
Tuning of Liver Sieve: The Interplay between Actin and Myosin Regulatory Light Chain Regulates Fenestration Size and Number in Murine Liver Sinusoidal Endothelial Cells
 , , ,
, , , 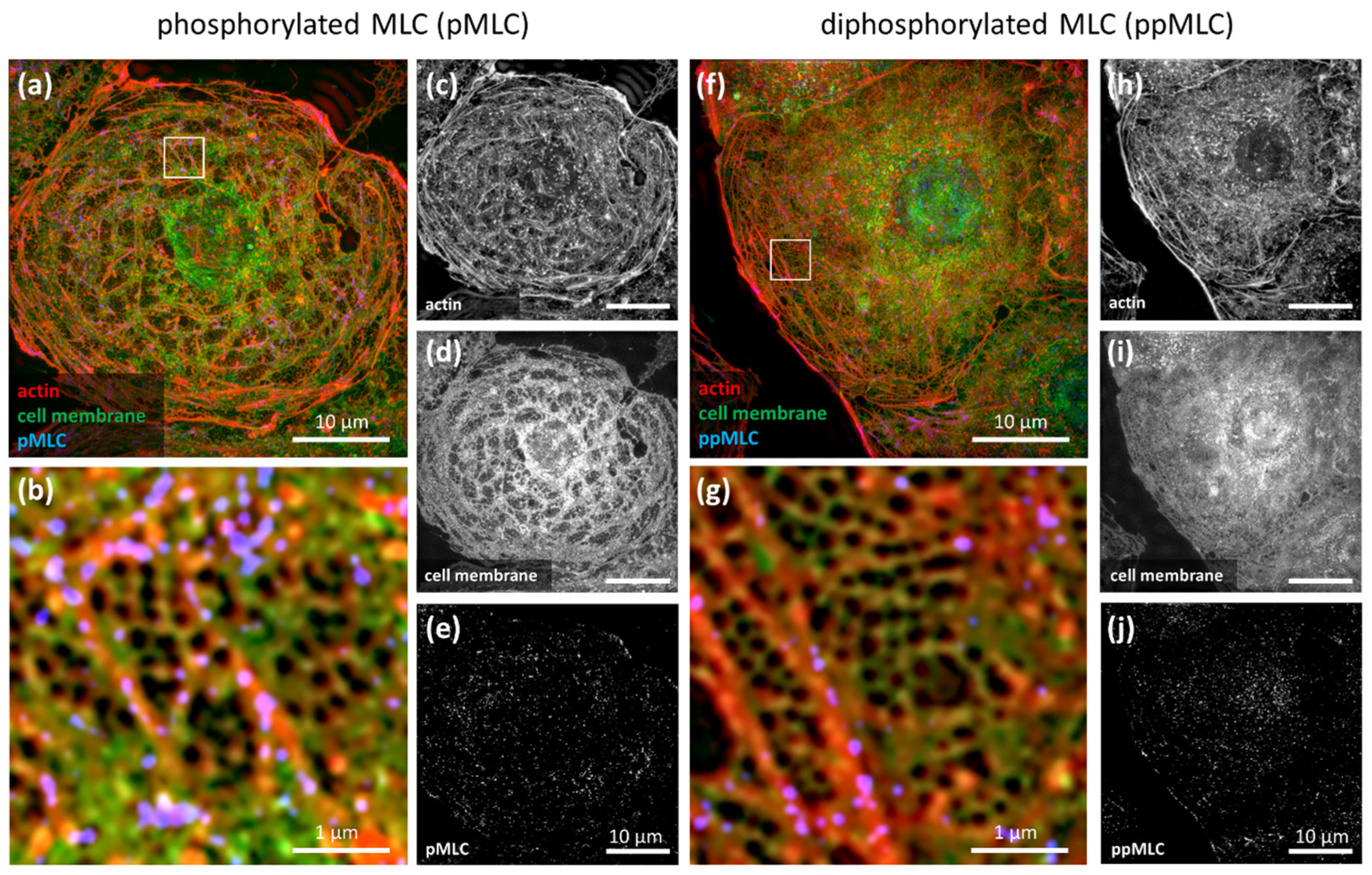{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
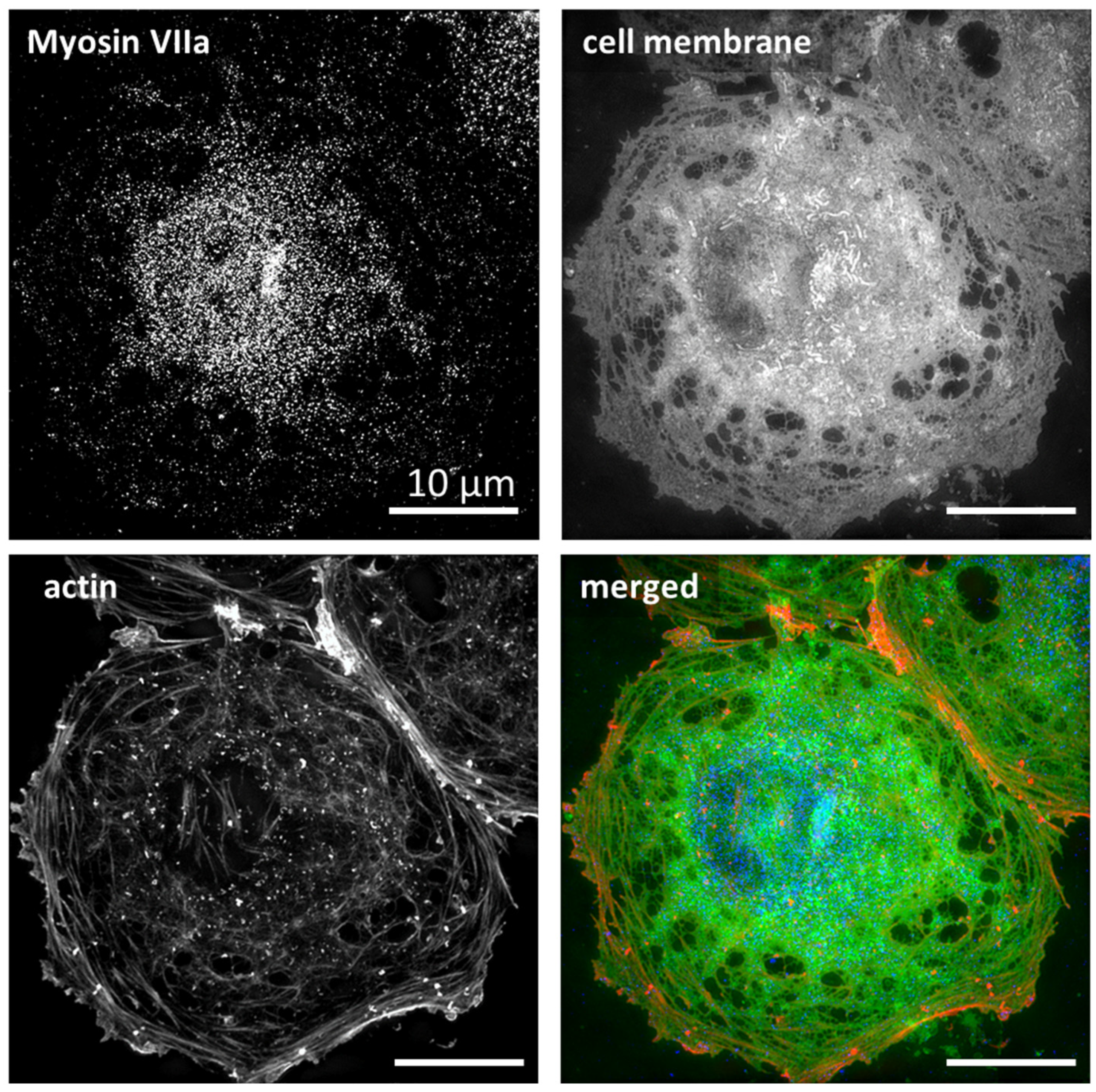{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
