
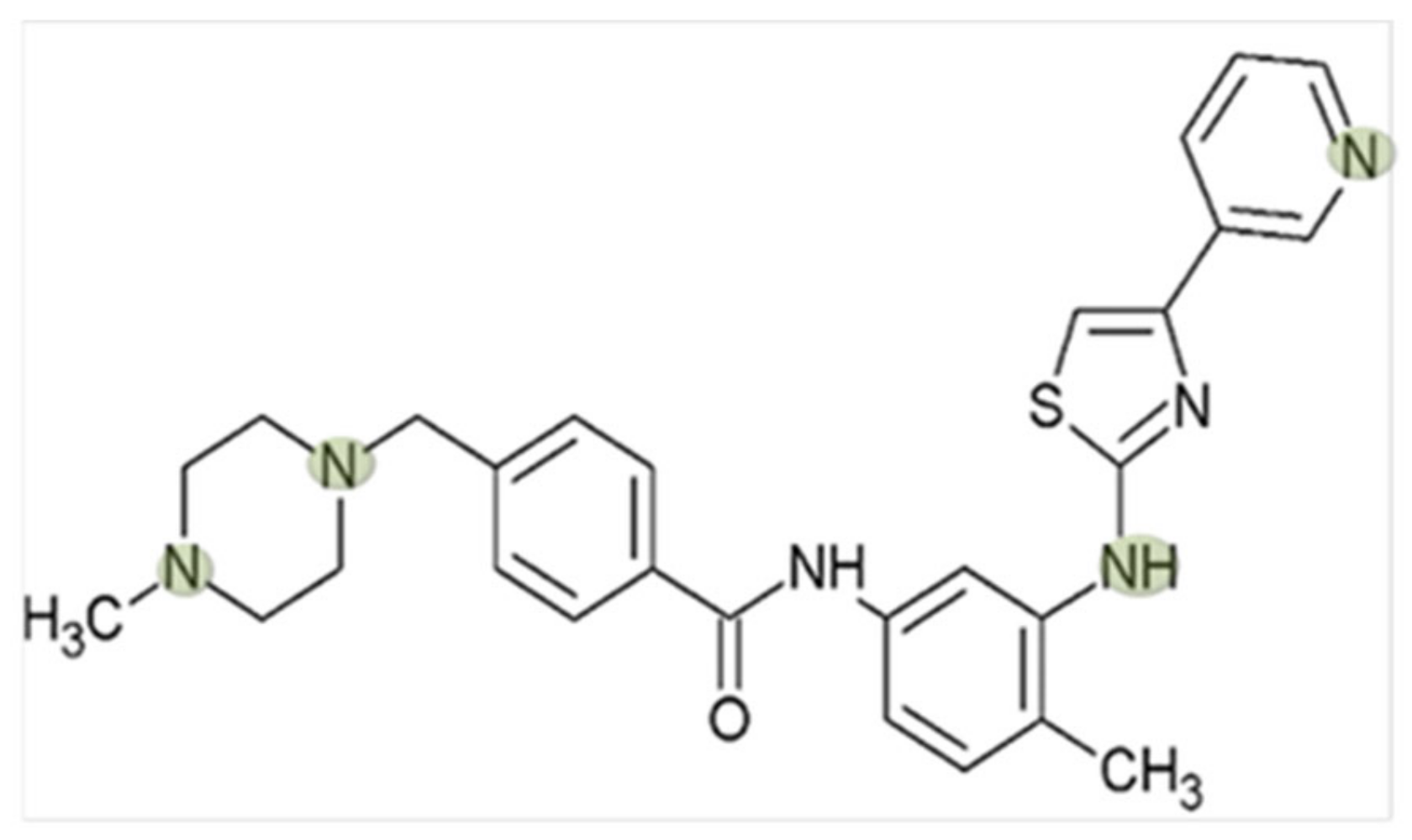
Interaction of Masitinib with Organic Cation Transporters
 and
and 
Abstract
:1. Introduction
2. Results
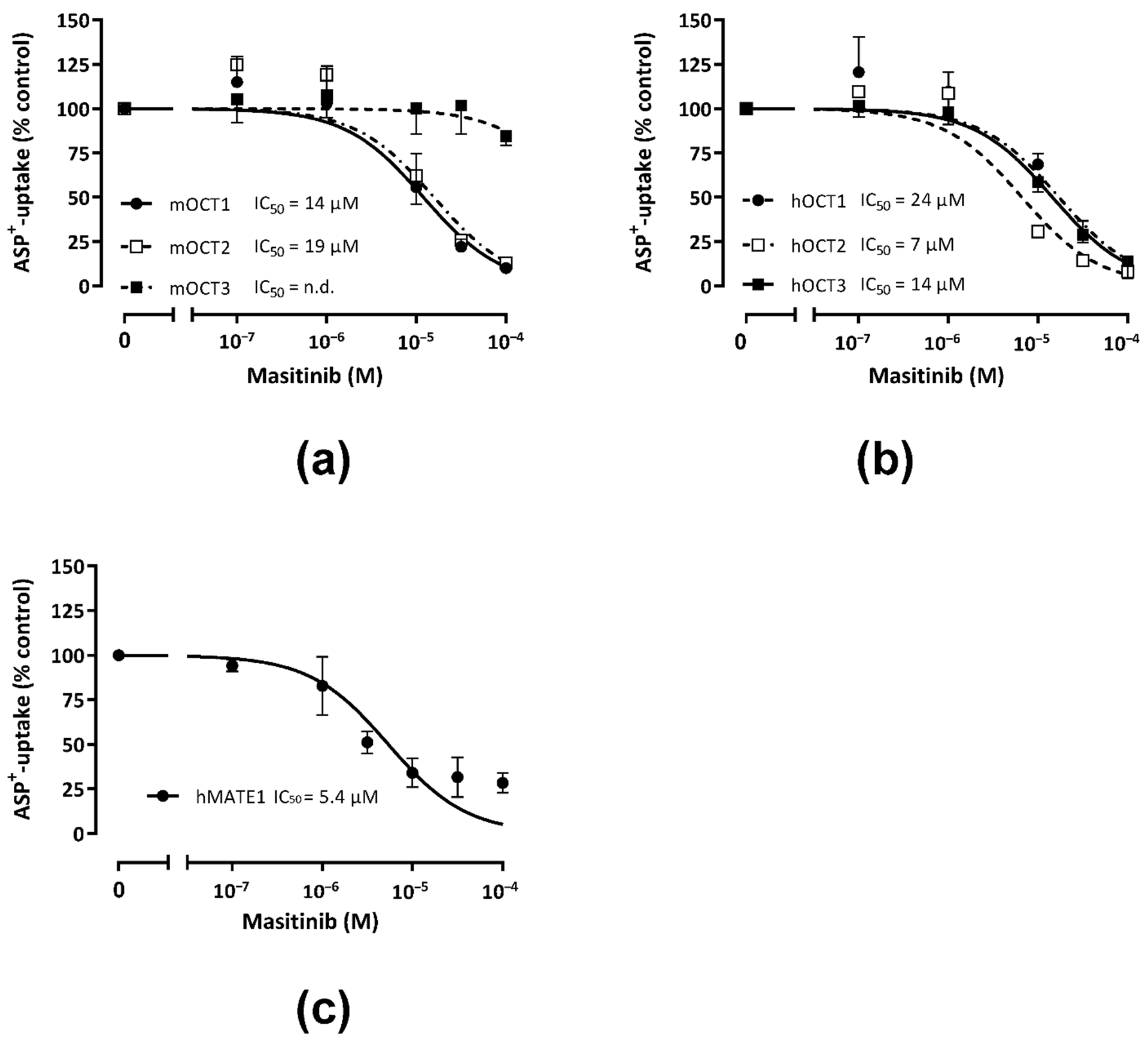
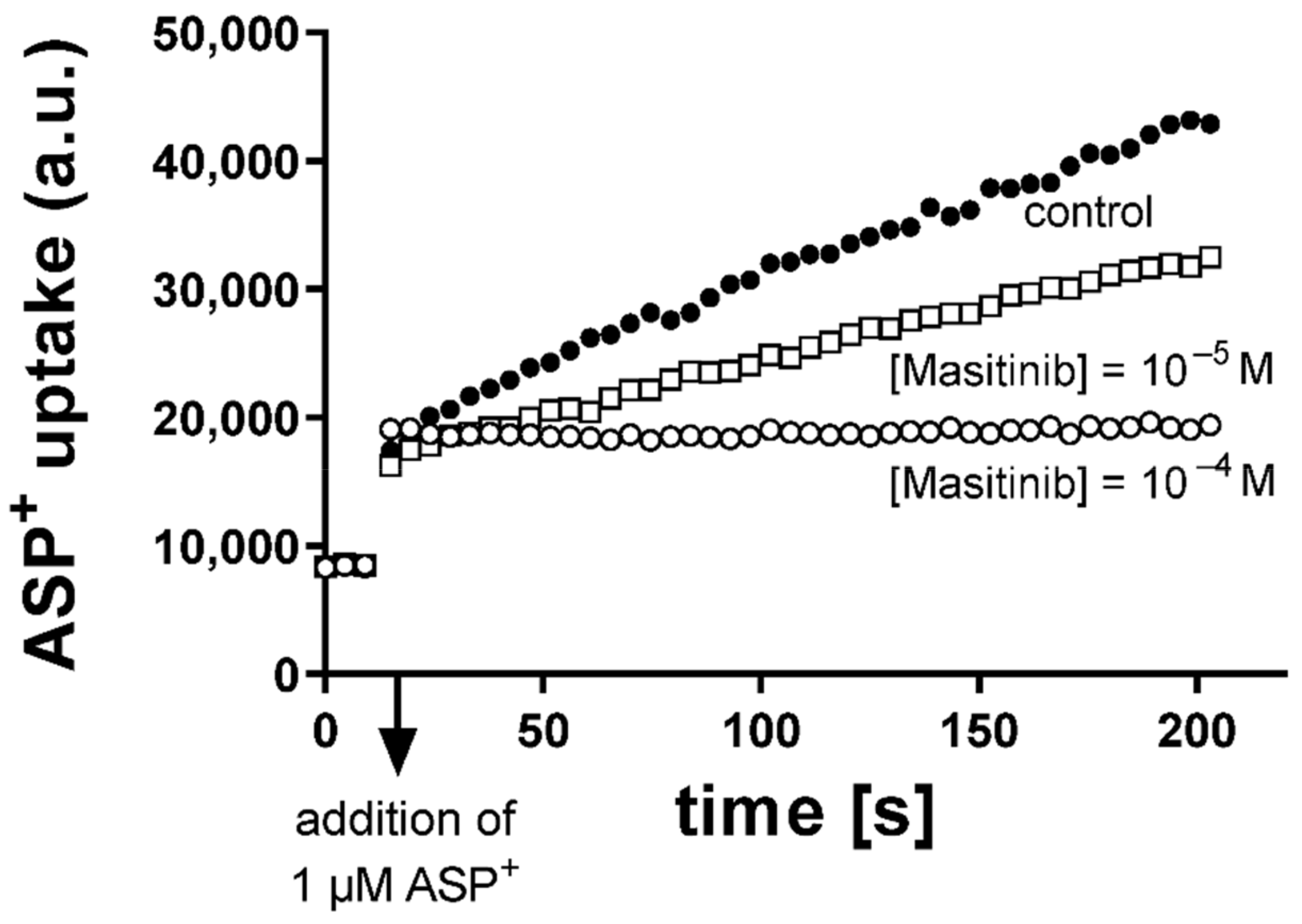
2.1. Inhibition of Uptake of the Fluorescent Organic Cation 4-(4-dimethylaminostyryl)-N-Methylpyridinium (ASP+) by Masitinib in Human Embryonic Kidney (HEK) 293 Cells Expressing Murine or Human OCT or hMATE1
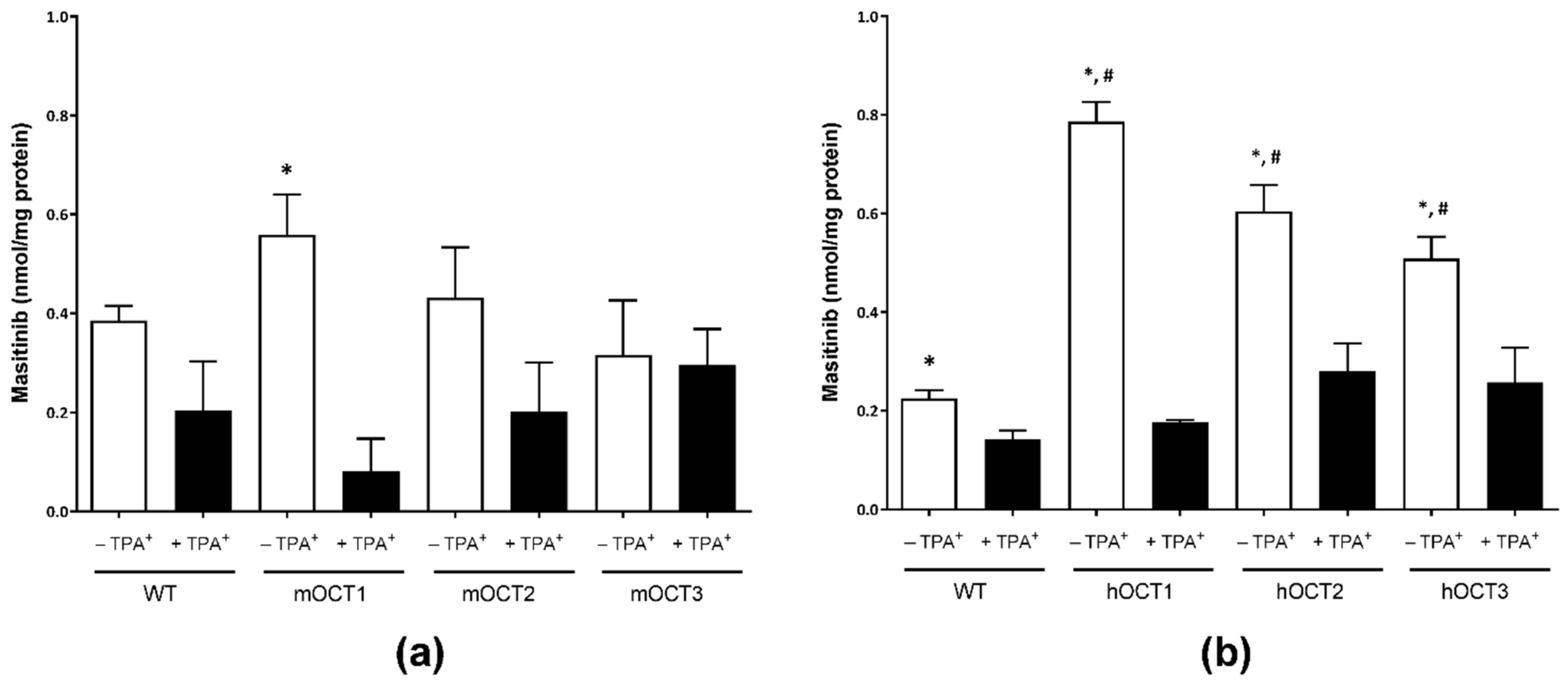
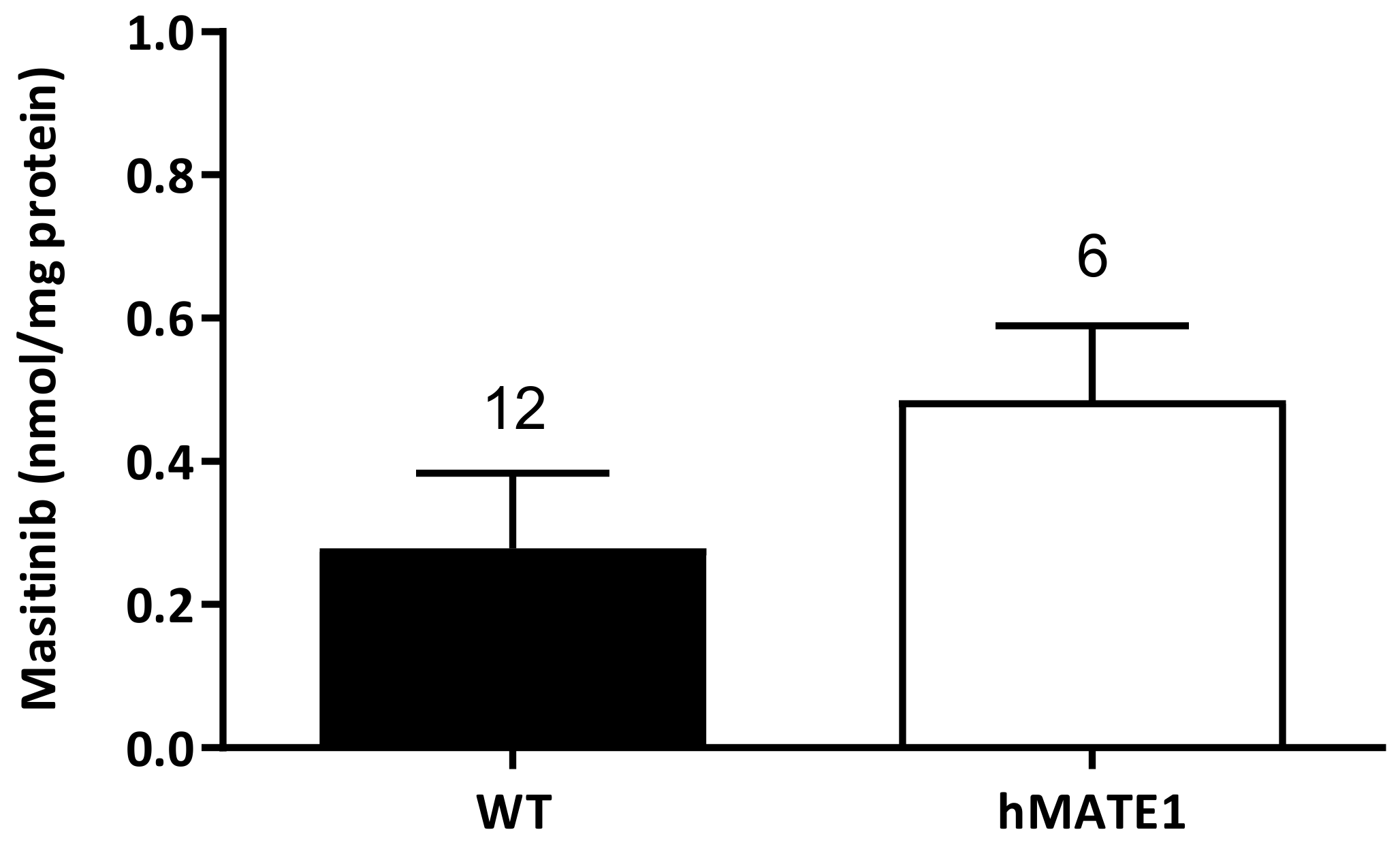
2.2. Accumulation of Masitinib Mediated by Murine and Human OCTs Stably Expressed in HEK293 Cells
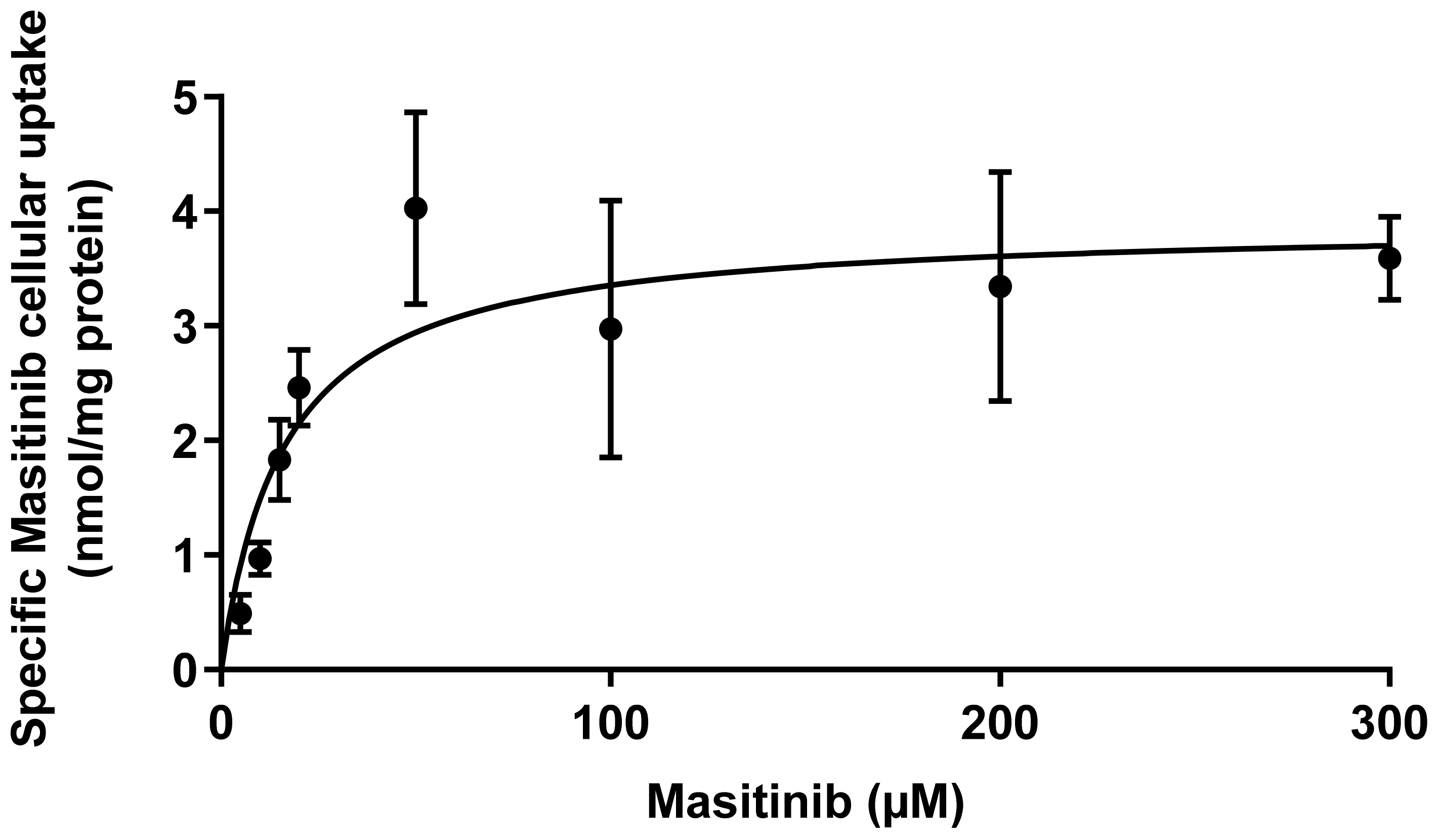
2.3. Saturation Kinetics
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. Fluorescence Measurements
4.3. Measurement of Masitinib in Cell Lysates by High Performance Liquid Chromatography (HPLC)
4.4. Preparation of Samples for Detection of Cellular Masitinib Accumulation by HPLC
4.5. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bhanumathy, K.; Balagopal, A.; Vizeacoumar, F.S.; Vizeacoumar, F.J.; Freywald, A.; Giambra, V. Protein Tyrosine Kinases: Their Roles and Their Targeting in Leukemia. Cancers 2021, 13, 184. [Google Scholar] [CrossRef] [PubMed]
- Wan, P.T.C.; Garnett, M.J.; Roe, S.M.; Lee, S.; Niculescu-Duvaz, D.; Good, V.M.; Project, C.G.; Jones, C.M.; Marshall, C.J.; Springer, C.J.; et al. Mechanism of Activation of the RAF-ERK Signaling Pathway by Oncogenic Mutations of B-RAF. Cell 2004, 116, 855–867. [Google Scholar] [CrossRef] [Green Version]
- London, C.A. Kinase Dysfunction and Kinase Inhibitors. Veter Dermatol. 2013, 24, 181-7.e39-40. [Google Scholar] [CrossRef] [PubMed]
- Cohen, P.; Cross, D.; Jänne, P.A. Kinase Drug Discovery 20 Years after Imatinib: Progress and Future Directions. Nat. Rev. Drug Discov. 2021, 20, 551–569. [Google Scholar] [CrossRef]
- Zhang, J.; Yang, P.L.; Gray, N.S. Targeting Cancer with Small Molecule Kinase Inhibitors. Nat. Rev. Cancer 2009, 9, 28–39. [Google Scholar] [CrossRef]
- Liang, X.; Yang, Q.; Wu, P.; He, C.; Yin, L.; Xu, F.; Yin, Z.; Yue, G.; Zou, Y.; Li, L.; et al. The Synthesis Review of the Approved Tyrosine Kinase Inhibitors for Anticancer Therapy in 2015–2020. Bioorg. Chem. 2021, 113, 105011. [Google Scholar] [CrossRef]
