

Durable CD8 T Cell Memory against SARS-CoV-2 by Prime/Boost and Multi-Dose Vaccination: Considerations on Inter-Dose Time Intervals
 ,
,
Abstract
:1. Introduction to COVID-19 and CD8 T Cells
2. Vaccine-Induced CD8 T Cell Responses: A General Overview
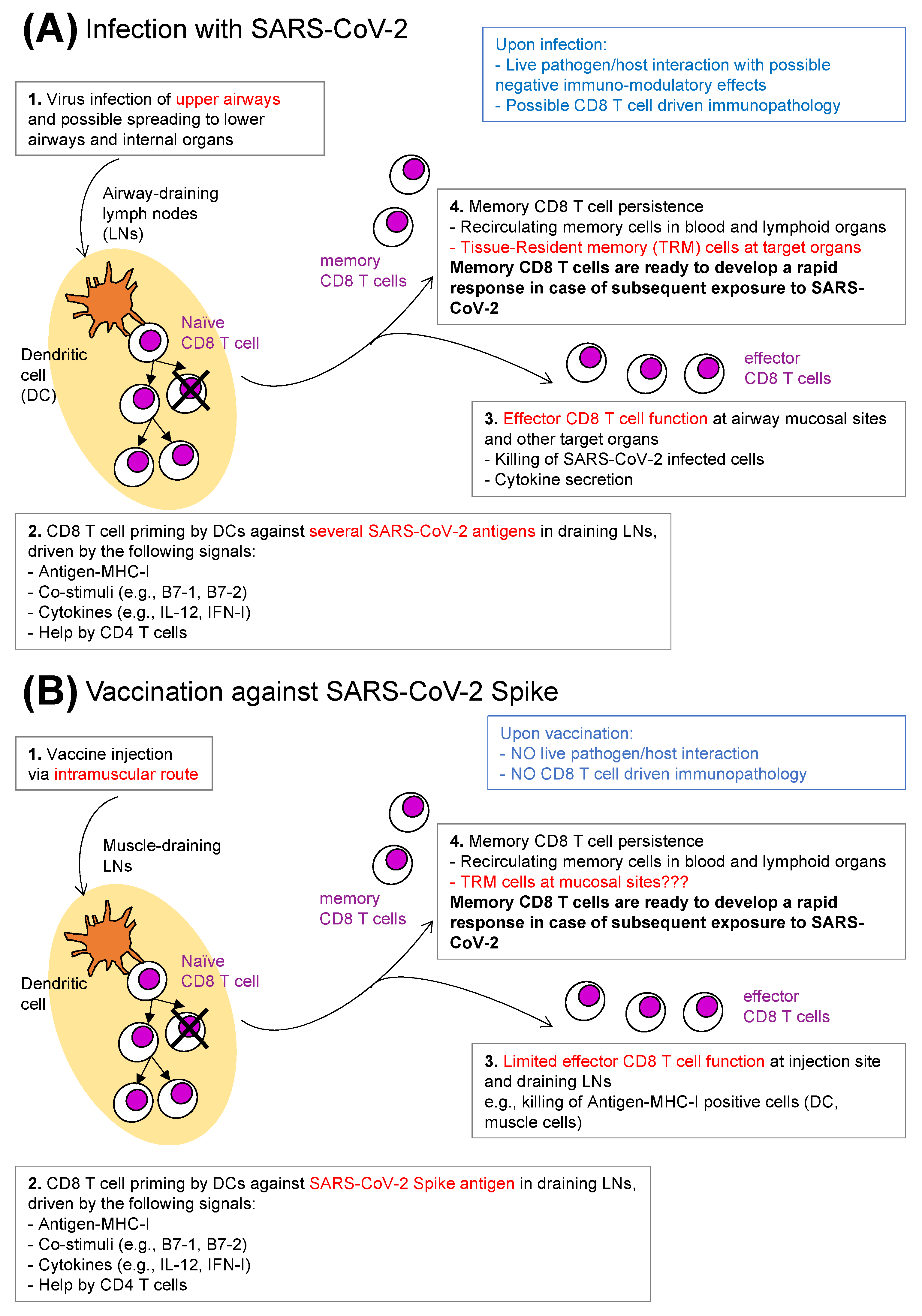
3. Anti-SARS-CoV-2 CD8 T Cells: Natural Infection versus Vaccination
4. CD8 T Cell Responses to Prime/Boost Vaccination