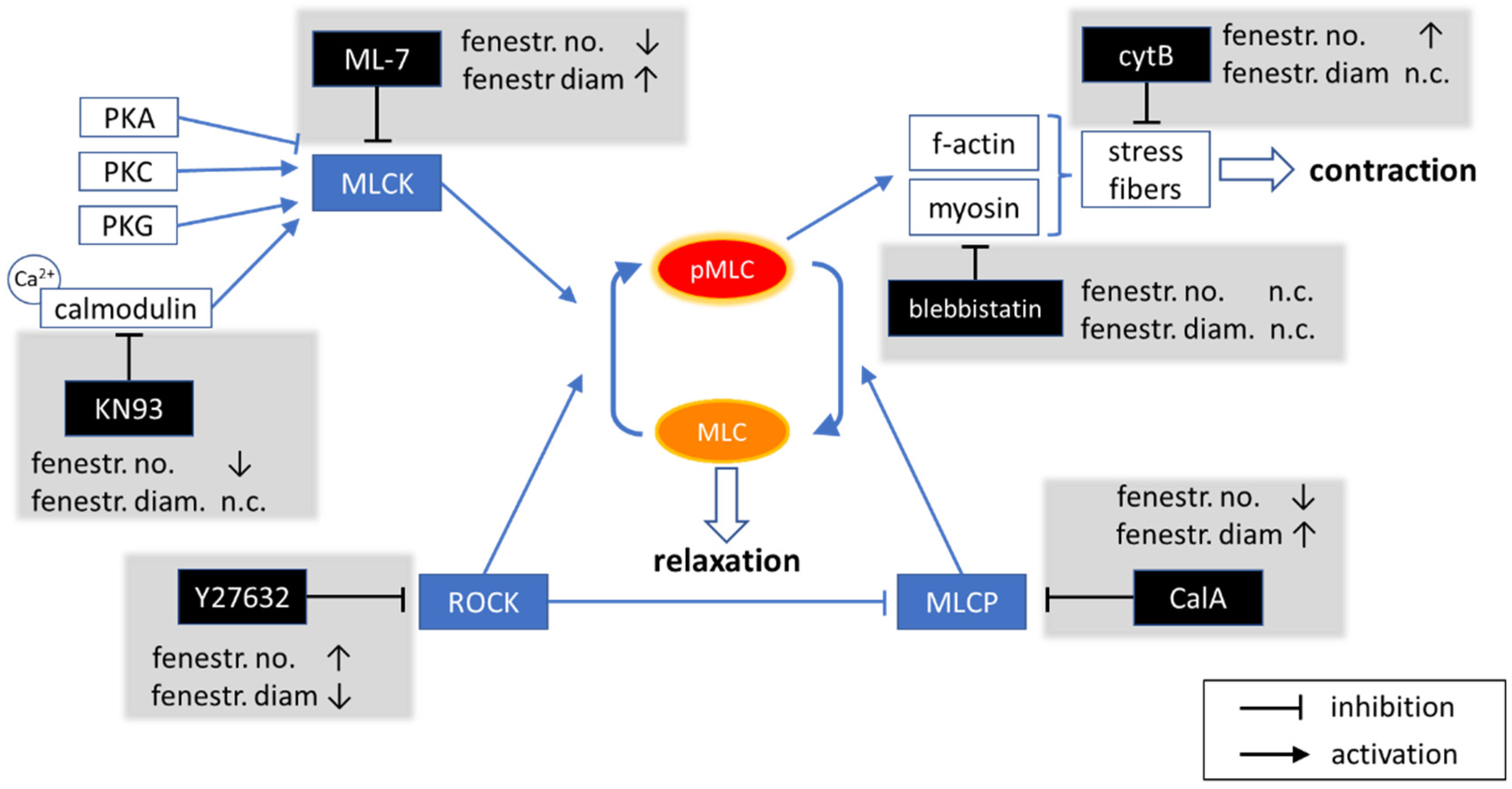
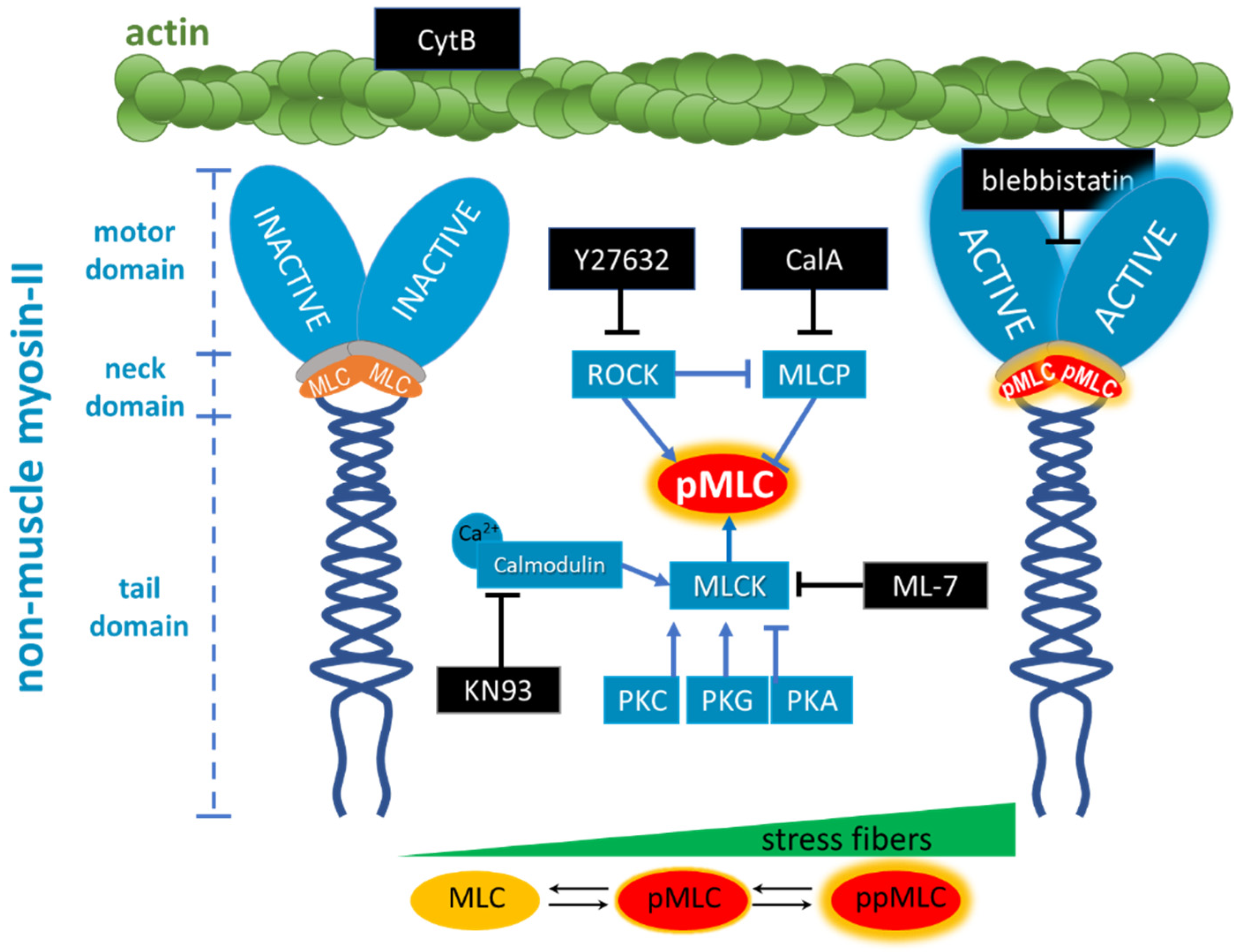
2.1. Modulation of MLC Phosphorylation
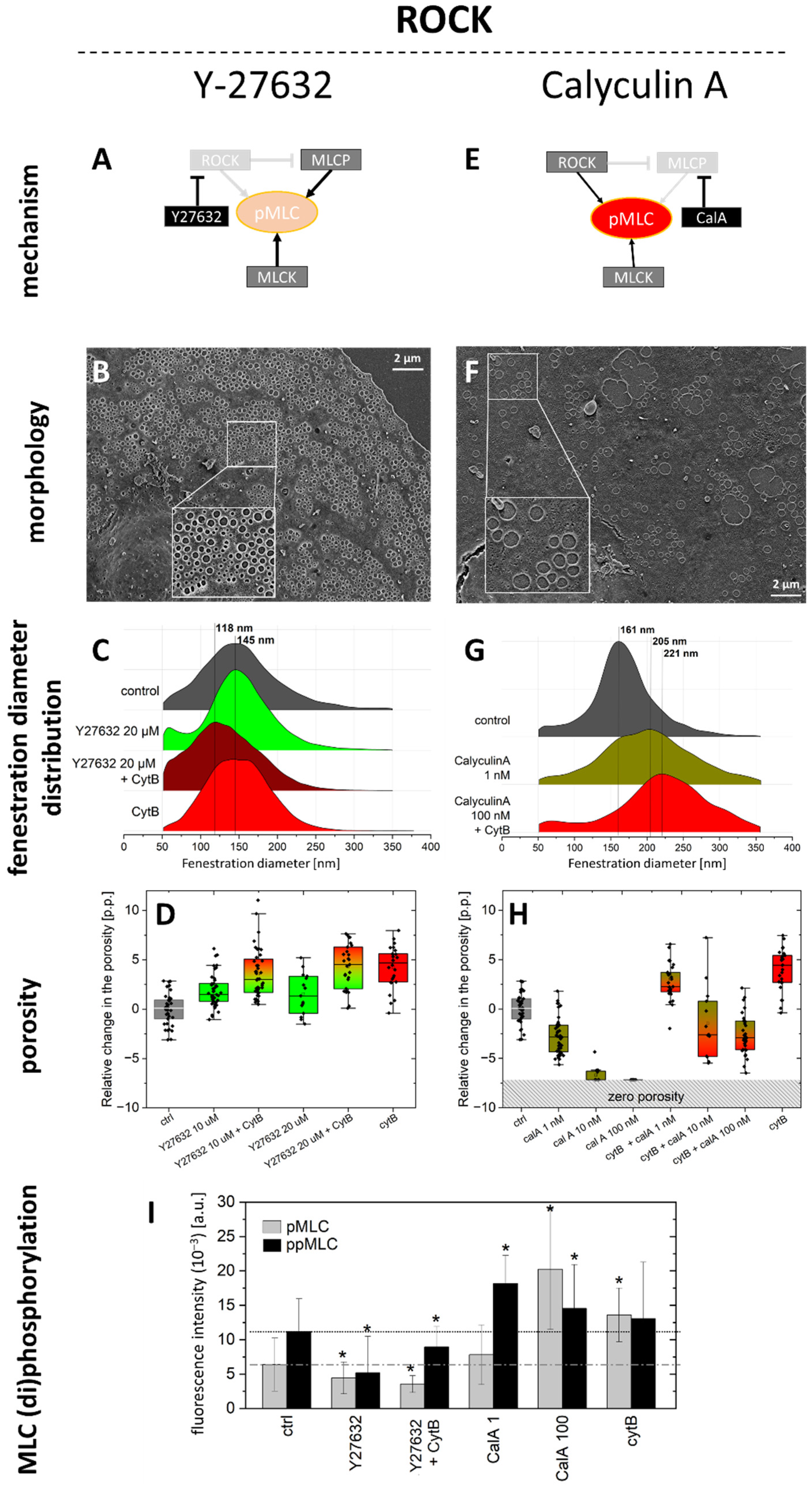
2.1.1. ROCK Pathway
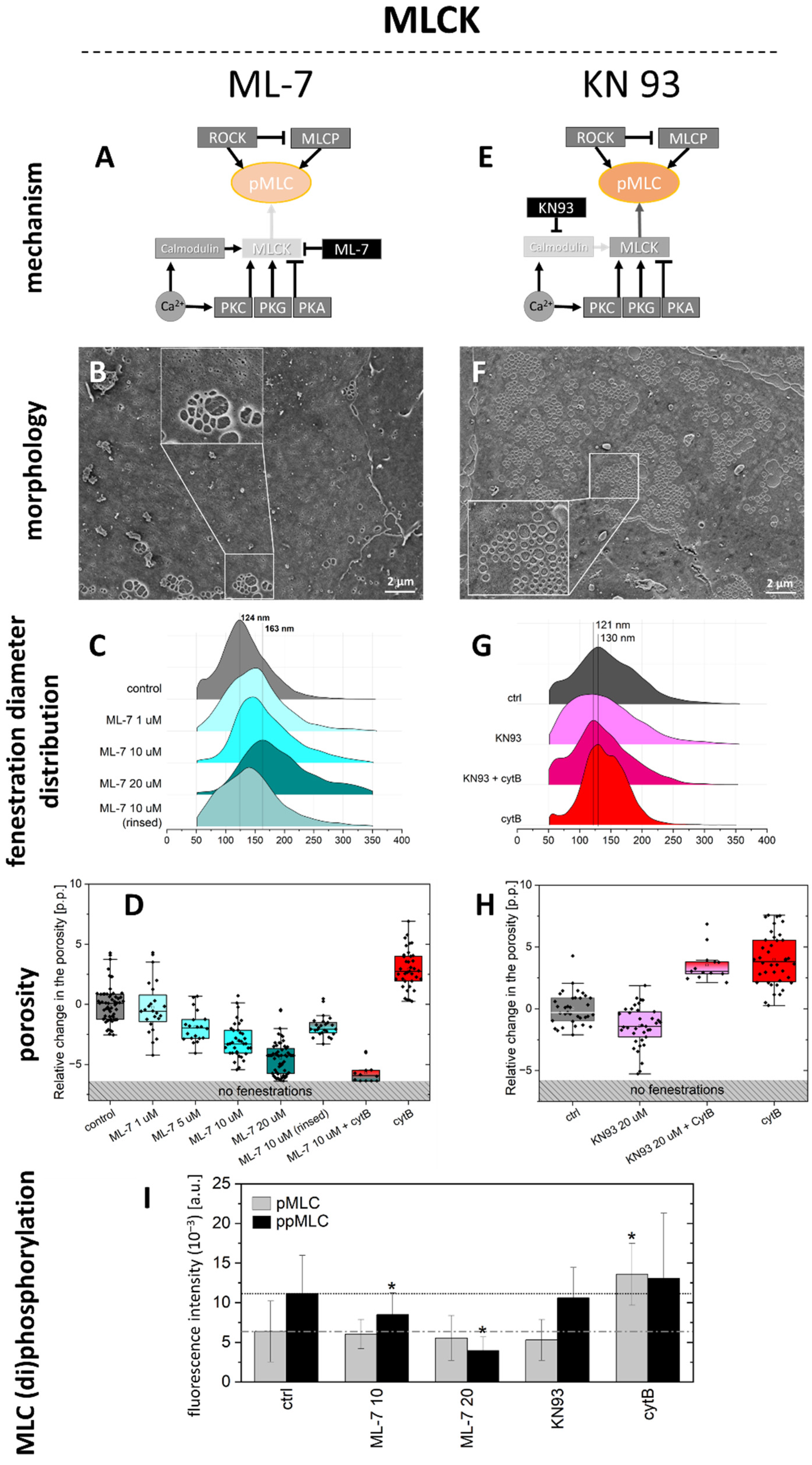
2.1.2. MLCK Pathway
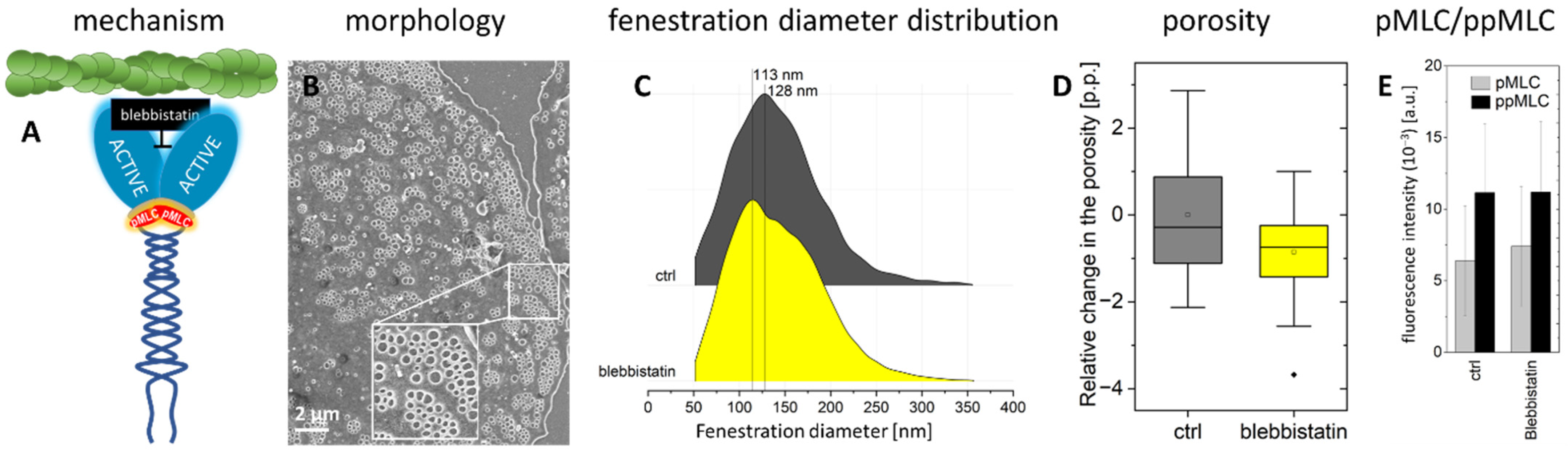
2.2. Actomyosin Contraction Uncoupling p/ppMLC with Blebbistatin
3. Discussion
4. Materials and Methods
4.1. Chemicals
4.2. Cell Isolation
4.3. Inhibitors
- Cytochalasin B (CytB) is used to depolymerise filamentous actin, and increases the number of fenestrations [15]. CytB is therefore used as a gold standard to easily induce fenestrations in LSEC (up to 300% increase in their number) within 15–60 min [13,50], without affecting the fenestration size [15,46]. We applied it to decrease the length of actin fibres. CytB thus decreases the overall force generated by activated myosin. We (pre)treated LSECs with CytB to enhance or abolish the effect of other inhibitors. We used 21 µM and 30 min of incubation in all experiments.
