
A Pan-RNase Inhibitor Enabling CRISPR-mRNA Platforms for Engineering of Primary Human Monocytes

 , , , , and
, , , , and
Abstract
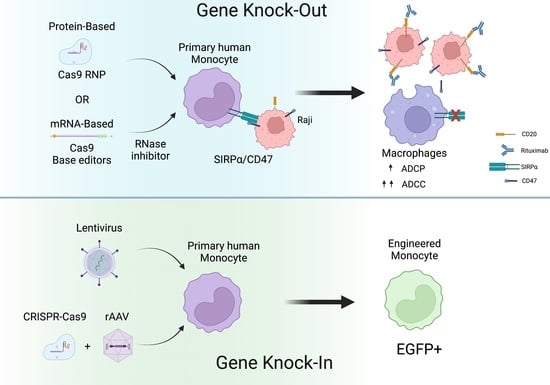
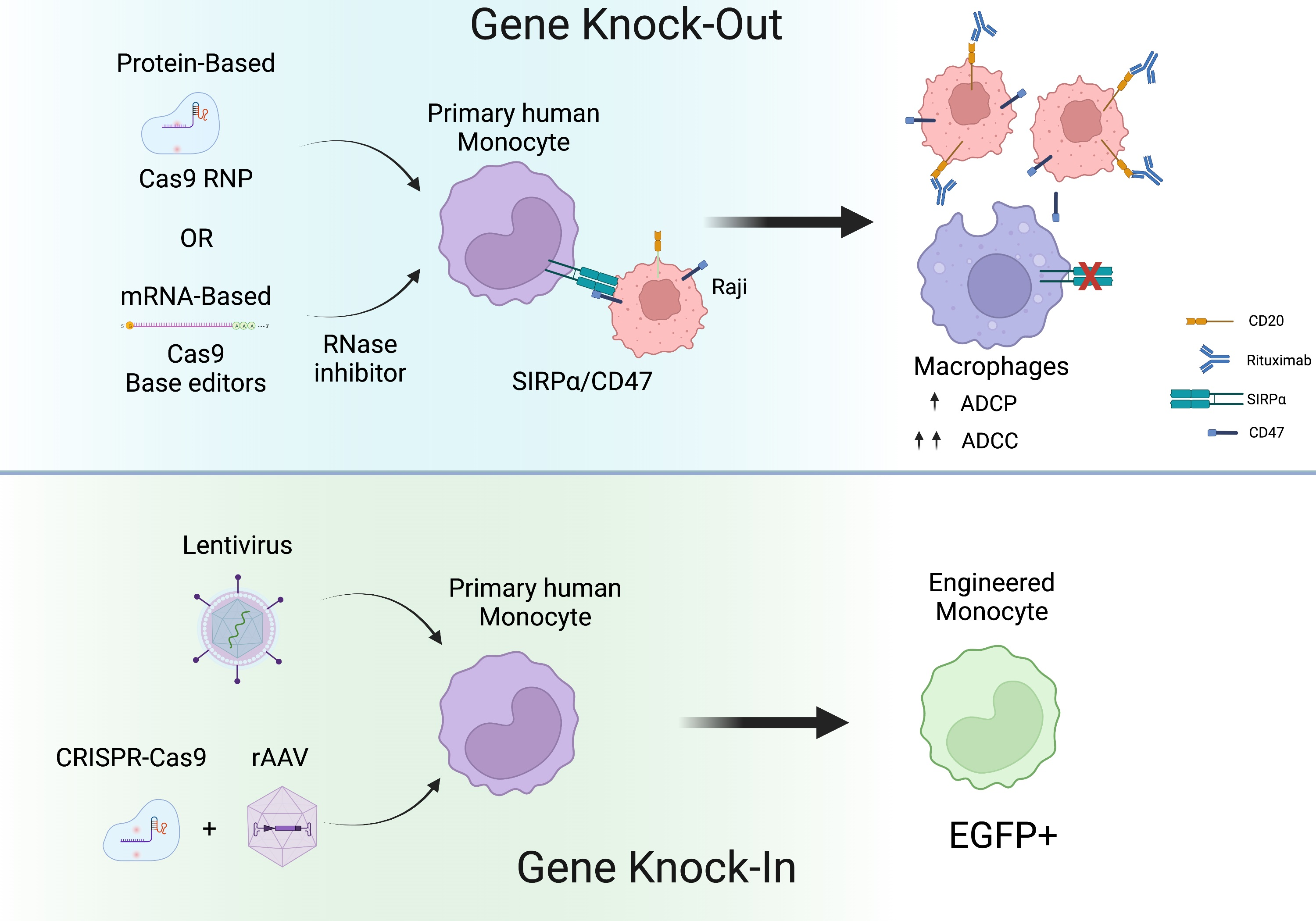
:
1. Introduction
2. Results
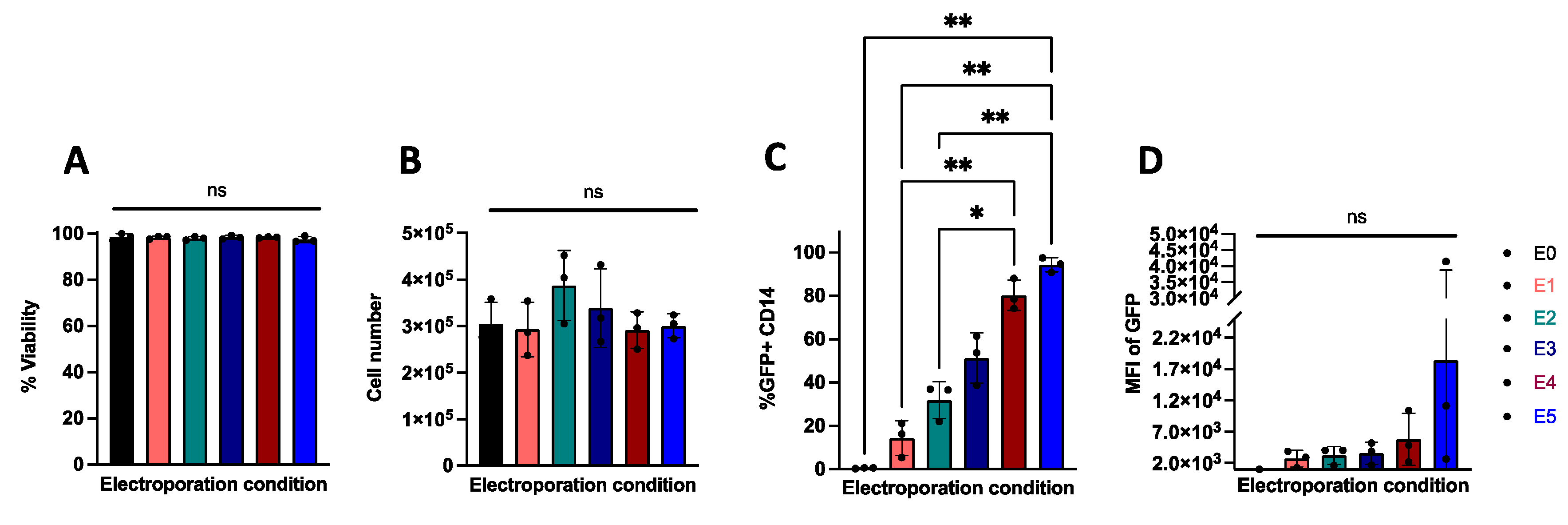
2.1. Electroporation Allows for Efficient Transfection of Nucleic Acids in Primary Human Monocytes
2.2. CRISPR-Cas9 RNPs Mediate Efficient Editing of Primary Human Monocytes
2.3. mRNA Encoding Cas9 Results in Inefficient Genome Editing in Primary Human Monocytes
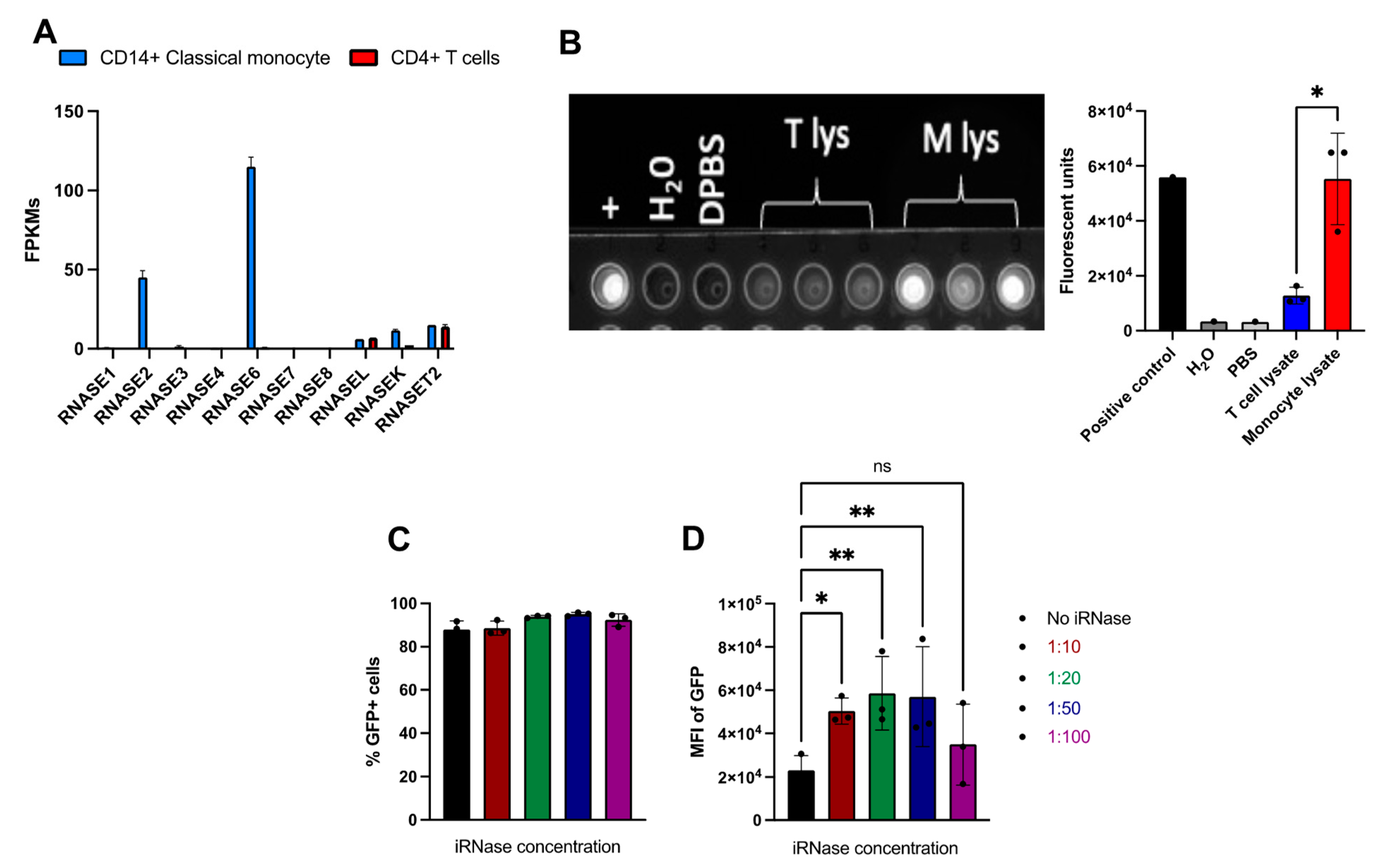
2.4. Use of RNase Inhibitor results in More Consistent and Efficient RNA-Based Gene Editing in Primary Human Monocytes
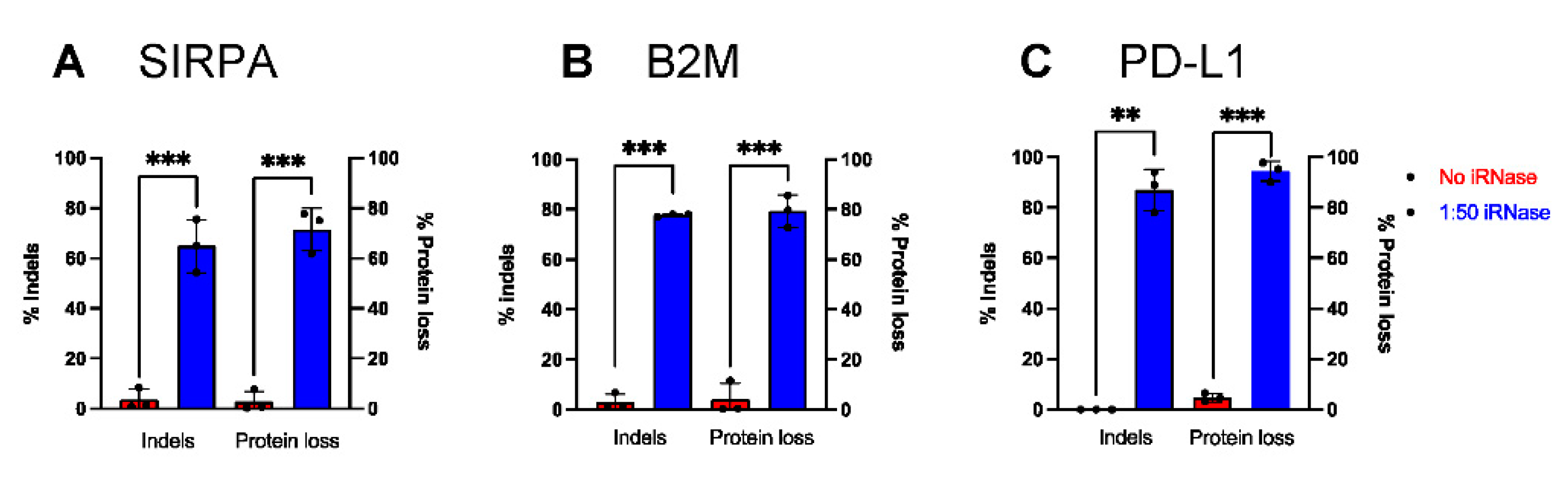
2.5. iRNase Enables other RNA-Based Genome Engineering Platforms in Primary Human Monocytes
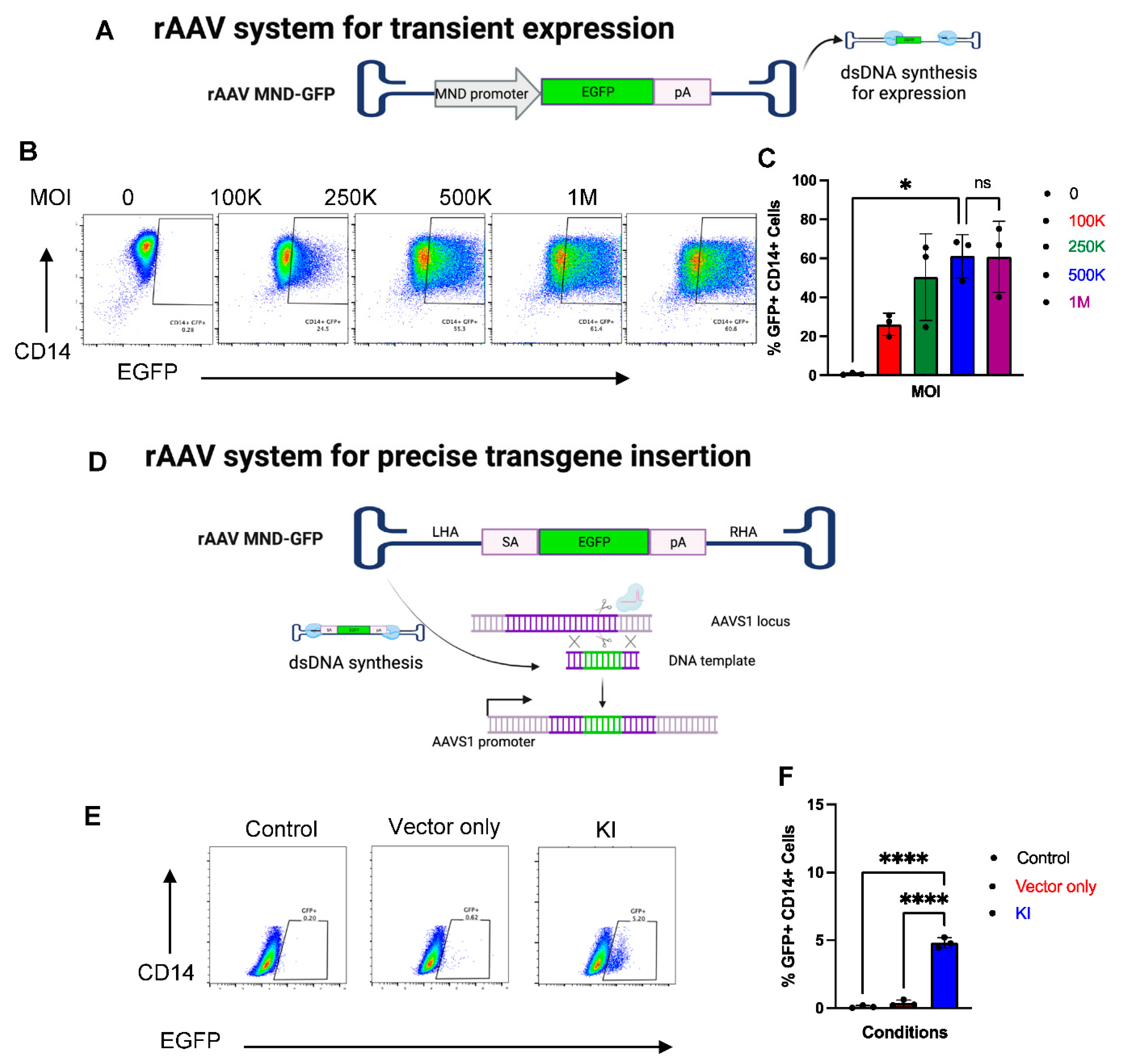
2.6. CRISPR-Cas9 and rAAV Mediated HDR for Site-Specific Transgene Insertion in Primary Human Monocytes
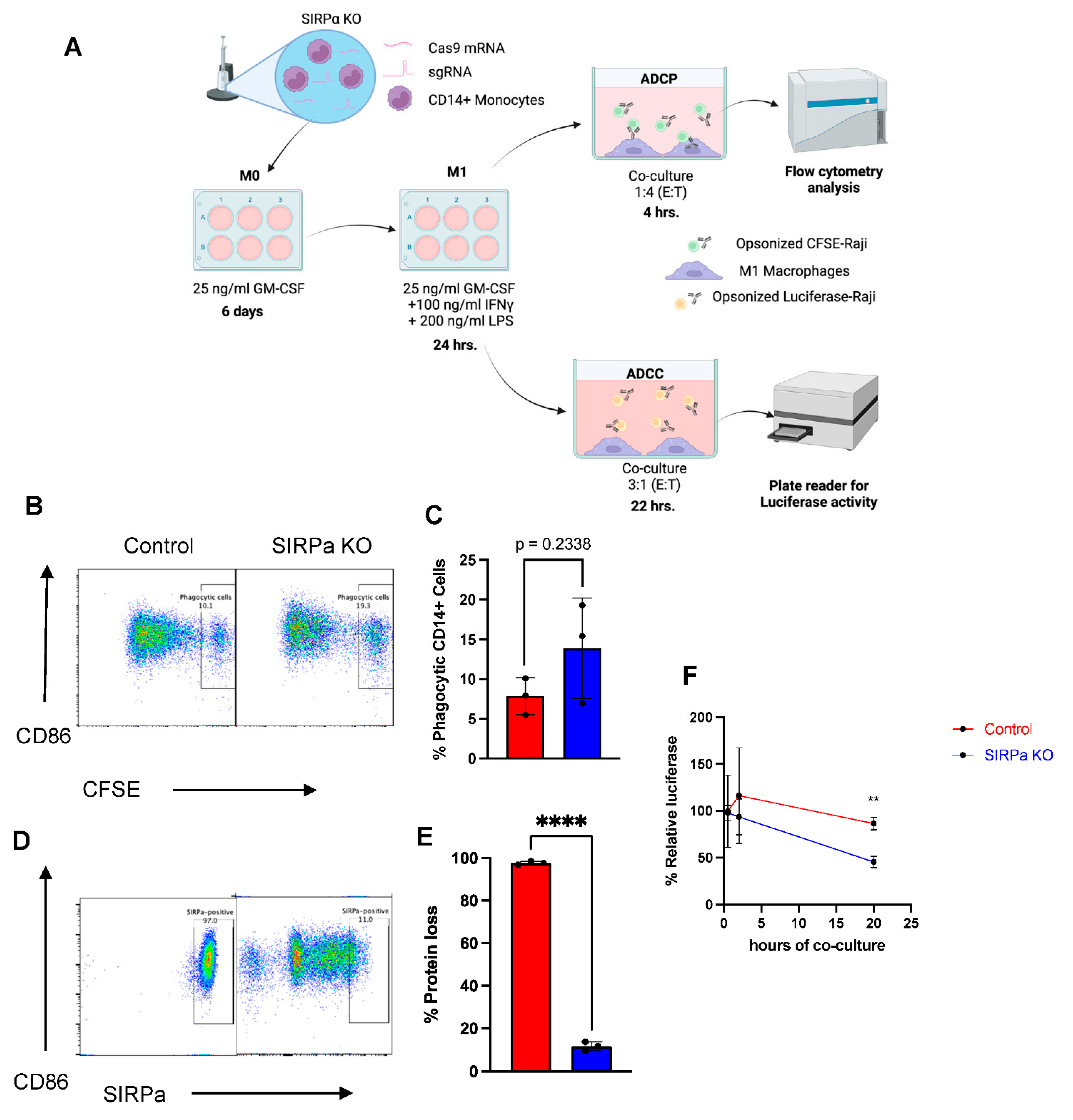
2.7. Genetically Engineered Primary Human Monocytes Have Enhanced Anti-Cancer Activity In Vitro
3. Discussion
4. Materials and Methods
4.1. Monocyte Isolation and Culture
4.2. M1 Polarization
4.3. Guide RNA Design and Cas9 RNP Preparation
4.4. Monocyte Electroporation
4.5. Quantification of Genome Editing
4.6. Flow Cytometry
4.7. RNase Alert Assay
4.8. rAAV Design and Transduction
4.9. Quantification of Phagocytosis
4.10. Quantification of Antibody-Dependent Cellular Cytoxicity (ADCC)
4.11. Statistical Analysis and Figure Construction
5. Patents
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sprangers, S.; de Vries, T.J.; Everts, V. Monocyte Heterogeneity: Consequences for Monocyte-Derived Immune Cells. J. Immunol. Res. 2016, 2016, 1475435. [Google Scholar] [CrossRef] [PubMed]
- Van Furth, R.; Cohn, Z.A. The origin and kinetics of mononuclear phagocytes. J. Exp. Med. 1968, 128, 415–435. [Google Scholar] [CrossRef] [PubMed]
- Steinman, R.M.; Lustig, D.S.; Cohn, Z.A. Identification of a novel cell type in peripheral lymphoid organs of mice: III. Functional properties in vivo. J. Exp. Med. 1974, 139, 1431–1445. [Google Scholar] [CrossRef] [PubMed]