- Purcaru, O.-S.; Artene, S.-A.; Barcan, E.; Silosi, C.A.; Stanciu, I.; Danoiu, S.; Tudorache, S.; Tataranu, L.G.; Dricu, A. The Interference between SARS-CoV-2 and Tyrosine Kinase Receptor Signaling in Cancer. Int. J. Mol. Sci. 2021, 22, 4830. [Google Scholar] [CrossRef]
- Cagno, V.; Magliocco, G.; Tapparel, C.; Daali, Y. The Tyrosine Kinase Inhibitor Nilotinib Inhibits SARS-CoV-2 in Vitro. Basic Clin. Pharmacol. Toxicol. 2021, 128, 621–624. [Google Scholar] [CrossRef]
- Schor, S.; Einav, S. Repurposing of Kinase Inhibitors as Broad-Spectrum Antiviral Drugs. DNA Cell Biol. 2018, 37, 63–69. [Google Scholar] [CrossRef]
- Dubreuil, P.; Letard, S.; Ciufolini, M.; Gros, L.; Humbert, M.; Castéran, N.; Borge, L.; Hajem, B.; Lermet, A.; Sippl, W.; et al. Masitinib (AB1010), a Potent and Selective Tyrosine Kinase Inhibitor Targeting KIT. PLoS ONE 2009, 4, e7258. [Google Scholar] [CrossRef]
- Zemke, D.; Yamini, B.; Yuzbasiyan-Gurkan, V. Mutations in the Juxtamembrane Domain of C- KIT Are Associated with Higher Grade Mast Cell Tumors in Dogs. Vet. Pathol. 2002, 39, 529–535. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hahn, K.A.; Legendre, A.M.; Shaw, N.G.; Phillips, B.; Ogilvie, G.K.; Prescott, D.M.; Atwater, S.W.; Carreras, J.K.; Lana, S.E.; Ladue, T.; et al. Evaluation of 12- and 24-Month Survival Rates after Treatment with Masitinib in Dogs with Nonresectable Mast Cell Tumors. Am. J. Vet. Res. 2010, 71, 1354–1361. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Daly, M.; Sheppard, S.; Cohen, N.; Nabity, M.; Moussy, A.; Hermine, O.; Wilson, H. Safety of Masitinib Mesylate in Healthy Cats. J. Vet. Intern. Med. 2011, 25, 297–302. [Google Scholar] [CrossRef] [PubMed]
- Palomo, V.; Nozal, V.; Rojas-Prats, E.; Gil, C.; Martinez, A. Protein Kinase Inhibitors for Amyotrophic Lateral Sclerosis Therapy. Br. J. Pharmacol. 2021, 178, 1316–1335. [Google Scholar] [CrossRef] [PubMed]