5. Prime/Boost Vaccination during COVID-19 Pandemic: Strengths and Limitations
6. Additional Boosters and Long-Lived Memory CD8 T Cells
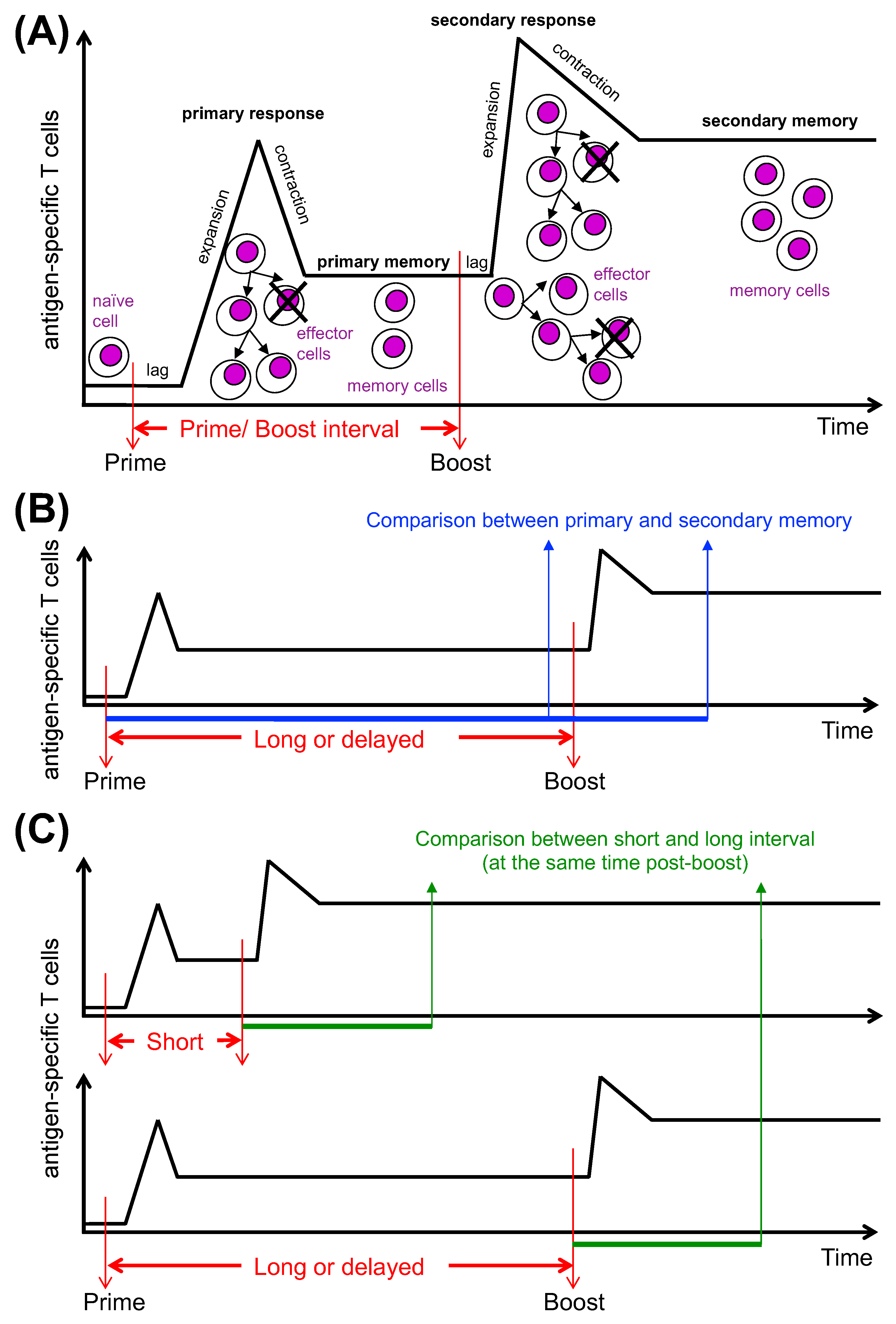
7. Time Interval between Vaccine Doses and CD8 T Cell Differentiation
8. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Appendix A
- What are the best correlates of protection against SARS-CoV-2 by the different arms of the adaptive immunity?
