matsuda, M.; Shimomura, I. Sulfonylurea agents exhibit peroxisome proliferator-activated receptor γ agonistic activity. J. Biol. Chem. 2005, 280, 23653–23659. [Google Scholar] [CrossRef]
- Chatterjee, S.; Khunti, K.; Davies, M.J. Type 2 diabetes. Lancet 2017, 389, 2239–2251. [Google Scholar] [CrossRef]
- Onishi, Y.; Fujisawa, T.; Sakaguchi, K.; Maeda, M. Effect of nateglinide on the size of LDL particles in patients with type 2 diabetes. Adv. Ther. 2006, 23, 549–555. [Google Scholar] [CrossRef]
- Ristic, S.; Collober-Maugeais, C.; Pecher, E.; Cressier, F. Comparison of nateglinide and gliclazide in combination with metformin, for treatment of patients with Type 2 diabetes mellitus inadequately controlled on maximum doses of metformin alone. Diabet. Med. 2006, 23, 757–762. [Google Scholar] [CrossRef]
- Pourhanifeh, M.H.; Hosseinzadeh, A.; Dehdashtian, E.; Hemati, K.; Mehrzadi, S. Melatonin: New insights on its therapeutic properties in diabetic complications. Diabetol. Metab. Syndr. 2020, 12, 30. [Google Scholar] [CrossRef]
- Baum, U. Diabetes therapy related to etiology. Indications, uses and side effects of new insulin sensitizers. MMW Fortschr. Med. 2000, 142, 27–87. [Google Scholar]
- Fuchtenbusch, M.; Standl, E.; Schatz, H. Clinical efficacy of new thiazolidinediones and glinides in the treatment of type 2 diabetes mellitus. Exp. Clin. Endocrinol. Diabetes 2000, 108, 151–163. [Google Scholar] [CrossRef] [PubMed]
- Scheen, A.J. Pharmacokinetic interactions with thiazolidinediones. Clin. Pharmacokinet. 2007, 46, 1–12. [Google Scholar] [CrossRef]
- Gupta, C.M.; Bhaduri, A.P.; Khanna, N.M.; Mukherjee, S.K. A Novel class of hypoglycemic agents; Synthesis & SAR in 2-substituted-4(3H)-quinazolinones, 2-substituted 4-hydroxymethylene [5,6]-pyrimidines & 3-substituted-4-oxopyridol [1,2-a]pyrimidines. Indian J. Chem. 1971, 9, 201–206. [Google Scholar]