- Paul, C.; Sans, B.; Suarez, F.; Casassus, P.; Barete, S.; Lanternier, F.; Grandpeix-Guyodo, C.; Dubreuil, P.; Palmérini, F.; Mansfield, C.D.; et al. Masitinib for the Treatment of Systemic and Cutaneous Mastocytosis with Handicap: A Phase 2a Study. Am. J. Hematol. 2010, 85, 921–925. [Google Scholar] [CrossRef]
- Piette, F.; Belmin, J.; Vincent, H.; Schmidt, N.; Pariel, S.; Verny, M.; Marquis, C.; Mely, J.; Hugonot-Diener, L.; Kinet, J.-P.; et al. Masitinib as an Adjunct Therapy for Mild-to-Moderate Alzheimer’s Disease: A Randomised, Placebo-Controlled Phase 2 Trial. Alzheimer’s Res. Ther. 2011, 3, 16. [Google Scholar] [CrossRef]
- Tebib, J.; Mariette, X.; Bourgeois, P.; Flipo, R.-M.; Gaudin, P.; le Loët, X.; Gineste, P.; Guy, L.; Mansfield, C.D.; Moussy, A.; et al. Masitinib in the Treatment of Active Rheumatoid Arthritis: Results of a Multicentre, Open-Label, Dose-Ranging, Phase 2a Study. Arthritis Res. Ther. 2009, 11, R95. [Google Scholar] [CrossRef] [Green Version]
- Vermersch, P.; Benrabah, R.; Schmidt, N.; Zéphir, H.; Clavelou, P.; Vongsouthi, C.; Dubreuil, P.; Moussy, A.; Hermine, O. Masitinib Treatment in Patients with Progressive Multiple Sclerosis: A Randomized Pilot Study. BMC Neurol. 2012, 12, 36. [Google Scholar] [CrossRef] [Green Version]
- Ettcheto, M.; Cano, A.; Sanchez-López, E.; Verdaguer, E.; Folch, J.; Auladell, C.; Camins, A. Masitinib for the Treatment of Alzheimer’s Disease. Neurodegener. Dis. Manag. 2021, 11, 263–276. [Google Scholar] [CrossRef]
- Vermersch, P.; Brieva-Ruiz, L.; Fox, R.J.; Paul, F.; Ramio-Torrenta, L.; Schwab, M.; Moussy, A.; Mansfield, C.; Hermine, O.; Maciejowski, M. Efficacy and Safety of Masitinib in Progressive Forms of Multiple Sclerosis. Neurol. Neuroimmunol. Neuroinflamm. 2022, 9, e1148. [Google Scholar] [CrossRef]
- Drayman, N.; DeMarco, J.K.; Jones, K.A.; Azizi, S.-A.; Froggatt, H.M.; Tan, K.; Maltseva, N.I.; Chen, S.; Nicolaescu, V.; Dvorkin, S.; et al. Masitinib Is a Broad Coronavirus 3CL Inhibitor That Blocks Replication of SARS-CoV-2. Science 2021, 373, 931–936. [Google Scholar] [CrossRef] [PubMed]
- Koepsell, H. The SLC22 Family with Transporters of Organic Cations, Anions and Zwitterions. Mol. Asp. Med. 2013, 34, 413–435. [Google Scholar] [CrossRef] [PubMed]