- What are the distinct effector mechanisms and molecules of CD8 T cell-mediated protection against SARS-CoV-2?
- What are the key molecular determinants (e.g., transcriptional, epigenetic, metabolic, etc.) for establishing and maintaining memory CD8 T cells against SARS-CoV-2?
- What are the key differences between anti-SARS-CoV-2 memory CD8 T cells induced by natural infection versus those induced by vaccination?
- What are the key features that make anti-SARS-CoV-2 memory CD8 T cells induced by a combination of vaccination and infection (hybrid immunity) stronger than that induced by vaccination only?
- Which SARS-CoV-2 antigenic epitopes are targeted by anti-SARS-CoV-2 memory CD8 T cells induced by natural infection, vaccination and hybrid immunity?
- What is the role of TRM versus recirculating memory CD8 T cells for protection against SARS-CoV-2? Which molecules and cells are required for establishing and maintaining each of the two cell types?
- What are the most effective routes of vaccination and vaccine platforms to induce protective memory CD8 T cells against SARS-CoV-2?
- What are the most effective vaccine platform combinations to prime and boost memory CD8 T cells against SARS-CoV-2? What are the most effective platforms for multiple boosters?
- What is the most effective prime/boost time interval to induce protective memory CD8 T cells against SARS-CoV-2? What is the most effective time interval between booster doses?
References
- Stanley, D.A.; Honko, A.N.; Asiedu, C.; Trefry, J.C.; Lau-Kilby, A.W.; Johnson, J.C.; Hensley, L.; Ammendola, V.; Abbate, A.; Grazioli, F.; et al. Chimpanzee adenovirus vaccine generates acute and durable protective immunity against ebolavirus challenge. Nat. Med. 2014, 20, 1126–1129. [Google Scholar] [CrossRef] [PubMed]
- Pardi, N.; Hogan, M.J.; Porter, F.W.; Weissman, D. mRNA vaccines—A new era in vaccinology. Nat. Rev. Drug Discov. 2018, 17, 261–279. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Graham, B.S. Rapid COVID-19 vaccine development. Science 2020, 368, 945. [Google Scholar] [CrossRef]
- Krammer, F. SARS-CoV-2 vaccines in development. Nature 2020, 586, 516–527. [Google Scholar] [CrossRef] [PubMed]
- Baden, L.R.; El Sahly, H.M.; Essink, B.; Kotloff, K.; Frey, S.; Novak, R.; Diemert, D.; Spector, S.A.; Rouphael, N.; Creech, C.B.; et al. Efficacy and Safety of the mRNA-1273 SARS-CoV-2 Vaccine. N. Engl. J. Med. 2021, 384, 403–416. [Google Scholar] [CrossRef] [PubMed]
- Ramasamy, M.N.; Minassian, A.M.; Ewer, K.J.; Flaxman, A.L.; Folegatti, P.M.; Owens, D.R.; Voysey, M.; Aley, P.K.; Angus, B.; Babbage, G.; et al. Safety and immunogenicity of ChAdOx1 nCoV-19 vaccine administered in a prime-boost regimen in young and old adults (COV002): A single-blind, randomised, controlled, phase 2/3 trial. Lancet 2021, 396, 1979–1993. [Google Scholar] [CrossRef]