- Deshpande, S.M.; Datta, K.C.; Singth, A.K. p-Toluenesulfonyl Derivatives of N,N′-bis-(4-quinolino-, 4-quinaldino-, 4-quinazolino and 9-acridino) poly-methylene diamines as Hypoglycemic Agent. J. Indian Chem. Soc. 1975, LII, 746–749. [Google Scholar]
- Murthy, G.R.; Reddy, A.M.; Redly, V.M. Synthesis and Hypoglycemic Activity of some quinazolinylureas. Indian Drugs 1987, 25, 19–22. [Google Scholar]
- Mukherjee, S.K. A novel hypoglycemic compound. Biochem. Pharmacol. 1973, 32, 1529–1531. [Google Scholar] [CrossRef]
- Mukherjee, S.K.; Hausain, S.T. Effect of 2-piperazino-4(3H)-quinazoline monoacetate on some aspects of carbohydrate metabolism of albino rats. Biochem. Pharmacol. 1973, 22, 2205–2206. [Google Scholar] [CrossRef]
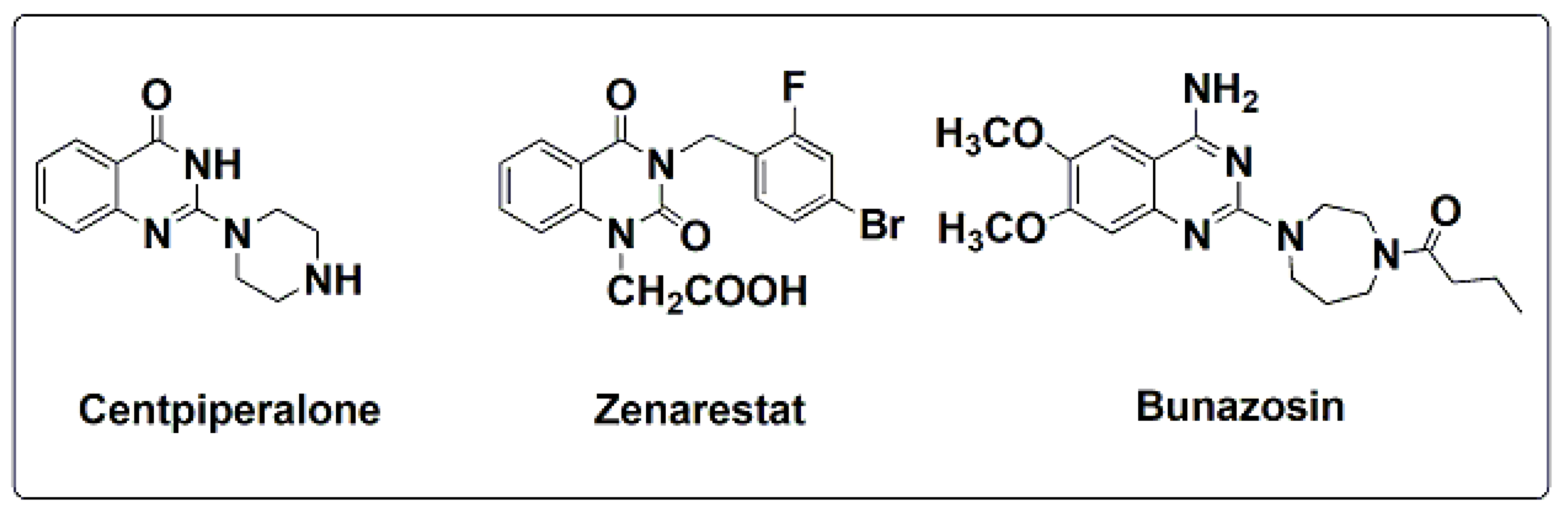
- Chatterjee, A.K.; Murthy, P.S.; Mukherjee, S.K. Effect of centipiperalone in insulin deficient diabetes. Indian J. Exp. Biol. 1980, 18, 1005–1008. [Google Scholar]
- Chatterjee, A.K.; Mukherjee, B.; Mukherjee, S.K. Effect of centipiperalone, a new hypoglycemic agent on insulin biosynthesis and release from isolated pancreatic islets of rat. Indian J. Exp. Biol. 1982, 20, 270–272. [Google Scholar]
- Tanaka, Y.; Sawamoto, T.; Suzuli, A.; Kimura, T. Pharmacokinetics of Zenerastate, An aldose reductase inhibitor, in male and female diabetic rats. Drug Metab. Disp. 1993, 21, 677–681. [Google Scholar]
- Sekiya, T.; Hiranuma, H.; Kanayama, T.; Hata, S. Pyrimidine derivatives III(1), Synthesis of hypoglycemic 4-alkoxy-2-piperazino-5,6-polymethylenepyrimidines. Eur. J. Med. Chem. 1980, 15, 317–322. [Google Scholar] [CrossRef]
- Sekiya, T.; Hata, S.; Yamada, S. Pyrimidine derivatives VII, Structure-Activity Relationship of hypoglycemic 4-amino-2-(1-piperazinyl) pyrimidines investigated by the adaptive last suares method. Chem. Pharm. Bull. Japan. 1983, 31, 2432–2437. [Google Scholar] [CrossRef]
- Saxena, A.M.; Murthy, P.S.; Mukherjee, S.K. Mode of action of three structurally different hypoglycemic agents. A Comp. Study Indian J. Exp. Biol. 1996, 34, 351–355. [Google Scholar]