- Geissmann, F.; Manz, M.G.; Jung, S.; Sieweke, M.H.; Merad, M.; Ley, K. Development of Monocytes, Macrophages, and Dendritic Cells. Science 2010, 327, 656–661. [Google Scholar] [CrossRef]
- León, B.; López-Bravo, M.; Ardavín, C. Monocyte-derived dendritic cells. Semin. Immunol. 2005, 17, 313–318. [Google Scholar] [CrossRef]
- Bosshart, H.; Heinzelmann, M. THP-1 cells as a model for human monocytes. Ann. Transl. Med. 2016, 4, 438. [Google Scholar] [CrossRef]
- Schildberger, A.; Rossmanith, E.; Eichhorn, T.; Strassl, K.; Weber, V. Monocytes, peripheral blood mononuclear cells, and THP-1 cells exhibit different cytokine expression patterns following stimulation with lipopolysaccharide. Mediat. Inflamm. 2013, 2013, 697972. [Google Scholar] [CrossRef]
- Wynn, T.A.; Chawla, A.; Pollard, J.W. Macrophage biology in development, homeostasis and disease. Nature 2013, 496, 445–455. [Google Scholar] [CrossRef]
- Vogel, D.Y.S.; Kooij, G.; Heijnen, P.D.A.M.; Breur, M.; Peferoen, L.A.N.; van der Valk, P.; de Vries, H.E.; Amor, S.; Dijkstra, C.D. GM-CSF promotes migration of human monocytes across the blood brain barrier. Eur. J. Immunol. 2015, 45, 1808–1819. [Google Scholar] [CrossRef]
- Shand, F.H.W.; Ueha, S.; Otsuji, M.; Koid, S.S.; Shichino, S.; Tsukui, T.; Kosugi-Kanaya, M.; Abe, J.; Tomura, M.; Ziogas, J.; et al. Tracking of intertissue migration reveals the origins of tumor-infiltrating monocytes. Proc. Natl. Acad. Sci. USA 2014, 111, 7771–7776. [Google Scholar] [CrossRef] [Green Version]
- Italiani, P.; Boraschi, D. From Monocytes to M1/M2 Macrophages: Phenotypical vs. Functional Differentiation. Front. Immunol. 2014, 5, 514. [Google Scholar] [CrossRef] [PubMed]
- Sponaas, A.M.; Do Rosario, A.P.F.; Voisine, C.; Mastelic, B.; Thompson, J.; Koernig, S.; Jarra, W.; Renia, L.; Mauduit, M.; Potocnik, A.J.; et al. Migrating monocytes recruited to the spleen play an important role in control of blood stage malaria. Blood 2009, 114, 5522–5531. [Google Scholar] [CrossRef] [PubMed]
- Vangetiid, S.; Strandinid, T.; Liu, S.; Tauriainen, J.; Raïsänen-Sokolowski, A.; Cabreraid, L.; Hassinenid, A.; Id, S.M.; Mustonen, J.; Vaheri, A.; et al. Monocyte subset redistribution from blood to kidneys in patients with Puumala virus caused hemorrhagic fever with renal syndrome. PLOS Pathog. 2021, 17, e1009400. [Google Scholar]