- Polack, F.P.; Thomas, S.J.; Kitchin, N.; Absalon, J.; Gurtman, A.; Lockhart, S.; Perez, J.L.; Pérez Marc, G.; Moreira, E.D.; Zerbini, C.; et al. Safety and Efficacy of the BNT162b2 mRNA Covid-19 Vaccine. N. Engl. J. Med. 2020, 383, 2603–2615. [Google Scholar] [CrossRef]
- Logunov, D.Y.; Dolzhikova, I.V.; Shcheblyakov, D.V.; Tukhvatulin, A.I.; Zubkova, O.V.; Dzharullaeva, A.S.; Kovyrshina, A.V.; Lubenets, N.L.; Grousova, D.M.; Erokhova, A.S.; et al. Safety and efficacy of an rAd26 and rAd5 vector-based heterologous prime-boost COVID-19 vaccine: An interim analysis of a randomised controlled phase 3 trial in Russia. Lancet 2021, 397, 671–681. [Google Scholar] [CrossRef]
- Lipsitch, M.; Krammer, F.; Regev-Yochay, G.; Lustig, Y.; Balicer, R.D. SARS-CoV-2 breakthrough infections in vaccinated individuals: Measurement, causes and impact. Nat. Rev. Immunol. 2022, 22, 57–65. [Google Scholar] [CrossRef]
- Vivaldi, G.; Jolliffe, D.A.; Faustini, S.; Shields, A.M.; Holt, H.; Perdek, N.; Talaei, M.; Tydeman, F.; Chambers, E.S.; Cai, W.; et al. Correlation between post-vaccination anti-Spike antibody titres and protection against breakthrough SARS-CoV-2 infection: A population-based longitudinal study. J. Infect. Dis. 2022. [Google Scholar] [CrossRef]
- Gupta, R.K. Will SARS-CoV-2 variants of concern affect the promise of vaccines. Nat. Rev. Immunol. 2021, 21, 340–341. [Google Scholar] [CrossRef] [PubMed]
- McLean, G.; Kamil, J.; Lee, B.; Moore, P.; Schulz, T.F.; Muik, A.; Sahin, U.; Türeci, Ö.; Pather, S. The Impact of Evolving SARS-CoV-2 Mutations and Variants on COVID-19 Vaccines. mBio 2022, 13, e0297921. [Google Scholar] [CrossRef] [PubMed]
- Weingarten-Gabbay, S.; Klaeger, S.; Sarkizova, S.; Pearlman, L.R.; Chen, D.Y.; Gallagher, K.M.E.; Bauer, M.R.; Taylor, H.B.; Dunn, W.A.; Tarr, C.; et al. Profiling SARS-CoV-2 HLA-I peptidome reveals T cell epitopes from out-of-frame ORFs. Cell 2021, 184, 3962–3980.e17. [Google Scholar] [CrossRef] [PubMed]
- Sette, A.; Crotty, S. Immunological memory to SARS-CoV-2 infection and COVID-19 vaccines. Immunol. Rev. 2022, 310, 27–46. [Google Scholar] [CrossRef] [PubMed]
- Goldblatt, D.; Alter, G.; Crotty, S.; Plotkin, S.A. Correlates of protection against SARS-CoV-2 infection and COVID-19 disease. Immunol. Rev. 2022, 310, 6–26. [Google Scholar] [CrossRef]
- Ahlers, J.D.; Belyakov, I.M. Memories that last forever: Strategies for optimizing vaccine T-cell memory. Blood 2010, 115, 1678–1689. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Butler, N.S.; Nolz, J.C.; Harty, J.T. Immunologic considerations for generating memory CD8 T cells through vaccination. Cell. Microbiol. 2011, 13, 925–933. [Google Scholar] [CrossRef] [Green Version]
- Majhen, D.; Calderon, H.; Chandra, N.; Fajardo, C.A.; Rajan, A.; Alemany, R.; Custers, J. Adenovirus-based vaccines for fighting infectious diseases and cancer: Progress in the field. Hum. Gene Ther. 2014, 25, 301–317. [Google Scholar] [CrossRef]
- Cagigi, A.; Loré, K. Immune Responses Induced by mRNA Vaccination in Mice, Monkeys and Humans. Vaccines 2021, 9, 61. [Google Scholar] [CrossRef]