- (−)-Blebbistatin blocks myosin-II ATPase activity. It was reported that the addition of 20 µM blebbistatin decreased fibre tension by ~80% and the effect was independent of MLC phosphorylation [51]. Blebbistatin arrests the myosin cycle in states either detached from actin, or only weakly bound to actin [52]. We applied blebbistatin to decouple MLC-dependent contraction of actomyosin. We tested concentrations of 5, 10, and 20 µM (1 h incubation), and selected 20 µM because the effect on the shape of actin filaments was the most prominent, without significant effects on fenestrations.
- ML-7 hydrochloride is a reversible and ATP-competitive MLCK inhibitor. By blocking phosphorylation of MLCK, it blocks both calcium-calmodulin-dependent and independent MLCK-induced phosphorylation of MLC. The phosphorylation of MLC can still be facilitated by ROCK. We tested 1, 5, 10, and 20 µM (1 h incubation), observing a dose-dependent effect on fenestrations. However, the highest concentration reduced cell viability after 2 h of treatment (Figure S1).
- KN93 is a cell-permeable, reversible, and competitive inhibitor of calmodulin-dependent kinase type II. It selectively blocks calcium-dependent phosphorylation of MLCK without disturbing the calcium-independent phosphorylation. KN93 was used at a concentration of 20 µM (1 h incubation).
- Y27632 dihydrochloride is a ROCK inhibitor that causes relaxation of myosin tension in two ways: by preventing the direct phosphorylation of MLC via ROCK and by preventing ROCK-dependent phosphorylation/inactivation of MLCP. MLCK still phosphorylates MLC, but MLCP is constantly active, causing an elevated rate of the dephosphorylation of MLC. Y27632 dihydrochloride was used at a concentration of 10 µM (1 h incubation).
- Calyculin A (CalA) is a potent inhibitor of protein phosphatase 1 and 2A. It inhibits MLCP and other phosphatases. Phosphorylation of MLC via ROCK and MLCK occurs normally, but MLCP does not reverse it. We tested 1, 10, and 100 nM for 30 min, as longer treatments were detrimental to cell viability.
4.4. Microscopy
4.4.1. Scanning Electron Microscopy (SEM)
4.4.2. Structured Illumination Microscopy (SIM)
4.5. Data Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- De Zanger, R.; Wisse, E.; Knook, D. The Filtration Effect of Rat Liver Fenestrated Sinusoidal Endothelium on the Passage of (Remnant) Chylomicrons to the Space of Disse. In Sinusoidal Liver Cells; Knook, D.L., Wisse, E., Eds.; Elsevier Biomedical Liver Press: Amsterdam, The Netherlands, 1982; pp. 69–76. [Google Scholar]
- Poisson, J.; Lemoinne, S.; Boulanger, C.; Durand, F.; Moreau, R.; Valla, D.; Rautou, P. Liver Sinusoidal Endothelial Cells: Physiology and Role in Liver Diseases. J. Hepatol. 2017, 66, 212–227. [Google Scholar] [CrossRef] [PubMed]
- Sørensen, K.K.; McCourt, P.; Berg, T.; Crossley, C.; Le Couteur, D.; Wake, K.; Smedsrød, B. The Scavenger Endothelial Cell: A New Player in Homeostasis and Immunity. Am. J. Physiol.—Regul. Integr. Comp. Physiol. 2012, 303, R1217–R1230. [Google Scholar] [CrossRef] [PubMed]
- Braet, F. How Molecular Microscopy Revealed New Insights into the Dynamics of Hepatic Endothelial Fenestrae in the Past Decade. Liver Int. 2004, 24, 532–539. [Google Scholar] [CrossRef]
- Butola, A.; Coucheron, D.A.; Szafranska, K.; Ahmad, A.; Mao, H.; Tinguely, J.C.; McCourt, P.; Senthilkumaran, P.; Mehta, D.S.; Agarwal, K.; et al. Multimodal On-Chip Nanoscopy and Quantitative Phase Imaging Reveals the Nanoscale Morphology of Liver Sinusoidal Endothelial Cells. Proc. Natl. Acad. Sci. USA 2021, 118, e2115323118. [Google Scholar] [CrossRef] [PubMed]
- Mori, T.; Okanoue, T.; Sawa, Y.; Hori, N.; Ohta, M.; Kagawa, K. Defenestration of the Sinusoidal Endothelial-Cell in a Rat Model of Cirrhosis. Hepatology 1993, 17, 891–897. [Google Scholar] [PubMed]
- Warren, A.; Bertolino, P.; Cogger, V.C.; McLean, A.J.; Fraser, R.; Le Couteur, D.G. Hepatic Pseudocapillarization in Aged Mice. Exp. Gerontol. 2005, 40, 807–812. [Google Scholar] [CrossRef]
- Hunt, N.J.; Lockwood, G.P.; Warren, A.; Mao, H.; Mccourt, P.A.G.G.; Le Couteur, D.G.; Cogger, V.C.; Hunt, X.N.J.; Lockwood, G.P.; Warren, A.; et al. Manipulating Fenestrations in Young and Old Liver Sinusoidal Endothelial Cells. Am. J. Physiol.—Gastrointest. Liver Physiol. 2019, 316, G144–G154. [Google Scholar] [CrossRef] [PubMed]
- Cogger, V.C.; Hunt, N.J.; Le Couteur, D.G. Fenestrations in the Liver Sinusoidal Endothelial Cell. In The Liver: Biology and Pathobiology, 5th ed.; Wiley-Blackwell: Hoboken, NJ, USA, 2020; pp. 435–443. ISBN 9780470723135. [Google Scholar]
- Mitchell, S.J.; Huizer-pajkos, A.; Cogger, V.C.; Mclachlan, A.J.; Le Couteur, D.G.; Jones, B.; De Cabo, R.; Hilmer, S.N. Age-Related Pseudocapillarization of the Liver Sinusoidal Endothelium Impairs the Hepatic Clearance of Acetaminophen in Rats. J. Gerontol. Biol. Sci. 2011, 66A, 400–408. [Google Scholar] [CrossRef]
- Wisse, E. An Electron Microscopic Study of the Fenestrated Endothelial Lining of Rat Liver Sinusoids. J. Ultrasructure Res. 1970, 31, 125–150. [Google Scholar]
- Szafranska, K.; Holte, C.F.; Kruse, L.D.; McCourt, P.; Zapotoczny, B. The Whole Story about Fenestrations in Liver Sinusoidal Endothelial Cells. Front. Physiol. 2021, 12, 735573. [Google Scholar] [CrossRef]
- Zapotoczny, B.; Szafranska, K.; Kus, E.; Braet, F.; Wisse, E.; Chlopicki, S.; Szymonski, M. Tracking Fenestrae Dynamics in Live Murine Liver Sinusoidal Endothelial Cells. Hepatology 2019, 69, 876–888. [Google Scholar] [CrossRef] [PubMed]
- Spector, I.; Braet, F.; Shochet, N.R.; Bubb, M.R. New Anti-Actin Drugs in the Study of the Organization and Function of the Actin Cytoskeleton. Microsc. Res. Tech. 1999, 47, 18–37. [Google Scholar] [CrossRef] [PubMed]
- Steffan, A.M.; Gendrault, J.L.; Kirn, A. Increase in the Number of Fenestrae in Mouse Endothelial Liver Cells by Altering the Cytoskeleton with Cytochalasin, B. Hepatology 1987, 7, 1230–1238. [Google Scholar] [CrossRef]
- Braet, F.; Spector, I.; De Zanger, R.; Wisse, E. A Novel Structure Involved in the Formation of Liver Endothelial Cell Fenestrae Revealed by Using the Actin Inhibitor Misakinolide. Proc. Natl. Acad. Sci. USA 1998, 95, 13635–13640. [Google Scholar] [CrossRef] [PubMed]
- Svistounov, D.; Warren, A.; Mcnerney, G.P.; Owen, D.M.; Zencak, D.; Le, D.G.; Zykova, S.N.; Crane, H.; Huser, T.; Quinn, R.J.; et al. The Relationship between Fenestrations, Sieve Plates and Rafts in Liver Sinusoidal Endothelial Cells. PLoS ONE 2012, 7, e46134. [Google Scholar] [CrossRef]
- Zapotoczny, B.; Braet, F.; Kus, E.; Ginda-Mäkelä, K.; Klejevskaja, B.; Campagna, R.; Chlopicki, S.; Szymonski, M. Actin-Spectrin Scaffold Supports Open Fenestrae in Liver Sinusoidal Endothelial Cells. Traffic 2019, 20, 932–942. [Google Scholar] [CrossRef]
- Braet, F.; Wisse, E. Structural and Functional Aspects of Liver Sinusoidal Endothelial Cell Fenestrae: A Review. Comp. Hepatol. 2002, 1, 1. [Google Scholar] [CrossRef]
- Oda, M.; Azuma, T.; Watanabe, N.; Nishizaki, Y.; Nishida, J.; Ishii, K.; Suzuki, H.; Kaneko, H.; Komatsu, H.; Tsukada, N.; et al. Regulatory Mechanism of Hepatic Microcirculation: Involvement of the Contraction and Dilatation of Sinusoids and Sinusoidal Endothelial Fenestrae. In Gastrointestinal Microcirculation; Karger Publishers: Basel, Switzerland, 1990; Volume 17, pp. 103–128. [Google Scholar] [CrossRef]
- Gatmaitan, Z.; Varticovski, L.; Ling, L.; Mikkelsen, R.; Steffan, A.M.; Arias, I.M. Studies on Fenestral Contraction in Rat Liver Endothelial Cells in Culture. Am. J. Pathol. 1996, 148, 2027–2041. [Google Scholar]
- Brauneis, U.; Gatmaitan, Z.; Arias, I.M. Serotonin Stimulates a Ca2+ Permanent Nonspecific Cation Channel in Hepatic Endothelial Cells. Biochem. Biophys. Res. Commun. 1992, 186, 1560–1566. [Google Scholar] [CrossRef]
- Yokomori, H.; Yoshimura, K.; Funakoshi, S.; Nagai, T.; Fujimaki, K.; Nomura, M.; Ishii, H.; Oda, M. Rho Modulates Hepatic Sinusoidal Endothelial Fenestrae via Regulation of the Actin Cytoskeleton in Rat Endothelial Cells. Lab. Investig. 2004, 84, 857–864. [Google Scholar] [CrossRef]
- Venkatraman, L.; Tucker-Kellogg, L. The CD47-Binding Peptide of Thrombospondin-1 Induces Defenestration of Liver Sinusoidal Endothelial Cells. Liver Int. 2013, 33, 1386–1397. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shen, Q.; Rigor, R.R.; Pivetti, C.D.; Wu, M.H.; Yuan, S.Y. Myosin Light Chain Kinase in Microvascular Endothelial Barrier Function. Cardiovasc. Res. 2010, 87, 272–280. [Google Scholar] [CrossRef] [PubMed]
- Kazakova, O.A.; Khapchaev, A.Y.; Shirinsky, V.P. MLCK and ROCK Mutualism in Endothelial Barrier Dysfunction: Joint Work of MLCK and ROCK in Endothelium. Biochimie 2020, 168, 83–91. [Google Scholar] [CrossRef]
- Schnittler, H. Contraction of Endothelial Cells: 40 Years of Research, but the Debate Still Lives. Histochem. Cell Biol. 2016, 146, 651–656. [Google Scholar] [CrossRef]
- Szafranska, K.; Holte, C.F.; Kruse, L.D.; Mao, H.; Øie, C.I.; Szymonski, M.; Zapotoczny, B.; McCourt, P.A.G. Quantitative Analysis Methods for Studying Fenestrations in Liver Sinusoidal Endothelial Cells. A Comparative Study. Micron 2021, 150, 103121. [Google Scholar] [CrossRef] [PubMed]
- Szafranska, K.; Neuman, T.; Baster, Z.; Rajfur, Z.; Szelest, O.; Christopher, H.; Kubisiak, A.; Kus, E.; Wolfson, D.L.; Chlopicki, S.; et al. From Fixed-Dried to Wet-Fixed to Live—Comparative Super-Resolution Microscopy of Liver Sinusoidal Endothelial Cell Fenestrations. Nanophotonics 2022, 11, 2253–2270. [Google Scholar] [CrossRef]
- Somlyo, A.P.; Somlyo, A.V. Ca2+ Sensitivity of Smooth Muscle and Nonmuscle Myosin II: Modulated by G Proteins, Kinases, and Myosin Phosphatase. Physiol. Rev. 2003, 83, 1325–1358. [Google Scholar] [CrossRef]
- Cai, H.; Liu, D.; Garcia, J.G.N. CaM Kinase II-Dependent Pathophysiological Signalling in Endothelial Cells. Cardiovasc. Res. 2008, 77, 30–34. [Google Scholar] [CrossRef]
- Johnson, C.N.; Pattanayek, R.; Potet, F.; Rebbeck, R.T.; Blackwell, D.J.; Nikolaienko, R.; Sequeira, V.; Le Meur, R.; Radwański, P.B.; Davis, J.P.; et al. The CaMKII Inhibitor KN93-Calmodulin Interaction and Implications for Calmodulin Tuning of NaV 1.5 and RyR2 Function. Cell Calcium 2019, 82, 102063. [Google Scholar] [CrossRef]
- Berger, S.L.; Leo-Macias, A.; Yuen, S.; Khatri, L.; Pfennig, S.; Zhang, Y.; Agullo-Pascual, E.; Caillol, G.; Zhu, M.S.; Rothenberg, E.; et al. Localized Myosin II Activity Regulates Assembly and Plasticity of the Axon Initial Segment. Neuron 2018, 97, 555–570.e6. [Google Scholar] [CrossRef]
- Shu, S.; Liu, X.; Korn, E.D. Blebbistatin and Blebbistatin-Inactivated Myosin II Inhibit Myosin II-Independent Processes in Dictyostelium. Proc. Natl. Acad. Sci. USA 2005, 102, 1472–1477. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Limouze, J.; Straight, A.F.; Mitchison, T.; Sellers, J.R. Specificity of Blebbistatin, an Inhibitor of Myosin II. J. Muscle Res. Cell Motil. 2004, 25, 337–341. [Google Scholar] [CrossRef]
- Park, I.; Han, C.; Jin, S.; Lee, B.; Choi, H.; Kwon, J.T.; Kim, D.; Kim, J.; Lifirsu, E.; Park, W.J.; et al. Myosin Regulatory Light Chains Are Required to Maintain the Stability of Myosin II and Cellular Integrity. Biochem. J. 2011, 434, 171–180. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Chen, Y.; Deng, Y.; Unarta, I.C.; Lu, Q.; Huang, X.; Zhang, M. Ca2+-Induced Rigidity Change of the Myosin VIIa IQ Motif-Single α Helix Lever Arm Extension. Structure 2017, 25, 579–591.e4. [Google Scholar] [CrossRef] [PubMed]
- Bhandari, S.; Li, R.; Simón-Santamaría, J.; McCourt, P.; Johansen, S.D.; Smedsrød, B.; Martinez-Zubiaurre, I.; Sørensen, K.K. Transcriptome and Proteome Profiling Reveal Complementary Scavenger and Immune Features of Rat Liver Sinusoidal Endothelial Cells and Liver Macrophages. BMC Mol. Cell Biol. 2020, 21, 85. [Google Scholar] [CrossRef]
- Bindewald, K.; Gündüz, D.; Härtel, F.; Peters, S.C.; Rodewald, C.; Nau, S.; Schäfer, M.; Neumann, J.; Piper, H.M.; Noll, T. Opposite Effect of CAMP Signaling in Endothelial Barriers of Different Origin. Am. J. Physiol.—Cell Physiol. 2004, 287, 1246–1255. [Google Scholar] [CrossRef]
- Ding, J.; Li, Z.; Li, L.; Ding, Y.; Wang, D.; Meng, S.; Zhou, Q.; Gui, S.; Wei, W.; Zhu, H.; et al. Myosin Light Chain Kinase Inhibitor ML7 Improves Vascular Endothelial Dysfunction and Permeability via the Mitogen-Activated Protein Kinase Pathway in a Rabbit Model of Atherosclerosis. Biomed. Pharmacother. 2020, 128, 110258. [Google Scholar] [CrossRef]
- Braet, F.; De Zanger, R.; Kalle, W.; Raap, A.; Tanke, H.; Wisse, E. Comparative Scanning, Transmission and Atomic Force Microscopy of the Microtubular Cytoskeleton in Fenestrated Liver Endothelial Cells. Scanning Microsc. Suppl. 1996, 10, 225–235; discussion 235–236. [Google Scholar]
- Mönkemöller, V.; Øie, C.; Hübner, W.; Huser, T.; McCourt, P. Multimodal Super-Resolution Optical Microscopy Visualizes the Close Connection between Membrane and the Cytoskeleton. Sci. Rep. 2015, 5, 16279. [Google Scholar] [CrossRef]
- Parizi, M.; Howard, E.W.; Tomasek, J.J. Regulation of LPA-Promoted Myofibroblast Contraction: Role of Rho, Myosin Light Chain Kinase, and Myosin Light Chain Phosphatase. Exp. Cell Res. 2000, 254, 210–220. [Google Scholar] [CrossRef]
- Martinez, I.; Nedredal, G.I.; Øie, C.I.; Warren, A.; Johansen, O.; Le Couteur, D.G.; Smedsrød, B. The Influence of Oxygen Tension on the Structure and Function of Isolated Liver Sinusoidal Endothelial Cells. Comp. Hepatol. 2008, 7, 4. [Google Scholar] [CrossRef] [PubMed]
- Fraser, R.; Dobbs, B.R.; Rogers, G.W.T. Lipoproteins and the Liver Sieve: The Role of the Fenestrated Sinusoidal Endothelium in Lipoprotein Metabolism, Atherosclerosis, and Cirrhosis. Hepatology 1995, 21, 863–874. [Google Scholar] [CrossRef] [PubMed]
- Di Martino, J.; Mascalchi, P.; Legros, P.; Lacomme, S.; Gontier, E.; Bioulac-Sage, P.; Balabaud, C.; Moreau, V.; Saltel, F. STED Microscopy: A Simplified Method for Liver Sinusoidal Endothelial Fenestrae Analysis. Biol. Cell 2018, 110, 159–168. [Google Scholar] [CrossRef] [PubMed]
- Rolfe, B.E.; Worth, N.F.; World, C.J.; Campbell, J.H.; Campbell, G.R. Rho and Vascular Disease. Atherosclerosis 2005, 183, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Preller, M.; Manstein, D.J. Myosin Motors: Structural Aspects and Functionality; Elsevier: Amsterdam, The Netherlands, 2017; ISBN 9780128096338. [Google Scholar]
- Matsui, T.S.; Deguchi, S. Spatially Selective Myosin Regulatory Light Chain Regulation Is Absent in Dedifferentiated Vascular Smooth Muscle Cells but Is Partially Induced by Fibronectin and Klf4. Am. J. Physiol.-Cell Physiol. 2019, 316, C509–C521. [Google Scholar] [CrossRef] [PubMed]
- Di Martino, J.; Mascalchi, P.; Legros, P.; Lacomme, S.; Gontier, E.; Bioulac-Sage, P.; Balabaud, C.; Moreau, V.; Saltel, F. Actin Depolymerization in Dedifferentiated Liver Sinusoidal Endothelial Cells Promotes Fenestrae Re-Formation. Hepatol. Commun. 2019, 3, 213–219. [Google Scholar] [CrossRef] [PubMed]
- Stewart, M.; Franks-Skiba, K.; Cooke, R. Myosin Regulatory Light Chain Phosphorylation Inhibits Shortening Velocities of Skeletal Muscle Fibers in the Presence of the Myosin Inhibitor Blebbistatin. J. Muscle Res. Cell Motil. 2009, 30, 17–27. [Google Scholar] [CrossRef]
- Kovács, M.; Tóth, J.; Hetényi, C.; Málnási-Csizmadia, A.; Seller, J.R. Mechanism of Blebbistatin Inhibition of Myosin II. J. Biol. Chem. 2004, 279, 35557–35563. [Google Scholar] [CrossRef]
- Schindelin, J.; Arganda-Carreras, I.; Frise, E.; Kaynig, V.; Longair, M.; Pietzsch, T.; Preibisch, S.; Rueden, C.; Saalfeld, S.; Schmid, B.; et al. Fiji: An Open-Source Platform for Biological-Image Analysis. Nat. Methods 2012, 9, 676–682. [Google Scholar] [CrossRef]
- Fraser, R.; Heslop, V.R.; Murray, F.E.M.; Day, W.A. Ultrastructural Studies of the Portal Transport of Fat in Chickens. Br. J. Exp. Pathol. 1986, 67, 783–791. [Google Scholar]
- Wright, P.L.; Smith, K.F.; Day, W.A.; Fraser, R. Small Liver Fenestrae May Explain the Susceptibility of Rabbits to Atherosclerosis. Arteriosclerosis 1983, 3, 344–348. [Google Scholar] [CrossRef] [PubMed]
- Clark, S.; Cook, H.B.; Oxner, R.; Angus, H.; George, P.; Fraser, R. Defenestration of Hepatic Sinusoids as a Cause of Hyperlipoproteinaemia in Alcoholics. Lancet 1988, 332, 1225–1227. [Google Scholar] [CrossRef]
- Blomhoff, R.; Eskild, W.; Berg, T. Endocytosis of formaldehyde-treated serum albumin via scavenger pathway in liver endothelial cells. Biochem. J. 1984, 218, 81–86. [Google Scholar] [CrossRef] [Green Version]
- Bhandari, S.; Larsen, A.K.; McCourt, P.; Smedsrød, B.; Sørensen, K.K. The Scavenger Function of Liver Sinusoidal Endothelial Cells in Health and Disease. Front. Physiol. 2021, 12, 1–23. [Google Scholar] [CrossRef]
- Simon-Santamaria, J.; Malovic, I.; Warren, A.; Oteiza, A.; Le Couteur, D.; Smedsrød, B.; McCourt, P.; Sørensen, K.K. Age-Related Changes in Scavenger Receptor–Mediated Endocytosis in Rat Liver Sinusoidal Endothelial Cells. J. Gerontol. Ser. A 2010, 65, 951–960. [Google Scholar] [CrossRef] [PubMed]
- McCuskey, R.S. Sinusoidal endothelial cells as an early target for hepatic toxicants. Clin. Hemorheol. Microcirc. 2006, 34, 5–10. [Google Scholar]
- Politz, O.; Gratchev, A.; McCOURT, P.A.; Schledzewski, K.; Guillot, P.; Johansson, S.; Svineng, G.; Franke, P.; Kannicht, C.; Kzhyshkowska, J.; et al. Stabilin-1 and -2 constitute a novel family of fasciclin-like hyaluronan receptor homologues. Biochem. J. 2002, 362, 155–164. [Google Scholar] [CrossRef] [PubMed]
- Hansen, B.; Longati, P.; Elvevold, K.; Nedredal, G.-I.; Schledzewski, K.; Olsen, R.; Falkowski, M.; Kzhyshkowska, J.; Carlsson, F.; Johansson, S.; et al. Stabilin-1 and stabilin-2 are both directed into the early endocytic pathway in hepatic sinusoidal endothelium via interactions with clathrin/AP-2, independent of ligand binding. Exp. Cell Res. 2005, 303, 160–173. [Google Scholar] [CrossRef]
- McCourt, P.A.; Smedsrød, B.H.; Melkko, J.; Johansson, S. Characterization of a Hyaluronan Receptor on Rat Sinusoidal Liver Endothelial Cells and Its Functional Re-lationship to Scavenger Receptors. Hepatology 1999, 30, 1276–1286. [Google Scholar] [CrossRef]
- Kus, E.; Kaczara, P.; Czyzynska-Cichon, I.; Szafranska, K.; Zapotoczny, B.; Kij, A.; Sowinska, A.; Kotlinowski, J.; Mateuszuk, L.; Czarnowska, E.; et al. LSEC Fenestrae Are Preserved Despite Pro-inflammatory Phenotype of Liver Sinusoidal Endothelial Cells in Mice on High Fat Diet. Front. Physiol. 2019, 10, 6. [Google Scholar] [CrossRef]
- Sebbagh, M.; Renvoizé, C.; Hamelin, J.; Riché, N.; Bertoglio, J.; Bréard, J. Caspase-3-mediated cleavage of ROCK I induces MLC phosphorylation and apoptotic membrane blebbing. Nat. Cell Biol. 2001, 3, 346–352. [Google Scholar] [CrossRef] [PubMed]
- Fazal, F.; Gu, L.; Ihnatovych, I.; Han, Y.; Hu, W.; Antic, N.; Carreira, F.; Blomquist, J.F.; Hope, T.J.; Ucker, D.S.; et al. Inhibiting Myosin Light Chain Kinase Induces Apoptosis In Vitro and In Vivo. Mol. Cell. Biol. 2005, 25, 6259–6266. [Google Scholar] [CrossRef] [PubMed] [Green Version]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zapotoczny, B.; Szafranska, K.; Lekka, M.; Ahluwalia, B.S.; McCourt, P. Tuning of Liver Sieve: The Interplay between Actin and Myosin Regulatory Light Chain Regulates Fenestration Size and Number in Murine Liver Sinusoidal Endothelial Cells. Int. J. Mol. Sci. 2022, 23, 9850. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23179850
Zapotoczny B, Szafranska K, Lekka M, Ahluwalia BS, McCourt P. Tuning of Liver Sieve: The Interplay between Actin and Myosin Regulatory Light Chain Regulates Fenestration Size and Number in Murine Liver Sinusoidal Endothelial Cells. International Journal of Molecular Sciences. 2022; 23(17):9850. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23179850
Chicago/Turabian StyleZapotoczny, Bartlomiej, Karolina Szafranska, Malgorzata Lekka, Balpreet Singh Ahluwalia, and Peter McCourt. 2022. "Tuning of Liver Sieve: The Interplay between Actin and Myosin Regulatory Light Chain Regulates Fenestration Size and Number in Murine Liver Sinusoidal Endothelial Cells" International Journal of Molecular Sciences 23, no. 17: 9850. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23179850