- Richards, D.M.; Hettinger, J.; Feuerer, M. Monocytes and Macrophages in Cancer: Development and Functions. Cancer Microenviron. 2013, 6, 179–191. [Google Scholar] [CrossRef] [PubMed]
- Haldar, M.; Murphy, K.M. Origin, development, and homeostasis of tissue-resident macrophages. Immunol. Rev. 2014, 262, 25–35. [Google Scholar] [CrossRef]
- Merad, M.; Manz, M.G. Dendritic cell homeostasis. Blood 2009, 113, 3418–3427. [Google Scholar] [CrossRef] [PubMed]
- Wculek, S.K.; Cueto, F.J.; Mujal, A.M.; Melero, I.; Krummel, M.F.; Sancho, D. Dendritic cells in cancer immunology and immunotherapy. Nat. Rev. Immunol. 2020, 20, 7–24. [Google Scholar] [CrossRef] [PubMed]
- Gleitz, H.F.; Liao, A.Y.; Cook, J.R.; Rowlston, S.F.; Forte, G.M.; D’Souza, Z.; O’Leary, C.; Holley, R.J.; Bigger, B.W. Brain-targeted stem cell gene therapy corrects mucopolysaccharidosis type II via multiple mechanisms. EMBO Mol. Med. 2018, 10, e8730. [Google Scholar] [CrossRef]
- Khan, K.D.; Emmanouilides, C.; Benson, D.M.; Hurst, D.; Garcia, P.; Michelson, G.; Milan, S.; Ferketich, A.K.; Piro, L.; Leonard, J.P.; et al. A phase 2 study of rituximab in combination with recombinant interleukin-2 for rituximab-refractory indolent non-Hodgkin’s lymphoma. Clin. Cancer Res. 2006, 12, 7046–7053. [Google Scholar] [CrossRef]
- Alpaugh, R.K.; Von Mehren, M.; Palazzo, I.; Atkins, M.B.; Sparano, J.A.; Schuchter, L.; Weiner, L.M.; Dutcher, J.P. Phase IB trial for malignant melanoma using R24 monoclonal antibody, interleukin-2/alpha-interferon. Med. Oncol. 1998, 15, 191–198. [Google Scholar] [CrossRef]
- Randolph, G.J.; Jakubzick, C.; Qu, C. Antigen presentation by monocytes and monocyte-derived cells. Curr. Opin. Immunol. 2008, 20, 52–60. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feng, M.; Jiang, W.; Kim, B.Y.S.; Zhang, C.C.; Fu, Y.X.; Weissman, I.L. Phagocytosis checkpoints as new targets for cancer immunotherapy. Nat. Rev. Cancer 2019, 19, 568–586. [Google Scholar] [CrossRef] [PubMed]
- Rupp, L.J.; Schumann, K.; Roybal, K.T.; Gate, R.E.; Ye, C.J.; Lim, W.A.; Marson, A. CRISPR/Cas9-mediated PD-1 disruption enhances anti-tumor efficacy of human chimeric antigen receptor T cells. Sci. Rep. 2017, 7, 737. [Google Scholar] [CrossRef] [PubMed]