- Yonezawa, A.; Inui, K. Importance of the Multidrug and Toxin Extrusion MATE/SLC47A Family to Pharmacokinetics, Pharmacodynamics/Toxicodynamics and Pharmacogenomics. Br. J. Pharmacol. 2011, 164, 1817–1825. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kimura, N.; Okuda, M.; Inui, K. Metformin Transport by Renal Basolateral Organic Cation Transporter HOCT2. Pharm. Res. 2005, 22, 255–259. [Google Scholar] [CrossRef]
- Zhou, M.; Xia, L.; Wang, J. Metformin Transport by a Newly Cloned Proton-Stimulated Organic Cation Transporter (Plasma Membrane Monoamine Transporter) Expressed in Human Intestine. Drug Metab. Dispos. 2007, 35, 1956–1962. [Google Scholar] [CrossRef] [Green Version]
- Ciarimboli, G.; Schröter, R.; Neugebauer, U.; Vollenbröker, B.; Gabriëls, G.; Brzica, H.; Sabolić, I.; Pietig, G.; Pavenstädt, H.; Schlatter, E.; et al. Kidney Transplantation Down-Regulates Expression of Organic Cation Transporters, Which Translocate β-Blockers and Fluoroquinolones. Mol. Pharm. 2013, 10, 2370–2380. [Google Scholar] [CrossRef]
- Misaka, S.; Knop, J.; Singer, K.; Hoier, E.; Keiser, M.; Müller, F.; Glaeser, H.; König, J.; Fromm, M.F. The Nonmetabolized β-Blocker Nadolol Is a Substrate of OCT1, OCT2, MATE1, MATE2-K, and P-Glycoprotein, but Not of OATP1B1 and OATP1B3. Mol. Pharm. 2016, 13, 512–519. [Google Scholar] [CrossRef]
- Harrach, S.; Edemir, B.; Schmidt-Lauber, C.; Pap, T.; Bertrand, J.; Ciarimboli, G. Importance of the Novel Organic Cation Transporter 1 for Tyrosine Kinase Inhibition by Saracatinib in Rheumatoid Arthritis Synovial Fibroblasts. Sci. Rep. 2017, 7, 1258. [Google Scholar] [CrossRef] [Green Version]
- Schmidt-Lauber, C.; Harrach, S.; Pap, T.; Fischer, M.; Victor, M.; Heitzmann, M.; Hansen, U.; Fobker, M.; Brand, S.-M.; Sindic, A.; et al. Transport Mechanisms and Their Pathology-Induced Regulation Govern Tyrosine Kinase Inhibitor Delivery in Rheumatoid Arthritis. PLoS ONE 2012, 7, e52247. [Google Scholar] [CrossRef] [Green Version]
- Harrach, S.; Schmidt-Lauber, C.; Pap, T.; Pavenstädt, H.; Schlatter, E.; Schmidt, E.; Berdel, W.E.; Schulze, U.; Edemir, B.; Jeromin, S.; et al. MATE1 Regulates Cellular Uptake and Sensitivity to Imatinib in CML Patients. Blood Cancer J. 2016, 6, e470. [Google Scholar] [CrossRef]