- Lazzaro, S.; Giovani, C.; Mangiavacchi, S.; Magini, D.; Maione, D.; Baudner, B.; Geall, A.J.; De Gregorio, E.; D’Oro, U.; Buonsanti, C. CD8 T-cell priming upon mRNA vaccination is restricted to bone-marrow-derived antigen-presenting cells and may involve antigen transfer from myocytes. Immunology 2015, 146, 312–326. [Google Scholar] [CrossRef]
- Steinman, R.M. Dendritic cells in vivo: A key target for a new vaccine science. Immunity 2008, 29, 319–324. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sallusto, F.; Schaerli, P.; Loetscher, P.; Schaniel, C.; Lenig, D.; Mackay, C.R.; Qin, S.; Lanzavecchia, A. Rapid and coordinated switch in chemokine receptor expression during dendritic cell maturation. Eur. J. Immunol. 1998, 28, 2760–2769. [Google Scholar] [CrossRef]
- Janeway, C.A.J. Approaching the asymptote? Evolution and revolution in immunology. Cold Spring Harb. Symp. Quant. Biol. 1989, 54 Pt 1, 1–13. Available online: http://0-www-ncbi-nlm-nih-gov.brum.beds.ac.uk/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=2700931 (accessed on 30 September 2022). [CrossRef] [PubMed]
- Matzinger, P. Tolerance, danger, and the extended family. Annu. Rev. Immunol. 1994, 12, 991–1045. [Google Scholar] [CrossRef]
- Sobolev, O.; Binda, E.; O’Farrell, S.; Lorenc, A.; Pradines, J.; Huang, Y.; Duffner, J.; Schulz, R.; Cason, J.; Zambon, M.; et al. Adjuvanted influenza-H1N1 vaccination reveals lymphoid signatures of age-dependent early responses and of clinical adverse events. Nat. Immunol. 2016, 17, 204–213. [Google Scholar] [CrossRef]
- Gallucci, S.; Lolkema, M.; Matzinger, P. Natural adjuvants: Endogenous activators of dendritic cells. Nat. Med. 1999, 5, 1249–1255. [Google Scholar] [CrossRef]
- Monin, L.; Laing, A.G.; Muñoz-Ruiz, M.; McKenzie, D.R.; Del Molino Del Barrio, I.; Alaguthurai, T.; Domingo-Vila, C.; Hayday, T.S.; Graham, C.; Seow, J.; et al. Safety and immunogenicity of one versus two doses of the COVID-19 vaccine BNT162b2 for patients with cancer: Interim analysis of a prospective observational study. Lancet Oncol. 2021, 22, 765–778. [Google Scholar] [CrossRef]
- Jameson, S.C.; Masopust, D. Understanding Subset Diversity in T Cell Memory. Immunity 2018, 48, 214–226. [Google Scholar] [CrossRef] [Green Version]
- Ridge, J.P.; Di Rosa, F.; Matzinger, P. A conditioned dendritic cell can be a temporal bridge between a CD4+ T-helper and a T-killer cell. Nature 1998, 393, 474–478. [Google Scholar] [CrossRef]
- Schoenberger, S.P.; Toes, R.E.; van der Voort, E.I.; Offringa, R.; Melief, C.J. T-cell help for cytotoxic T lymphocytes is mediated by CD40-CD40L interactions. Nature 1998, 393, 480–483. [Google Scholar] [CrossRef]
- Bennett, S.R.; Carbone, F.R.; Karamalis, F.; Flavell, R.A.; Miller, J.F.; Heath, W.R. Help for cytotoxic-T-cell responses is mediated by CD40 signalling. Nature 1998, 393, 478–480. [Google Scholar] [CrossRef] [PubMed]
- Lanzavecchia, A. Immunology. Licence to kill. Nature 1998, 393, 413–414. [Google Scholar] [CrossRef]
- Opferman, J.T.; Ober, B.T.; Ashton-Rickardt, P.G. Linear differentiation of cytotoxic effectors into memory T lymphocytes. Science 1999, 283, 1745–1748. Available online: http://0-www-ncbi-nlm-nih-gov.brum.beds.ac.uk/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=10073942 (accessed on 30 September 2022). [CrossRef] [PubMed]
- Bannard, O.; Kraman, M.; Fearon, D.T. Secondary replicative function of CD8+ T cells that had developed an effector phenotype. Science 2009, 323, 505–509. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, J.T.; Palanivel, V.R.; Kinjyo, I.; Schambach, F.; Intlekofer, A.M.; Banerjee, A.; Longworth, S.A.; Vinup, K.E.; Mrass, P.; Oliaro, J.; et al. Asymmetric T lymphocyte division in the initiation of adaptive immune responses. Science 2007, 315, 1687–1691. [Google Scholar] [CrossRef]
- Teixeiro, E.; Daniels, M.A.; Hamilton, S.E.; Schrum, A.G.; Bragado, R.; Jameson, S.C.; Palmer, E. Different T cell receptor signals determine CD8+ memory versus effector development. Science 2009, 323, 502–505. [Google Scholar] [CrossRef]
- Moskophidis, D.; Lechner, F.; Pircher, H.; Zinkernagel, R.M. Virus persistence in acutely infected immunocompetent mice by exhaustion of antiviral cytotoxic effector T cells. Nature 1993, 362, 758–761. [Google Scholar] [CrossRef]
- Hashimoto, M.; Kamphorst, A.O.; Im, S.J.; Kissick, H.T.; Pillai, R.N.; Ramalingam, S.S.; Araki, K.; Ahmed, R. CD8 T Cell Exhaustion in Chronic Infection and Cancer: Opportunities for Interventions. Annu. Rev. Med. 2018, 69, 301–318. [Google Scholar] [CrossRef]
- Adam, L.; Rosenbaum, P.; Quentric, P.; Parizot, C.; Bonduelle, O.; Guillou, N.; Corneau, A.; Dorgham, K.; Miyara, M.; Luyt, C.E.; et al. CD8+PD-L1+CXCR3+ polyfunctional T cell abundances are associated with survival in critical SARS-CoV-2-infected patients. JCI Insight 2021, 6, e151571. [Google Scholar] [CrossRef]
- Roukens, A.H.E.; Pothast, C.R.; König, M.; Huisman, W.; Dalebout, T.; Tak, T.; Azimi, S.; Kruize, Y.; Hagedoorn, R.S.; Zlei, M.; et al. Prolonged activation of nasal immune cell populations and development of tissue-resident SARS-CoV-2-specific CD8+ T cell responses following COVID-19. Nat. Immunol. 2022, 23, 23–32. [Google Scholar] [CrossRef]
- Agerer, B.; Koblischke, M.; Gudipati, V.; Montaño-Gutierrez, L.F.; Smyth, M.; Popa, A.; Genger, J.W.; Endler, L.; Florian, D.M.; Mühlgrabner, V.; et al. SARS-CoV-2 mutations in MHC-I-restricted epitopes evade CD8+ T cell responses. Sci. Immunol. 2021, 6, eabg6461. [Google Scholar] [CrossRef] [PubMed]
- Vibholm, L.K.; Nielsen, S.S.F.; Pahus, M.H.; Frattari, G.S.; Olesen, R.; Andersen, R.; Monrad, I.; Andersen, A.H.F.; Thomsen, M.M.; Konrad, C.V.; et al. SARS-CoV-2 persistence is associated with antigen-specific CD8 T-cell responses. EBioMedicine 2021, 64, 103230. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Beltran, W.F.; Lam, E.C.; Astudillo, M.G.; Yang, D.; Miller, T.E.; Feldman, J.; Hauser, B.M.; Caradonna, T.M.; Clayton, K.L.; Nitido, A.D.; et al. COVID-19-neutralizing antibodies predict disease severity and survival. Cell 2021, 184, 476–488.e11. [Google Scholar] [CrossRef] [PubMed]
- Littlefield, K.M.; Watson, R.O.; Schneider, J.M.; Neff, C.P.; Yamada, E.; Zhang, M.; Campbell, T.B.; Falta, M.T.; Jolley, S.E.; Fontenot, A.P.; et al. SARS-CoV-2-specific T cells associate with inflammation and reduced lung function in pulmonary post-acute sequalae of SARS-CoV-2. PLoS Pathog. 2022, 18, e1010359. [Google Scholar] [CrossRef]
- Laing, A.G.; Lorenc, A.; Del Molino Del Barrio, I.; Das, A.; Fish, M.; Monin, L.; Muñoz-Ruiz, M.; McKenzie, D.R.; Hayday, T.S.; Francos-Quijorna, I.; et al. A dynamic COVID-19 immune signature includes associations with poor prognosis. Nat. Med. 2020, 26, 1623–1635. [Google Scholar] [CrossRef]
- Sekine, T.; Perez-Potti, A.; Rivera-Ballesteros, O.; Strålin, K.; Gorin, J.B.; Olsson, A.; Llewellyn-Lacey, S.; Kamal, H.; Bogdanovic, G.; Muschiol, S.; et al. Robust T Cell Immunity in Convalescent Individuals with Asymptomatic or Mild COVID-19. Cell 2020, 183, 158–168.e14. [Google Scholar] [CrossRef]
- Zuo, J.; Dowell, A.C.; Pearce, H.; Verma, K.; Long, H.M.; Begum, J.; Aiano, F.; Amin-Chowdhury, Z.; Hoschler, K.; Brooks, T.; et al. Robust SARS-CoV-2-specific T cell immunity is maintained at 6 months following primary infection. Nat. Immunol. 2021, 22, 620–626. [Google Scholar] [CrossRef]
- Khan, M.S.; Kim, E.; McPherson, A.; Weisel, F.J.; Huang, S.; Kenniston, T.W.; Percivalle, E.; Cassaniti, I.; Baldanti, F.; Meisel, M.; et al. Adenovirus-vectored SARS-CoV-2 vaccine expressing S1-N fusion protein. Antib. Ther. 2022, 5, 177–191. [Google Scholar] [CrossRef]
- Jeyanathan, M.; Afkhami, S.; Smaill, F.; Miller, M.S.; Lichty, B.D.; Xing, Z. Immunological considerations for COVID-19 vaccine strategies. Nat. Rev. Immunol. 2020, 20, 615–632. [Google Scholar] [CrossRef]
- Zhang, Z.; Mateus, J.; Coelho, C.H.; Dan, J.M.; Moderbacher, C.R.; Gálvez, R.I.; Cortes, F.H.; Grifoni, A.; Tarke, A.; Chang, J.; et al. Humoral and cellular immune memory to four COVID-19 vaccines. Cell 2022, 185, 2434–2451.e17. [Google Scholar] [CrossRef]
- Oberhardt, V.; Luxenburger, H.; Kemming, J.; Schulien, I.; Ciminski, K.; Giese, S.; Csernalabics, B.; Lang-Meli, J.; Janowska, I.; Staniek, J.; et al. Rapid and stable mobilization of CD8+ T cells by SARS-CoV-2 mRNA vaccine. Nature 2021, 597, 268–273. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.; Agrawal, S.; Sandoval, A.; Su, H.; Tran, M.; Demirdag, Y. SARS-CoV-2-Specific and Functional Cytotoxic CD8 Cells in Primary Antibody Deficiency: Natural Infection and Response to Vaccine. J. Clin. Immunol. 2022, 42, 914–922. [Google Scholar] [CrossRef] [PubMed]
- Antolí, A.; Rocamora-Blanch, G.; Framil, M.; Mas-Bosch, V.; Navarro, S.; Bermudez, C.; Martinez-Yelamos, S.; Dopico, E.; Calatayud, L.; