

Italian Precision Medicine in Pediatric Oncology: Moving beyond Actionable Alterations
 , , , , , , and
, , , , , , and
Abstract
:1. Introduction
2. Results
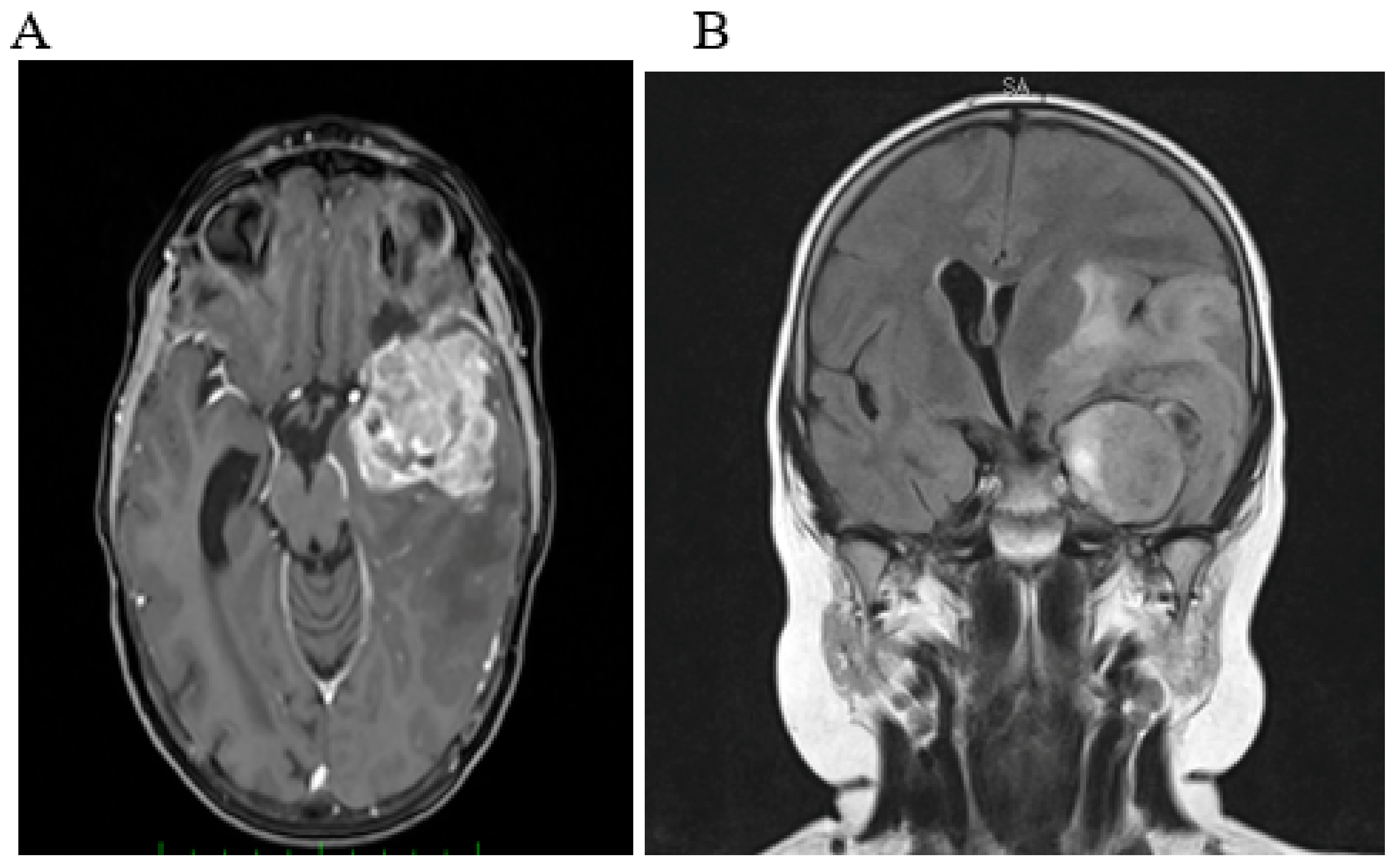
2.1. Case 1
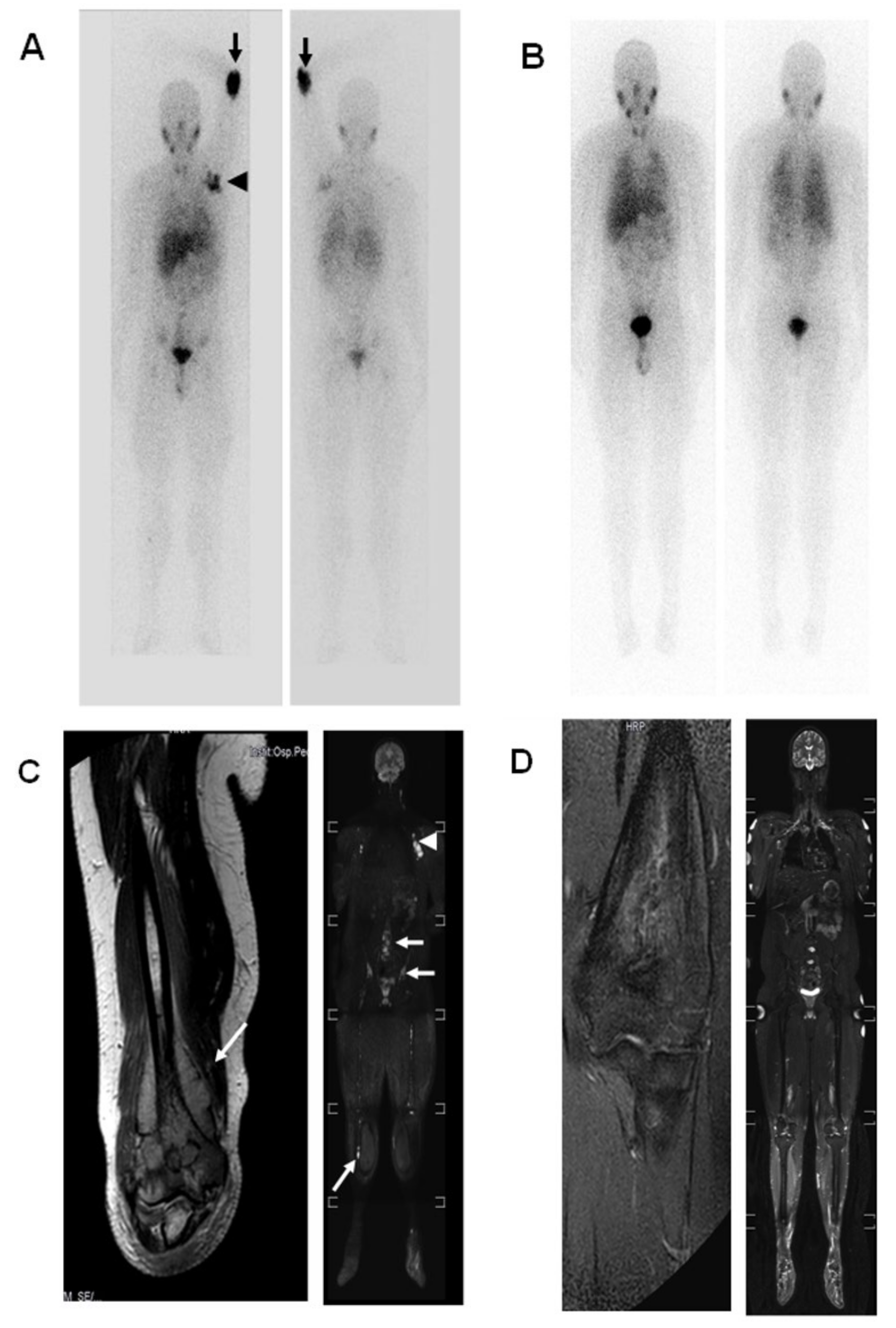
2.2. Case 2
3. Discussion
4. Materials and Methods
4.1. Patients and MTB Organization
4.2. Preparation and Characterization of Neuroblastoma Samples
4.3. Data Interpretation and Reporting
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Brodeur, G.M. Neuroblastoma: Biological insights into a clinical enigma. Nat. Rev. Cancer 2003, 3, 203–216. [Google Scholar] [CrossRef] [PubMed]
- Johnsen, J.I.; Dyberg, C.; Wickström, M. Neuroblastoma-A Neural Crest Derived Embryonal Malignancy. Front. Mol. Neurosci. 2019, 12, 9. [Google Scholar] [CrossRef] [PubMed]
- Qiu, B.; Matthay, K.K. Advancing therapy for neuroblastoma. Nat. Rev. Clin. Oncol. 2022, 19, 515–533. [Google Scholar] [CrossRef] [PubMed]
- Lasorsa, V.A.; Formicola, D.; Pignataro, P.; Cimmino, F.; Calabrese, F.M.; Mora, J.; Esposito, M.R.; Pantile, M.; Zanon, C.; Mariano, M.D.; et al. Exome and deep sequencing of clinically aggressive neuroblastoma reveal somatic mutations that affect key pathways involved in cancer progression. Oncotarget 2016, 7, 21840–21852. [Google Scholar] [CrossRef] [PubMed]
- Gröbner, S.N.; Worst, B.C.; Weischenfeldt, J.; Buchhalter, I.; Kleinheinz, K.; Rudneva, V.A.; Johann, P.D.; Balasubramanian, G.P.; Segura-Wang, M.; Brabetz, S.; et al. The landscape of genomic alterations across childhood cancers. Nature 2018, 555, 321–327. [Google Scholar] [CrossRef]
- Ma, X.; Liu, Y.; Liu, Y.; Alexandrov, L.B.; Edmonson, M.N.; Gawad, C.; Zhou, X.; Li, Y.; Rusch, M.C.; Easton, J.; et al. Pan-cancer genome and transcriptome analyses of 1699 paediatric leukaemias and solid tumours. Nature 2018, 555, 371–376. [Google Scholar] [CrossRef]
- Pugh, T.J.; Morozova, O.; Attiyeh, E.F.; Asgharzadeh, S.; Wei, J.S.; Auclair, D.; Carter, S.L.; Cibulskis, K.; Hanna, M.; Kiezun, A.; et al. The genetic landscape of high-risk neuroblastoma. Nat. Genet. 2013, 45, 279–284. [Google Scholar] [CrossRef]