- Ibrahim, K.M.; Eissa, H.I.; Abdallah, E.A.; Metwaly, M.A.; Radwan, M.; ElSohly, M. Design, synthesis, molecular modeling and anti-hyperglycemic evaluation of novel quinoxaline derivatives as potential PPARγ and SUR agonists. Biorg. Med. Chem. 2017, 25, 1496–1513. [Google Scholar] [CrossRef]
- Ibrahim, K.M.; Eissa, H.I.; Alesawy, S.M.; Metwaly, M.A.; Radwan, A.M.; ElSohly, A.M. Design, synthesis, molecular modeling and anti-hyperglycemic evaluation of quinazolin-4 (3H)-one derivatives as potential PPARγ and SUR agonists. Biorg. Med. Chem. 2017, 25, 4723–4744. [Google Scholar] [CrossRef]
- Wolffenbuttel, B.H.; Graal, M.B. New treatments for patients with type 2 diabetes mellitus. Postgrad Med. J. 1996, 72, 657–662. [Google Scholar] [CrossRef]
- Ramsey, J.D.; Ripps, H.; Qian, H. Streptozotocin-induced diabetes modulates GABA receptor activity of rat retinal neurons. Exp. Eye Res. 2007, 85, 413–422. [Google Scholar] [CrossRef]
- Bajare, S.; Anthony, J.; Nair, A.; Marita, R.; Damre, A.; Patel, D.; Rao, C.; Sivaramakrishnan, H.; Deka, N. Synthesis of N-(5-chloro-6-(quinolin-3-yloxy) pyridin-3-yl)benzenesulfonamide derivatives as non-TZD peroxisome proliferator-activated receptor γ(PPARγ) agonist. Eur. J. Med. Chem. 2012, 58, 355–360. [Google Scholar] [CrossRef]
- Alaa, A.M.; El-Azab, A.S.; Attia, S.M.; Al-Obaid, A.M.; Al-Omar, M.A.; El-Subbagh, H.I. Synthesis and biological evaluation of some novel cyclic-imides as hypoglycaemic, anti-hyperlipidemic agents. Eur. J. Med. Chem. 2011, 46, 4324–4329. [Google Scholar]
- Molecular Operating Environment (MOE). Chemical Computing Group. Available online: http://www.chemcomp.com (accessed on 10 May 2019).
- Swiss Institute of Bioinformatics (SwissADME). Available online: http://www.swissADME.ch (accessed on 10 March 2022).
- Toxicity Estimation Software Tool (T.E.S.T). Available online: http://www.epa.gov (accessed on 5 April 2022).
- Stumvoll, M.; Mitrakou, A.; Pimenta, W.; Jenssen, T.; Yki-Järvinen, H.; Van, H.T.; Renn, W.; Gerich, J. Use of the oral glucose tolerance test to assess insulin release and insulin sensitivity. Diabetes Care 2000, 23, 295–301. [Google Scholar] [CrossRef] [Green Version]
















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Rats and Treatment | Blood Glucose Level (mg/dL) | Change of the Value before Treatment | % Change | RP | ||
|---|---|---|---|---|---|---|
| Before Treatment | After Treatment | After Withdrawal | ||||
| Normal Rats | 102 ± 2.21 | 110 ± 3.22 | 100± 4.55 | |||
| Diabetic Rats | 406 ± 63.4 | 415 ± 25.6 | 430 ± 23.78 | |||
| Glibenclamide | 336 ± 23 | 150 ** ± 30 | 183.3 ** ± 24.1 | 152.7 | 45.5 | 100 |
| III-1 | 177 ± 22.2 | 167 ± 42 | 183.3 ± 61.55 | 6.3 | 3.5 | 8 |
| IV-1 | 298 ± 35.4 | 124 ** ± 12 | 130.8 ** ± 24.5 | 167.2 | 56.1 | 123 |
| VI-1-a | 164 ± 2.71 | 113 ** ± 5.7 | 129.8 ** ± 4.42 | 34.2 | 21.3 | 47 |
| IV-2 | 245 ± 51.9 | 108 ** ± 4.4 | 121 ** ± 4.509 | 124 | 50.6 | 111 |
| VI-2-a | 326 ± 50.6 | 128 ± 25 | 159 ** ± 39.96 | 167 | 51.2 | 113 |
| VI-2-b | 360 ± 41.3 | 189 ** ± 49 | 315 ± 24.11 | 45 | 12.5 | 27 |
| IV-3 | 205 ± 30.4 | 107 ** ± 8.8 | 119.7 ** ± 13.8 | 85.3 | 41.6 | 91 |
| VI-3-b | 306 ± 42.1 | 184 ** ± 40 | 295.2 ± 25.47 | 10.8 | 3.52 | 8 |
| VI-3-c | 203 ± 44.9 | 148 ± 41 | 160 ± 43.26 | 57 | 21.2 | 47 |
| IV-4 | 325 ± 14.5 | 93 ** ± 3.7 | 325 ± 58.43 | 0.00 | 0.00 | 00 |
| VI-4-b | 368 ± 5.55 | 229 ** ± 9 | 429.2 ± 45.5 | 61 | 16.6 | 36 |
| VI-4-c | 349 ± 52.1 | 114 ** ± 9.4 | 272.3 ± 41.63 | 76.7 | 21.9 | 48 |
| IV-5 | 384 ± 21.2 | 246 ** ± 22 | 430 ± 56.41 | 46 | 11.9 | 26 |
| VI-5-a | 348 ± 33 | 187 ** ± 25 | 327.8 ± 42.57 | 21 | 6 | 13 |
| VI-5-b | 381 ± 82.9 | 319 ± 87 | 355.6 ± 33.38 | 25.4 | 6.7 | 14 |
| IV-6 | 282 ± 38.6 | 107 ** ± 9.3 | 146.3 ** ± 12.2 | 135.7 | 48.1 | 106 |
| VI-6-a | 464 ± 40.9 | 101 ** ± 9.8 | 424.2 ± 35.33 | 39.8 | 8.6 | 19 |
| VI-6-b | 194 ± 7.09 | 133 ** ± 24 | 166.3 ± 34.26 | 27.7 | 14.4 | 32 |
| V | 334 ± 52.4 | 87 ** ± 6.8 | 167.5 ** ± 28.5 | 166.5 | 49.8 | 109 |
| VII | 188 ± 24.6 | 102 ** ± 4.4 | 111 ** ± 4 | 77 | 40.9 | 89 |
| Comp. No. | Binding Free Energy (Kcal/mol) |
|---|---|
| Glibenclamide | −22.89 |
| III-1 | −14.89 |
| IV-1 | −21.84 |
| IV-2 | −22.52 |
| IV-3 | −21.76 |
| IV-4 | −17.08 |
| IV-5 | −12.48 |
| IV-6 | −13.45 |
| V | −17.89 |
| VI-1-a | −20.23 |
| VI-2-a | −24.01 |
| VI-2-b | −20.79 |
| VI-3-b | −12.53 |
| VI-3-c | −17.48 |
| VI-4-b | −22.61 |
| VI-4-c | −22.51 |
| VI-5-a | −9.92 |
| VI-5-b | −22.82 |
| VI-6-a | −15.99 |
| VI-6-b | −13.81 |
| VII | −15.32 |
| Compound No. | C log P a | Log S b | Drug Likeness | Lipiniski Violation | GIT Absorption | Molecular Weight | Toxicity Risk c |
|---|---|---|---|---|---|---|---|
| Glinenclamide | 3.5 | −5.48 | yes | 0 | Low | 494.00 | Negative |
| III-1 | 2.38 | −4.27 | yes | 0 | Good | 423.48 | Negative |
| IV-1 | 2.17 | −4.51 | yes | 0 | Good | 405.47 | Negative |
| IV-2 | 1.94 | −3.08 | yes | 0 | Good | 343.40 | Negative |
| IV-3 | 2.07 | −3.38 | yes | 0 | Good | 357.43 | Negative |
| IV-4 | 2.14 | −3.60 | yes | 0 | Good | 371.45 | Negative |
| IV-5 | 2.30 | −3.81 | yes | 0 | Good | 371.45 | Negative |
| IV-6 | 2.42 | −4.47 | yes | 0 | Good | 419.50 | Negative |
| V | 2.30 | −4.07 | yes | 0 | Good | 377.42 | Negative |
| VI-1-a | 3.47 | −6.30 | yes | 1 | Poor | 524.59 | Negative |
| VI-2-a | 3.22 | −5.19 | yes | 0 | Good | 476.55 | Negative |
| VI-2-b | 2.88 | −3.98 | yes | 0 | Good | 442.53 | Negative |
| VI-3-b | 3.25 | −5.26 | yes | 0 | Good | 482.60 | Negative |
| VI-3-c | 2.62 | −4.29 | yes | 0 | Good | 456.56 | Negative |
| VI-4-b | 3.22 | −5.50 | yes | 1 | Poor | 496.62 | Negative |
| VI-4-c | 2.89 | −4.52 | yes | 0 | Good | 470.58 | Negative |
| VI-5-a | 4.04 | −5.69 | yes | 0 | Poor | 496.62 | Negative |
| VI-5-b | 3.01 | −4.72 | yes | 0 | Good | 470.58 | Negative |
| VI-6-a | 3.76 | −6.27 | yes | 1 | Poor | 538.62 | Negative |
| VI-6-b | 3.49 | −6.35 | yes | 1 | Good | 544.66 | Negative |
| VII | 3.73 | −6.12 | yes | 1 | Poor | 502.58 | Negative |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
El-Zahabi, M.A.; Bamanie, F.H.; Ghareeb, S.; Alshaeri, H.K.; Alasmari, M.M.; Moustafa, M.; Al-Marzooki, Z.; Zayed, M.F. Design, Synthesis, Molecular Modeling and Anti-Hyperglycemic Evaluation of Quinazoline-Sulfonylurea Hybrids as Peroxisome Proliferator-Activated Receptor Gamma (PPARγ) and Sulfonylurea Receptor (SUR) Agonists. Int. J. Mol. Sci. 2022, 23, 9605. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23179605
El-Zahabi MA, Bamanie FH, Ghareeb S, Alshaeri HK, Alasmari MM, Moustafa M, Al-Marzooki Z, Zayed MF. Design, Synthesis, Molecular Modeling and Anti-Hyperglycemic Evaluation of Quinazoline-Sulfonylurea Hybrids as Peroxisome Proliferator-Activated Receptor Gamma (PPARγ) and Sulfonylurea Receptor (SUR) Agonists. International Journal of Molecular Sciences. 2022; 23(17):9605. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23179605
Chicago/Turabian StyleEl-Zahabi, Mohamed Ayman, Faida H. Bamanie, Salah Ghareeb, Heba K. Alshaeri, Moudi M. Alasmari, Mohamed Moustafa, Zohair Al-Marzooki, and Mohamed F. Zayed. 2022. "Design, Synthesis, Molecular Modeling and Anti-Hyperglycemic Evaluation of Quinazoline-Sulfonylurea Hybrids as Peroxisome Proliferator-Activated Receptor Gamma (PPARγ) and Sulfonylurea Receptor (SUR) Agonists" International Journal of Molecular Sciences 23, no. 17: 9605. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23179605