- Ellawatty, W.E.A.; Masuo, Y.; Fujita, K.-I.; Yamazaki, E.; Ishida, H.; Arakawa, H.; Nakamichi, N.; Abdelwahed, R.; Sasaki, Y.; Kato, Y. Organic Cation Transporter 1 Is Responsible for Hepatocellular Uptake of the Tyrosine Kinase Inhibitor Pazopanib. Drug Metab. Dispos. 2018, 46, 33–40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Swift, B.; Nebot, N.; Lee, J.K.; Han, T.; Proctor, W.R.; Thakker, D.R.; Lang, D.; Radtke, M.; Gnoth, M.J.; Brouwer, K.L.R. Sorafenib Hepatobiliary Disposition: Mechanisms of Hepatic Uptake and Disposition of Generated Metabolites. Drug Metab. Dispos. 2013, 41, 1179–1186. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Minematsu, T.; Giacomini, K.M. Interactions of Tyrosine Kinase Inhibitors with Organic Cation Transporters and Multidrug and Toxic Compound Extrusion Proteins. Mol. Cancer Ther. 2011, 10, 531–539. [Google Scholar] [CrossRef] [Green Version]
- Koepsell, H. Organic Cation Transporters in Health and Disease. Pharmacol. Rev. 2020, 72, 253–319. [Google Scholar] [CrossRef]
- Harrach, S.; Ciarimboli, G. Role of Transporters in the Distribution of Platinum-Based Drugs. Front. Pharmacol. 2015, 6, 85. [Google Scholar] [CrossRef] [Green Version]
- Wright, S.H.; Dantzler, W.H. Molecular and Cellular Physiology of Renal Organic Cation and Anion Transport. Physiol. Rev. 2004, 84, 987–1049. [Google Scholar] [CrossRef]
- Terada, T.; Masuda, S.; Asaka, J.-I.; Tsuda, M.; Katsura, T.; Inui, K. Molecular Cloning, Functional Characterization and Tissue Distribution of Rat H+/Organic Cation Antiporter MATE1. Pharm. Res. 2006, 23, 1696–1701. [Google Scholar] [CrossRef]
- Nies, A.T.; Koepsell, H.; Damme, K.; Schwab, M. Organic Cation Transporters (OCTs, MATEs), In Vitro and In Vivo Evidence for the Importance in Drug Therapy. In Handbook of Experimental Pharmacology; Springer: Berlin/Heidelberg, Germany, 2011; pp. 105–167. [Google Scholar]
- Tamai, I.; Nakanishi, T.; Kobayashi, D.; China, K.; Kosugi, Y.; Nezu, J.; Sai, Y.; Tsuji, A. Involvement of OCTN1 (SLC22A4) in PH-Dependent Transport of Organic Cations. Mol. Pharm. 2004, 1, 57–66. [Google Scholar] [CrossRef] [PubMed]
- Amphoux, A.; Millan, M.J.; Cordi, A.; Bönisch, H.; Vialou, V.; Mannoury la Cour, C.; Dupuis, D.S.; Giros, B.; Gautron, S. Inhibitory and Facilitory Actions of Isocyanine Derivatives at Human and Rat Organic Cation Transporters 1, 2 and 3: A Comparison to Human Alpha 1- and Alpha 2-Adrenoceptor Subtypes. Eur. J. Pharmacol. 2010, 634, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Keller, T.; Gorboulev, V.; Mueller, T.D.; Dötsch, V.; Bernhard, F.; Koepsell, H. Rat Organic Cation Transporter 1 Contains Three Binding Sites for Substrate 1-Methyl-4-Phenylpyridinium per Monomer. Mol. Pharmacol. 2019, 95, 169–182. [Google Scholar] [CrossRef]
- Rochat, B. Importance of Influx and Efflux Systems and Xenobiotic Metabolizing Enzymes in Intratumoral Disposition of Anticancer Agents. Curr. Cancer Drug Targets 2009, 9, 652–674. [Google Scholar] [CrossRef] [PubMed]
- Grimminger, F.; Schermuly, R.T.; Ghofrani, H.A. Targeting Non-Malignant Disorders with Tyrosine Kinase Inhibitors. Nat. Rev. Drug Discov. 2010, 9, 956–970. [Google Scholar] [CrossRef] [PubMed]
- Kantarjian, H.; Sawyers, C.; Hochhaus, A.; Guilhot, F.; Schiffer, C.; Gambacorti-Passerini, C.; Niederwieser, D.; Resta, D.; Capdeville, R.; Zoellner, U.; et al. Hematologic and Cytogenetic Responses to Imatinib Mesylate in Chronic Myelogenous Leukemia. N. Engl. J. Med. 2002, 346, 645–652. [Google Scholar] [CrossRef] [PubMed]
- Shao, D.; Massoud, E.; Anand, U.; Parikh, A.; Cowley, E.; Clarke, D.; Agu, R.U. Organic Cation Transporters in Human Nasal Primary Culture: Expression and Functional Activity. Ther. Deliv. 2013, 4, 439–451. [Google Scholar] [CrossRef] [PubMed]
- Snieder, B.; Brast, S.; Grabner, A.; Buchholz, S.; Schröter, R.; Spoden, G.A.; Florin, L.; Salomon, J.; Albrecht, T.; Barz, V.; et al. Identification of the Tetraspanin CD9 as an Interaction Partner of Organic Cation Transporters 1 and 2. SLAS Discov. 2019, 24, 904–914. [Google Scholar] [CrossRef] [PubMed]
- Selo, M.A.; Sake, J.A.; Ehrhardt, C.; Salomon, J.J. Organic Cation Transporters in the Lung—Current and Emerging (Patho)Physiological and Pharmacological Concepts. Int. J. Mol. Sci. 2020, 21, 9168. [Google Scholar] [CrossRef]
- Han, T.; Everett, R.S.; Proctor, W.R.; Ng, C.M.; Costales, C.L.; Brouwer, K.L.R.; Thakker, D.R. Organic Cation Transporter 1 (OCT1/MOct1) Is Localized in the Apical Membrane of Caco-2 Cell Monolayers and Enterocytes. Mol. Pharmacol. 2013, 84, 182–189. [Google Scholar] [CrossRef] [Green Version]
- Soria, J.C.; Massard, C.; Magné, N.; Bader, T.; Mansfield, C.D.; Blay, J.Y.; Bui, B.N.; Moussy, A.; Hermine, O.; Armand, J.P. Phase 1 Dose-Escalation Study of Oral Tyrosine Kinase Inhibitor Masitinib in Advanced and/or Metastatic Solid Cancers. Eur. J. Cancer 2009, 45, 2333–2341. [Google Scholar] [CrossRef]
- Lee, W.-K.; Reichold, M.; Edemir, B.; Ciarimboli, G.; Warth, R.; Koepsell, H.; Thévenod, F. Organic Cation Transporters OCT1, 2, and 3 Mediate High-Affinity Transport of the Mutagenic Vital Dye Ethidium in the Kidney Proximal Tubule. Am. J. Physiol. Renal. Physiol. 2009, 296, F1504–F1513. [Google Scholar] [CrossRef] [Green Version]
- Schlatter, E.; Klassen, P.; Massmann, V.; Holle, S.K.; Guckel, D.; Edemir, B.; Pavenstädt, H.; Ciarimboli, G. Mouse Organic Cation Transporter 1 Determines Properties and Regulation of Basolateral Organic Cation Transport in Renal Proximal Tubules. Pflugers Arch. 2014, 466, 1581–1589. [Google Scholar] [CrossRef]
- Massmann, V.; Edemir, B.; Schlatter, E.; Al-Monajjed, R.; Harrach, S.; Klassen, P.; Holle, S.K.; Sindic, A.; Dobrivojevic, M.; Pavenstädt, H.; et al. The Organic Cation Transporter 3 (OCT3) as Molecular Target of Psychotropic Drugs: Transport Characteristics and Acute Regulation of Cloned Murine OCT3. Pflugers Arch. 2014, 466, 517–527. [Google Scholar] [CrossRef] [PubMed]
- Hucke, A.; Park, G.Y.; Bauer, O.B.; Beyer, G.; Köppen, C.; Zeeh, D.; Wehe, C.A.; Sperling, M.; Schröter, R.; Kantauskaitè, M.; et al. Interaction of the New Monofunctional Anticancer Agent Phenanthriplatin With Transporters for Organic Cations. Front. Chem. 2018, 6, 180. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilde, S.; Schlatter, E.; Koepsell, H.; Edemir, B.; Reuter, S.; Pavenstädt, H.; Neugebauer, U.; Schröter, R.; Brast, S.; Ciarimboli, G. Calmodulin-Associated Post-Translational Regulation of Rat Organic Cation Transporter 2 in the Kidney Is Gender Dependent. Cell Mol. Life Sci. 2009, 66, 1729–1740. [Google Scholar] [CrossRef] [PubMed]
- Amrhein, J.; Drynda, S.; Schlatt, L.; Karst, U.; Lohmann, C.H.; Ciarimboli, G.; Bertrand, J. Tofacitinib and Baricitinib Are Taken up by Different Uptake Mechanisms Determining the Efficacy of Both Drugs in RA. Int. J. Mol. Sci. 2020, 21, 6632. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| IC50 (µM) (logIC50 ± SEM) for Masitinib and Number of Replicates (n) Measured in 3 Independent Experiments | ||
|---|---|---|
| Transporters | Murine | Human |
| OCT1 | 14 (−4.8 ± 0.3) n = 20–50 | 24 (−4.6 ± 0.2) n = 19–39 |
| OCT2 | 19 (−4.7 ± 0.3) n = 23–43 | 7 (−5.1 ± 0.1) n = 20–42 |
| OCT3 | n.d. 1 n = 19–31 | 14 (−4.8 ± 0.1) n = 20–42 |
| MATE1 | 5 (−5.3 ± 0.3) n = 18–37 | |
| Time (min) | A % (0.1% Formic Acid) | B % (Acetonitrile) | Flow Velocity (µL/min) |
|---|---|---|---|
| 0 | 100 | 0 | 150 |
| 1 | 100 | 0 | 150 |
| 10 | 50 | 50 | 150 |
| 11 | 0 | 100 | 150 |
| 15 | 100 | 0 | 150 |
| 20 | 100 | 0 | 150 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Harrach, S.; Haag, J.; Steinbüchel, M.; Schröter, R.; Neugebauer, U.; Bertrand, J.; Ciarimboli, G. Interaction of Masitinib with Organic Cation Transporters. Int. J. Mol. Sci. 2022, 23, 14189. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms232214189
Harrach S, Haag J, Steinbüchel M, Schröter R, Neugebauer U, Bertrand J, Ciarimboli G. Interaction of Masitinib with Organic Cation Transporters. International Journal of Molecular Sciences. 2022; 23(22):14189. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms232214189
Chicago/Turabian StyleHarrach, Saliha, Jasmin Haag, Martin Steinbüchel, Rita Schröter, Ute Neugebauer, Jessica Bertrand, and Giuliano Ciarimboli. 2022. "Interaction of Masitinib with Organic Cation Transporters" International Journal of Molecular Sciences 23, no. 22: 14189. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms232214189