- Molenaar, J.J.; Koster, J.; Zwijnenburg, D.A.; Sluis, P.v.; Valentijn, L.J.; Ploeg, I.v.d.; Hamdi, M.; Nes, J.v.; Westerman, B.A.; Arkel, J.v.; et al. Sequencing of neuroblastoma identifies chromothripsis and defects in neuritogenesis genes. Nature 2012, 483, 589–593. [Google Scholar] [CrossRef]
- Ponzoni, M.; Bachetti, T.; Corrias, M.V.; Brignole, C.; Pastorino, F.; Calarco, E.; Bensa, V.; Giusto, E.; Ceccherini, I.; Perri, P. Recent advances in the developmental origin of neuroblastoma: An overview. J. Exp. Clin. Cancer Res. 2022, 41, 92. [Google Scholar] [CrossRef]
- Padovan-Merhar, O.M.; Raman, P.; Ostrovnaya, I.; Kalletla, K.; Rubnitz, K.R.; Sanford, E.M.; Ali, S.M.; Miller, V.A.; Mossé, Y.P.; Granger, M.P.; et al. Enrichment of Targetable Mutations in the Relapsed Neuroblastoma Genome. PLoS Genet. 2016, 12, e1006501. [Google Scholar] [CrossRef] [Green Version]
- Tonini, G.P.; Capasso, M. Genetic predisposition and chromosome instability in neuroblastoma. Cancer Metastasis Rev. 2020, 39, 275–285. [Google Scholar] [CrossRef] [PubMed]
- Forrest, S.J.; Geoerger, B.; Janeway, K.A. Precision medicine in pediatric oncology. Curr. Opin. Pediatr. 2018, 30, 17–24. [Google Scholar] [CrossRef] [PubMed]
- Sharma, P.; Nag, A. CUL4A ubiquitin ligase: A promising drug target for cancer and other human diseases. Open Biol. 2014, 4, 130217. [Google Scholar] [CrossRef]
- Ren, S.; Xu, C.; Cui, Z.; Yu, Y.; Xu, W.; Wang, F.; Lu, J.; Wei, M.; Lu, X.; Gao, X.; et al. Oncogenic CUL4A determines the response to thalidomide treatment in prostate cancer. J. Mol. Med. 2012, 90, 1121–1132. [Google Scholar] [CrossRef]
- Robison, N.J.; Yeo, K.K.; Berliner, A.P.; Malvar, J.; Sheard, M.A.; Margol, A.S.; Seeger, R.C.; Rushing, T.; Finlay, J.L.; Sposto, R.; et al. Phase I trial of dasatinib, lenalidomide, and temozolomide in children with relapsed or refractory central nervous system tumors. J. Neuro-Oncol. 2018, 138, 199–207. [Google Scholar] [CrossRef] [PubMed]
- Berg, S.L.; Cairo, M.S.; Russell, H.; Ayello, J.; Ingle, A.M.; Lau, H.; Chen, N.; Adamson, P.C.; Blaney, S.M. Safety, pharmacokinetics, and immunomodulatory effects of lenalidomide in children and adolescents with relapsed/refractory solid tumors or myelodysplastic syndrome: A Children′s Oncology Group Phase I Consortium report. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2011, 29, 316–323. [Google Scholar] [CrossRef]