- Shang, W.; Wang, F.; Zhu, Q.; Wang, L.; Wang, H. CRISPR/Cas9-Based Genetic Screening to Study T-Cell Function. Methods Mol. Biol. 2020, 2111, 59–70. [Google Scholar]
- Steinhart, Z.; Pavlovic, Z.; Chandrashekhar, M.; Hart, T.; Wang, X.; Zhang, X.; Robitaille, M.; Brown, K.R.; Jaksani, S.; Overmeer, R.; et al. Genome-wide CRISPR screens reveal a Wnt–FZD5 signaling circuit as a druggable vulnerability of RNF43-mutant pancreatic tumors. Nat. Med. 2017, 23, 60–68. [Google Scholar] [CrossRef] [PubMed]
- Tabdanov, E.D.; Rodríguez-Merced, N.J.; Cartagena-Rivera, A.X.; Puram, V.V.; Callaway, M.K.; Ensminger, E.A.; Pomeroy, E.J.; Yamamoto, K.; Lahr, W.S.; Webber, B.R.; et al. Engineering T cells to enhance 3D migration through structurally and mechanically complex tumor microenvironments. Nat. Commun. 2021, 12, 2815. [Google Scholar] [CrossRef]
- Webber, B.R.; Lonetree, C.; Kluesner, M.G.; Johnson, M.J.; Pomeroy, E.J.; Diers, M.D.; Lahr, W.S.; Draper, G.M.; Slipek, N.J.; Smeester, B.A.; et al. Highly efficient multiplex human T cell engineering without double-strand breaks using Cas9 base editors. Nat. Commun. 2019, 10, 5222. [Google Scholar]
- Pomeroy, E.J.; Hunzeker, J.T.; Kluesner, M.G.; Lahr, W.S.; Smeester, B.A.; Crosby, M.R.; Lonetree, C.; Yamamoto, K.; Bendzick, L.; Miller, J.S.; et al. A Genetically Engineered Primary Human Natural Killer Cell Platform for Cancer Immunotherapy. Mol. Ther. 2020, 28, 52–63. [Google Scholar] [CrossRef]
- Huang, R.-S.; Shih, H.-A.; Lai, M.-C.; Chang, Y.-J.; Lin, S. Enhanced NK-92 Cytotoxicity by CRISPR Genome Engineering Using Cas9 Ribonucleoproteins. Front. Immunol. 2020, 11, 1008. [Google Scholar] [CrossRef]
- Lambert, M.; Leijonhufvud, C.; Segerberg, F.; Melenhorst, J.J.; Carlsten, M. CRISPR/Cas9-Based Gene Engineering of Human Natural Killer Cells: Protocols for Knockout and Readouts to Evaluate Their Efficacy. Methods Mol. Biol. 2020, 2121, 213–239. [Google Scholar]
- Johnson, M.J.; Laoharawee, K.; Lahr, W.S.; Webber, B.R.; Moriarity, B.S. Engineering of Primary Human B cells with CRISPR/Cas9 Targeted Nuclease. Sci. Rep. 2018, 8, 12144. [Google Scholar] [CrossRef] [PubMed]
- Moffett, H.F.; Harms, C.K.; Fitzpatrick, K.S.; Tooley, M.R.; Boonyaratanakornkit, J.; Taylor, J.J. B cells engineered to express pathogen-specific antibodies protect against infection. Sci. Immunol. 2019, 4, eaax0644. [Google Scholar] [CrossRef] [PubMed]
- Luo, B.; Zhan, Y.; Luo, M.; Dong, H.; Liu, J.; Lin, Y.; Zhang, J.; Wang, G.; Verhoeyen, E.; Zhang, Y.; et al. Engineering of α-PD-1 antibody-expressing long-lived plasma cells by CRISPR/Cas9-mediated targeted gene integration. Cell Death Dis. 2020, 11, 973. [Google Scholar] [CrossRef] [PubMed]
- Hiatt, J.; Cavero, D.A.; McGregor, M.J.; Zheng, W.; Budzik, J.M.; Roth, T.L.; Haas, K.M.; Wu, D.; Rathore, U.; Meyer-Franke, A.; et al. Efficient generation of isogenic primary human myeloid cells using CRISPR-Cas9 ribonucleoproteins. Cell Rep. 2021, 35, 109105. [Google Scholar] [CrossRef]
- Brinkman, E.K.; Chen, T.; Amendola, M.; van Steensel, B. Easy quantitative assessment of genome editing by sequence trace decomposition. Nucleic Acids Res. 2014, 42, e168. [Google Scholar] [CrossRef] [PubMed]
- Willingham, S.B.; Volkmer, J.-P.; Gentles, A.J.; Sahoo, D.; Dalerba, P.; Mitra, S.S.; Wang, J.; Contreras-Trujillo, H.; Martin, R.; Cohen, J.D.; et al. The CD47-signal regulatory protein alpha (SIRPα) interaction is a therapeutic target for human solid tumors. Proc. Natl. Acad. Sci. USA 2012, 109, 6662–6667. [Google Scholar] [CrossRef]
- Liu, Q.; Wen, W.; Tang, L.; Qin, C.J.; Lin, Y.; Zhang, H.L.; Wu, H.; Ashton, C.; Wu, H.P.; Ding, J.; et al. Inhibition of SIRPα in dendritic cells potentiates potent antitumor immunity. Oncoimmunology 2016, 5, e1183850. [Google Scholar] [CrossRef]
- Tseng, D.; Volkmer, J.P.; Willingham, S.B.; Contreras-Trujillo, H.; Fathman, J.W.; Fernhoff, N.B.; Seita, J.; Inlay, M.A.; Weiskopf, K.; Miyanishi, M.; et al. Anti-CD47 antibody-mediated phagocytosis of cancer by macrophages primes an effective antitumor T-cell response. Proc. Natl. Acad. Sci. USA 2013, 110, 11103–11108. [Google Scholar] [CrossRef]
- Ring, N.G.; Herndler-Brandstetter, D.; Weiskopf, K.; Shan, L.; Volkmer, J.P.; George, B.M.; Lietzenmayer, M.; McKenna, K.M.; Naik, T.J.; McCarty, A.; et al. Anti-SIRPα antibody immunotherapy enhances neutrophil and macrophage antitumor activity. Proc. Natl. Acad. Sci. USA 2017, 114, E10578–E10585. [Google Scholar] [CrossRef]
- Rees, H.A.; Liu, D.R. Base editing: Precision chemistry on the genome and transcriptome of living cells. Nat. Rev. Genet. 2018, 19, 770–788. [Google Scholar] [CrossRef]
- Anzalone, A.V.; Randolph, P.B.; Davis, J.R.; Sousa, A.A.; Koblan, L.W.; Levy, J.M.; Chen, P.J.; Wilson, C.; Newby, G.A.; Raguram, A.; et al. Search-and-replace genome editing without double-strand breaks or donor DNA. Nature 2019, 576, 149–157. [Google Scholar] [CrossRef] [PubMed]
- Moradian, H.; Roch, T.; Lendlein, A.; Gossen, M. mRNA Transfection-Induced Activation of Primary Human Monocytes and Macrophages: Dependence on Carrier System and Nucleotide Modification. Sci. Rep. 2020, 10, 1–15. [Google Scholar] [CrossRef]
- Sahin, U.; Karikó, K.; Türeci, Ö. mRNA-based therapeutics—Developing a new class of drugs. Nat. Rev. Drug Discov. 2014, 13, 759–780. [Google Scholar] [CrossRef]
- Dyer, K.D.; Rosenberg, H.F. The RNase a superfamily: Generation of diversity and innate host defense. Mol. Divers. 2006, 10, 585–597. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koczera, P.; Martin, L.; Marx, G.; Schuerholz, T. The ribonuclease a superfamily in humans: Canonical RNases as the buttress of innate immunity. Int. J. Mol. Sci. 2016, 17, 1278. [Google Scholar] [CrossRef] [PubMed]
- Lu, L.; Li, J.; Moussaoui, M.; Boix, E. Immune modulation by human secreted RNases at the extracellular space. Front. Immunol. 2018, 9, 1012. [Google Scholar] [CrossRef]
- Chen, L.; Ge, B.; Casale, F.P.; Vasquez, L.; Kwan, T.; Garrido-Martín, D.; Watt, S.; Yan, Y.; Kundu, K.; Ecker, S.; et al. Genetic Drivers of Epigenetic and Transcriptional Variation in Human Immune Cells. Cell 2016, 167, 1398–1414.e24. [Google Scholar] [CrossRef]
- Peterson, C.W.; Venkataraman, R.; Reddy, S.S.; Pande, D.; Enstrom, M.R.; Radtke, S.; Humbert, O.; Kiem, H.P. Intracellular RNase activity dampens zinc finger nuclease-mediated gene editing in hematopoietic stem and progenitor cells. Mol. Ther. Methods Clin. Dev. 2022, 24, 30–39. [Google Scholar] [CrossRef]
- Protector RNase Inhibitor Sigma-Aldrich. Available online: https://www.sigmaaldrich.com/US/en/product/roche/rnainhro?gclid=CjwKCAiA4KaRBhBdEiwAZi1zzrZ4yaNErkyn6UsX8mbPrirGM4TKgeT2OWXKJ1vNHFHRrBOJc7tUxRoCRikQAvD_BwE (accessed on 3 July 2020).
- Vrtačnik, P.; Kos, Š.; Bustin, S.A.; Marc, J.; Ostanek, B. Influence of trypsinization and alternative procedures for cell preparation before RNA extraction on RNA integrity. Anal. Biochem. 2014, 463, 38–44. [Google Scholar] [CrossRef]
- Komor, A.C.; Kim, Y.B.; Packer, M.S.; Zuris, J.A.; Liu, D.R. Programmable editing of a target base in genomic DNA without double-stranded DNA cleavage. Nature 2016, 533, 420. [Google Scholar] [CrossRef]
- Gaudelli, N.M.; Komor, A.C.; Rees, H.A.; Packer, M.S.; Badran, A.H.; Bryson, D.I.; Liu, D.R. Programmable base editing of A•T to G•C in genomic DNA without DNA cleavage. Nature 2017, 551, 464. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Venet, F.; Wang, Y.L.; Lepape, A.; Yuan, Z.; Chen, Y.; Swan, R.; Kherouf, H.; Monneret, G.; Chung, C.S.; et al. PD-1 expression by macrophages plays a pathologic role in altering microbial clearance and the innate inflammatory response to sepsis. Proc. Natl. Acad. Sci. USA 2009, 106, 6303. [Google Scholar] [CrossRef] [PubMed]
- Kluesner, M.G.; Lahr, W.S.; Lonetree, C.; Smeester, B.A.; Qiu, X.; Slipek, N.J.; Claudio Vázquez, P.N.; Pitzen, S.P.; Pomeroy, E.J.; Vignes, M.J.; et al. CRISPR-Cas9 cytidine and adenosine base editing of splice-sites mediates highly-efficient disruption of proteins in primary and immortalized cells. Nat. Commun. 2021, 12, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, D.N.; Roth, T.L.; Li, P.J.; Chen, P.A.; Apathy, R.; Mamedov, M.R.; Vo, L.T.; Tobin, V.R.; Goodman, D.; Shifrut, E.; et al. Polymer-stabilized Cas9 nanoparticles and modified repair templates increase genome editing efficiency. Nat. Biotechnol. 2019, 38, 44–49. [Google Scholar] [CrossRef]
- Roth, T.L.; Puig-Saus, C.; Yu, R.; Shifrut, E.; Carnevale, J.; Li, P.J.; Hiatt, J.; Saco, J.; Krystofinski, P.; Li, H.; et al. Reprogramming human T cell function and specificity with non-viral genome targeting. Nature 2018, 559, 405–409. [Google Scholar] [CrossRef]
- Ussher, J.E.; Taylor, J.A. Optimized transduction of human monocyte-derived dendritic cells by recombinant adeno-associated virus serotype 6. Hum. Gene Ther. 2010, 21, 1675–1686. [Google Scholar] [CrossRef]
- Challita, P.M.; Skelton, D.; el-Khoueiry, A.; Yu, X.J.; Weinberg, K.; Kohn, D.B. Multiple modifications in cis elements of the long terminal repeat of retroviral vectors lead to increased expression and decreased DNA methylation in embryonic carcinoma cells. J. Virol. 1995, 69, 748–755. [Google Scholar] [CrossRef]
- Hayashi, H.; Kubo, Y.; Izumida, M.; Matsuyama, T. Efficient viral delivery of Cas9 into human safe harbor. Sci. Rep. 2020, 10, 1–14. [Google Scholar] [CrossRef]
- Laoharawee, K.; Johnson, M.J.; Lahr, W.S.; Peterson, J.J.; Webber, B.R.; Moriarity, B.S. Genome Engineering of Primary Human B Cells Using CRISPR/Cas9. J. Vis. Exp. 2020, 165, e61855. [Google Scholar] [CrossRef]
- Chao, M.P.; Alizadeh, A.A.; Tang, C.; Myklebust, J.H.; Varghese, B.; Gill, S.; Jan, M.; Cha, A.C.; Chan, C.K.; Tan, B.T.; et al. Anti-CD47 antibody synergizes with rituximab to promote phagocytosis and eradicate non-Hodgkin lymphoma. Cell 2010, 142, 699. [Google Scholar] [CrossRef]
- Uccellini, M.B.; Aslam, S.; Liu, S.T.H.; Alam, F.; García-Sastre, A. Development of a Macrophage-Based ADCC Assay. Vaccines 2021, 9, 660. [Google Scholar] [CrossRef] [PubMed]
- Auffray, C.; Sieweke, M.H.; Geissmann, F. Blood Monocytes: Development, Heterogeneity, and Relationship with Dendritic Cells. Annu. Rev. Immunol. 2009, 27, 669–692. [Google Scholar] [CrossRef] [PubMed]
- Yona, S.; Kim, K.-W.; Wolf, Y.; Mildner, A.; Varol, D.; Breker, M.; Strauss-Ayali, D.; Viukov, S.; Guilliams, M.; Misharin, A.; et al. Fate Mapping Reveals Origins and Dynamics of Monocytes and Tissue Macrophages under Homeostasis. Immunity 2013, 38, 79–91. [Google Scholar] [CrossRef] [PubMed]
- Brooks, R.C.; Hasley, P.B.; Jasti, H.; Macpherson, D. Comparison of gene expression profiles between human and mouse monocyte subsets. Ann. Intern. Med. 2012, 156, 649–654. [Google Scholar] [CrossRef]
- Ang, Z.; Er, J.Z.; Tan, N.S.; Lu, J.; Liou, Y.-C.; Grosse, J.; Ding, J.L. Human and mouse monocytes display distinct signalling and cytokine profiles upon stimulation with FFAR2/FFAR3 short-chain fatty acid receptor agonists. Sci. Rep. 2016, 6, 34145. [Google Scholar] [CrossRef]
- Jang, H.K.; Jo, D.H.; Lee, S.N.; Cho, C.S.; Jeong, Y.K.; Jung, Y.; Yu, J.; Kim, J.H.; Woo, J.S.; Bae, S. High-purity production and precise editing of DNA base editing ribonucleoproteins. Sci. Adv. 2021, 7, eabg2661. [Google Scholar] [CrossRef]
- Osborn, M.J.; Webber, B.R.; Knipping, F.; Lonetree, C.L.; Tennis, N.; DeFeo, A.P.; McElroy, A.N.; Starker, C.G.; Lee, C.; Merkel, S.; et al. Evaluation of TCR Gene Editing Achieved by TALENs, CRISPR/Cas9, and megaTAL Nucleases. Mol. Ther. 2016, 24, 570–581. [Google Scholar] [CrossRef]
- Liu, M.; Rehman, S.; Tang, X.; Gu, K.; Fan, Q.; Chen, D.; Ma, W. Methodologies for Improving HDR Efficiency. Front. Genet. 2019, 9, 691. [Google Scholar] [CrossRef]
- Yao, X.; Wang, X.; Hu, X.; Liu, Z.; Liu, J.; Zhou, H.; Shen, X.; Wei, Y.; Huang, Z.; Ying, W.; et al. Homology-mediated end joining-based targeted integration using CRISPR/Cas9. Cell Res. 2017, 27, 801–814. [Google Scholar] [CrossRef]
- Aziz, A.; Soucie, E.; Sarrazin, S.; Sieweke, M.H. MafB/c-Maf Deficiency Enables Self-Renewal of Differentiated Functional Macrophages. Science 2009, 326, 867–871. [Google Scholar] [CrossRef]
- Klichinsky, M.; Ruella, M.; Shestova, O.; Lu, X.M.; Best, A.; Zeeman, M.; Schmierer, M.; Gabrusiewicz, K.; Anderson, N.R.; Petty, N.E.; et al. Human chimeric antigen receptor macrophages for cancer immunotherapy. Nat. Biotechnol. 2020, 38, 947. [Google Scholar] [CrossRef] [PubMed]
- Oldenborg, P.A.; Zheleznyak, A.; Fang, Y.F.; Lagenaur, C.F.; Gresham, H.D.; Lindberg, F.P. Role of CD47 as a marker of self on red blood cells. Science 2000, 288, 2051–2054. [Google Scholar] [CrossRef] [PubMed]
- Wettersten, H.I.; Weis, S.M.; Pathria, P.; Schalscha TVon Minami, T.; Varner, J.A.; Cheresh, D.A. Arming tumor-associated macrophages to reverse epithelial cancer progression. Cancer Res. 2019, 79, 5048–5059. [Google Scholar] [CrossRef] [PubMed]
- Dubois, N.C.; Craft, A.M.; Sharma, P.; Elliott, D.A.; Stanley, E.G.; Elefanty, A.G.; Gramolini, A.; Keller, G. SIRPA is a specific cell-surface marker for isolating cardiomyocytes derived from human pluripotent stem cells. Nat. Biotechnol. 2011, 29, 1011. [Google Scholar] [CrossRef] [Green Version]
- Hendel, A.; Bak, R.O.; Clark, J.T.; Kennedy, A.B.; Ryan, D.E.; Roy, S.; Steinfeld, I.; Lunstad, B.D.; Kaiser, R.J.; Wilkens, A.B.; et al. Chemically modified guide RNAs enhance CRISPR-Cas genome editing in human primary cells. Nat. Biotechnol. 2015, 33, 985–989. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Protocol Name | Voltage (Volts) | Bandwidth (Milliseconds) | Number of Pulses |
|---|---|---|---|
| E0 | 0 | 0 | 0 |
| E1 | 1300 | 10 | 3 |
| E2 | 1400 | 10 | 3 |
| E3 | 1500 | 30 | 1 |
| E4 | 1600 | 10 | 3 |
| E5 | 1700 | 20 | 2 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Laoharawee, K.; Johnson, M.J.; Lahr, W.S.; Sipe, C.J.; Kleinboehl, E.; Peterson, J.J.; Lonetree, C.-l.; Bell, J.B.; Slipek, N.J.; Crane, A.T.; et al. A Pan-RNase Inhibitor Enabling CRISPR-mRNA Platforms for Engineering of Primary Human Monocytes. Int. J. Mol. Sci. 2022, 23, 9749. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23179749
Laoharawee K, Johnson MJ, Lahr WS, Sipe CJ, Kleinboehl E, Peterson JJ, Lonetree C-l, Bell JB, Slipek NJ, Crane AT, et al. A Pan-RNase Inhibitor Enabling CRISPR-mRNA Platforms for Engineering of Primary Human Monocytes. International Journal of Molecular Sciences. 2022; 23(17):9749. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23179749
Chicago/Turabian StyleLaoharawee, Kanut, Matthew J. Johnson, Walker S. Lahr, Christopher J. Sipe, Evan Kleinboehl, Joseph J. Peterson, Cara-lin Lonetree, Jason B. Bell, Nicholas J. Slipek, Andrew T. Crane, and et al. 2022. "A Pan-RNase Inhibitor Enabling CRISPR-mRNA Platforms for Engineering of Primary Human Monocytes" International Journal of Molecular Sciences 23, no. 17: 9749. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23179749