- Warren, K.E.; Goldman, S.; Pollack, I.F.; Fangusaro, J.; Schaiquevich, P.; Stewart, C.F.; Wallace, D.; Blaney, S.M.; Packer, R.; MacDonald, T.; et al. Phase I trial of lenalidomide in pediatric patients with recurrent, refractory, or progressive primary CNS tumors: Pediatric Brain Tumor Consortium study PBTC-018. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2011, 29, 324–329. [Google Scholar] [CrossRef]
- Mody, R.; Zhao, L.; Yanik, G.A.; Opipari, V. Phase I study of bortezomib in combination with irinotecan in patients with relapsed/refractory high-risk neuroblastoma. Pediatr. Blood Cancer 2017, 64, e26563. [Google Scholar] [CrossRef]
- Wong, M.; Mayoh, C.; Lau, L.M.S.; Khuong-Quang, D.-A.; Pinese, M.; Kumar, A.; Barahona, P.; Wilkie, E.E.; Sullivan, P.; Bowen-James, R.; et al. Whole genome, transcriptome and methylome profiling enhances actionable target discovery in high-risk pediatric cancer. Nat. Med. 2020, 26, 1742–1753. [Google Scholar] [CrossRef]
- Rokita, J.L.; Rathi, K.S.; Cardenas, M.F.; Upton, K.A.; Jayaseelan, J.; Cross, K.L.; Pfeil, J.; Egolf, L.E.; Way, G.P.; Farrel, A.; et al. Genomic Profiling of Childhood Tumor Patient-Derived Xenograft Models to Enable Rational Clinical Trial Design. Cell Rep. 2019, 29, 1675–1689. [Google Scholar] [CrossRef]
- Vogelstein, B.; Papadopoulos, N.; Velculescu, V.E.; Zhou, S.; Diaz, L.A.; Kinzler, K.W. Cancer genome landscapes. Science 2013, 339, 1546–1558. [Google Scholar] [CrossRef] [PubMed]
- Schramm, A.; Köster, J.; Assenov, Y.; Althoff, K.; Peifer, M.; Mahlow, E.; Odersky, A.; Beisser, D.; Ernst, C.; Henssen, A.G.; et al. Mutational dynamics between primary and relapse neuroblastomas. Nat. Genet. 2015, 47, 872–877. [Google Scholar] [CrossRef] [PubMed]
- Schleiermacher, G.; Javanmardi, N.; Bernard, V.; Leroy, Q.; Cappo, J.; Frio, T.R.; Pierron, G.; Lapouble, E.; Combaret, V.; Speleman, F.; et al. Emergence of new ALK mutations at relapse of neuroblastoma. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2014, 32, 2727–2734. [Google Scholar] [CrossRef] [PubMed]
- Garaventa, A.; Parodi, S.; Bernardi, B.D.; Dau, D.; Manzitti, C.; Conte, M.; Casale, F.; Viscardi, E.; Bianchi, M.; D’Angelo, P.; et al. Outcome of children with neuroblastoma after progression or relapse. A retrospective study of the Italian neuroblastoma registry. Eur. J. Cancer 2009, 45, 2835–2842. [Google Scholar] [CrossRef] [PubMed]
- Eleveld, T.F.; Oldridge, D.A.; Bernard, V.; Koster, J.; Daage, L.C.; Diskin, S.J.; Schild, L.; Bentahar, N.B.; Bellini, A.; Chicard, M.; et al. Relapsed neuroblastomas show frequent RAS-MAPK pathway mutations. Nat. Genet. 2015, 47, 864–871. [Google Scholar] [CrossRef]
- Brady, S.W.; Liu, Y.; Ma, X.; Gout, A.M.; Hagiwara, K.; Zhou, X.; Wang, J.; Macias, M.; Chen, X.; Easton, J.; et al. Pan-neuroblastoma analysis reveals age- and signature-associated driver alterations. Nat. Commun. 2020, 11, 5183. [Google Scholar] [CrossRef]
- Van Allen, E.M.; Wagle, N.; Stojanov, P.; Perrin, D.L.; Cibulskis, K.; Marlow, S.; Jane-Valbuena, J.; Friedrich, D.C.; Kryukov, G.; Carter, S.L.; et al. Whole-exome sequencing and clinical interpretation of formalin-fixed, paraffin-embedded tumor samples to guide precision cancer medicine. Nat. Med. 2014, 20, 682–688. [Google Scholar] [CrossRef]
- Moreno, L.; Barone, G.; DuBois, S.G.; Molenaar, J.; Fischer, M.; Schulte, J.; Eggert, A.; Schleiermacher, G.; Speleman, F.; Chesler, L.; et al. Accelerating drug development for neuroblastoma: Summary of the Second Neuroblastoma Drug Development Strategy forum from Innovative Therapies for Children with Cancer and International Society of Paediatric Oncology Europe Neuroblastoma. Eur. J. Cancer 2020, 136, 52–68. [Google Scholar] [CrossRef]
- Takagi, M.; Ogawa, C.; Aoki-Nogami, Y.; Iehara, T.; Ishibashi, E.; Imai, M.; Kihara, T.; Nobori, K.; Hasebe, K.; Mizutani, S.; et al. Phase I clinical study of oral olaparib in pediatric patients with refractory solid tumors: Study protocol. BMC Pediatr. 2019, 19, 31. [Google Scholar] [CrossRef]
- Liu, P.Y.; Sokolowski, N.; Guo, S.T.; Siddiqi, F.; Atmadibrata, B.; Telfer, T.J.; Sun, Y.; Zhang, L.; Yu, D.; Mccarroll, J.; et al. The BET bromodomain inhibitor exerts the most potent synergistic anticancer effects with quinone-containing compounds and anti-microtubule drugs. Oncotarget 2016, 7, 79217–79232. [Google Scholar] [CrossRef] [Green Version]
- Yang, L.Q.; Hu, H.Y.; Han, Y.; Tang, Z.Y.; Gao, J.; Zhou, Q.Y.; Liu, Y.X.; Chen, H.S.; Xu, T.N.; Ao, L.; et al. CpG-binding protein CFP1 promotes ovarian cancer cell proliferation by regulating BST2 transcription. Cancer Gene Ther. 2022, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Jang, S.M.; Zhang, Y.; Utani, K.; Fu, H.; Redon, C.E.; Marks, A.B.; Smith, O.K.; Redmond, C.J.; Baris, A.M.; Tulchinsky, D.A.; et al. The replication initiation determinant protein (RepID) modulates replication by recruiting CUL4 to chromatin. Nat. Commun. 2018, 9, 2782. [Google Scholar] [CrossRef] [PubMed]
- Berlanga, P.; Pasqualini, C.; Pötschger, U.; Sangüesa, C.; Castellani, M.R.; Cañete, A.; Luksch, R.; Elliot, M.; Schreier, G.; Kropf, M.; et al. Central nervous system relapse in high-risk stage 4 neuroblastoma: The HR-NBL1/SIOPEN trial experience. Eur. J. Cancer 2021, 144, 1–8. [Google Scholar] [CrossRef]
- DuBois, S.G.; Kalika, Y.; Lukens, J.N.; Brodeur, G.M.; Seeger, R.C.; Atkinson, J.B.; Haase, G.M.; Black, C.T.; Perez, C.; Shimada, H.; et al. Metastatic sites in stage IV and IVS neuroblastoma correlate with age, tumor biology, and survival. J. Pediatric Hematol. Oncol. 1999, 21, 181–189. [Google Scholar] [CrossRef]
- Stefanowicz, J.; Iżycka-Świeszewska, E.; Szurowska, E.; Bień, E.; Szarszewski, A.; Liberek, A.; Stempniewicz, M.; Kloc, W.; Adamkiewicz-Drożyńska, E. Brain metastases in paediatric patients: Characteristics of a patient series and review of the literature. Folia Neuropathol. 2011, 49, 271–281. [Google Scholar]
- Wiens, A.L.; Hattab, E.M. The pathological spectrum of solid CNS metastases in the pediatric population. J. Neurosurg. Pediatr. 2014, 14, 129–135. [Google Scholar] [CrossRef]
- Matthay, K.K.; Brisse, H.; Couanet, D.; Couturier, J.; Bénard, J.; Mosseri, V.; Edeline, V.; Lumbroso, J.; Valteau-Couanet, D.; Michon, J.; et al. Central nervous system metastases in neuroblastoma: Radiologic, clinical, and biologic features in 23 patients. Cancer 2003, 98, 155–165. [Google Scholar] [CrossRef] [PubMed]
- Tripathi, N.; Jo, Y.; Tripathi, A.; Sayegh, N.; Li, H.; Nussenzveig, R.; Haaland, B.; Thomas, V.M.; Gupta, S.; Maughan, B.L.; et al. Genomic landscape of locally advanced or metastatic urothelial carcinoma. In Urologic Oncology: Seminars and Original Investigations; Elsevier: Amsterdam, The Netherlands, 2022. [Google Scholar]
- Liu, T.T.; You, H.L.; Weng, S.W.; Wei, Y.C.; Eng, H.L.; Huang, W.T. Recurrent Amplification at 13q34 Targets at CUL4A, IRS2, and TFDP1 As an Independent Adverse Prognosticator in Intrahepatic Cholangiocarcinoma. PLoS ONE 2015, 10, e0145388. [Google Scholar] [CrossRef] [PubMed]
- Duan, X.; Li, H.; Wang, M.; Ju, S.; Li, F.; Chen, P.; Lu, H.; Han, X.; Ren, J. PSMC2/ITGA6 axis plays critical role in the development and progression of hepatocellular carcinoma. Cell Death Discov. 2021, 7, 217. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Xiong, H.; Zuo, Y.; Hu, S.; Zhu, C.; Min, A. PSMC2 knockdown inhibits the progression of oral squamous cell carcinoma by promoting apoptosis via PI3K/Akt pathway. Cell Cycle 2022, 21, 477–488. [Google Scholar] [CrossRef]
- Liu, T.; Zhang, J.; Chen, H.; Bianba, T.; Pan, Y.; Wang, X.; Jiang, Y.; Yang, Z. PSMC2 promotes the progression of gastric cancer via induction of RPS15A/mTOR pathway. Oncogenesis 2022, 11, 12. [Google Scholar] [CrossRef]
- Field-Smith, A.; Morgan, G.J.; Davies, F.E. Bortezomib (Velcadetrade mark) in the Treatment of Multiple Myeloma. Ther. Clin. Risk Manag. 2006, 2, 271–279. [Google Scholar] [CrossRef] [PubMed]
- Krytska, K.; Ryles, H.T.; Sano, R.; Raman, P.; Infarinato, N.R.; Hansel, T.D.; Makena, M.R.; Song, M.M.; Reynolds, C.P.; Mossé, Y.P. Crizotinib Synergizes with Chemotherapy in Preclinical Models of Neuroblastoma. Clin. Cancer Res. 2016, 22, 948–960. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Wu, B.; Baruchel, S. Oral Metronomic Topotecan Sensitizes Crizotinib Antitumor Activity in ALK(F1174L) Drug-Resistant Neuroblastoma Preclinical Models. Transl. Oncol. 2017, 10, 604–611. [Google Scholar] [CrossRef] [PubMed]
- Hart, L.S.; Rader, J.; Raman, P.; Batra, V.; Russell, M.R.; Tsang, M.; Gagliardi, M.; Chen, L.; Martinez, D.; Li, Y.; et al. Preclinical Therapeutic Synergy of MEK1/2 and CDK4/6 Inhibition in Neuroblastoma. Clin. Cancer Res. 2017, 23, 1785–1796. [Google Scholar] [CrossRef] [Green Version]



{kind=link}
{kind=link}
| Chr | Position | REF | ALT | Gene | Mutation Characteristics | COSMIC | SIFT | CADD | CancerVar | Drug | Interaction Types |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Case 1 | NA | ||||||||||
| chr2 | 240,139,318 | G | C | OTOS | exon4, c.C122G, p.P41R | D | 25 | 0.8762 | CISPLATIN | NA | |
| chr6 | 136,651,004 | G | T | MAP3K5 | exon11, c.C1768A, p.H590N | T | 24.6 | 0.9925 | HYDROXYUREA | NA | |
| chr13 | 113,219,000 | G | C | CUL4A | exon3, c.G320C, p.R107P | D | 28.5 | 0.9925 | LENALIDOMIDE, THALIDOMIDE, POMALIDOMIDE | inhibitor | |
| chr16 | 49,789,532 | C | T | ZNF423 | exon2, c.G31A, p.A11T | D | 23.2 | 0.9925 | TAMOXIFEN | NA | |
| Case 2 | |||||||||||
| chr2 | 141,143,477 | C | A | LRP1B | exon67, c.G10516T, p.D3506Y | D | 34 | 0.702 | DOXORUBICIN | NA | |
| chr3 | 134,920,343 | C | A | EPHB1 | exon12, c.C2158A, p.Q720K | D | 32 | 0.842 | VANDETANIB | inhibitor | |
| chr4 | 72,623,854 | C | A | GC | exon7, c.G736T, p.A246S | D | 29.5 | 0.996 | CETUXIMAB, CETUXIMAB | NA | |
| chr7 | 93,065,322 | G | C | CALCR | exon11, c.C1091G, p.S364C | D | 25.8 | 0.9632 | PRAMLINTIDE | agonist | |
| chr7 | 103,003,194 | A | G | PSMC2 | exon6, c.A484G, p.T162A | ID=COSM484567 | D | 24.6 | 0.9959 | IXAZOMIB CITRATE, CARFILZOMIB, BORTEZOMIB | inhibitor |
| chr20 | 43,703,716 | A | G | STK4 | exon11, c.A1363G, p.M455V | D | 25.1 | 0.2034 | BOSUTINIB | inhibitor |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pastorino, F.; Capasso, M.; Brignole, C.; Giglio, S.; Bensa, V.; Cantalupo, S.; Lasorsa, V.A.; Tondo, A.; Mura, R.; Sementa, A.R.; et al. Italian Precision Medicine in Pediatric Oncology: Moving beyond Actionable Alterations. Int. J. Mol. Sci. 2022, 23, 11236. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms231911236
Pastorino F, Capasso M, Brignole C, Giglio S, Bensa V, Cantalupo S, Lasorsa VA, Tondo A, Mura R, Sementa AR, et al. Italian Precision Medicine in Pediatric Oncology: Moving beyond Actionable Alterations. International Journal of Molecular Sciences. 2022; 23(19):11236. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms231911236
Chicago/Turabian StylePastorino, Fabio, Mario Capasso, Chiara Brignole, Serena Giglio, Veronica Bensa, Sueva Cantalupo, Vito Alessandro Lasorsa, Annalisa Tondo, Rossella Mura, Angela Rita Sementa, and et al. 2022. "Italian Precision Medicine in Pediatric Oncology: Moving beyond Actionable Alterations" International Journal of Molecular Sciences 23, no. 19: 11236. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms231911236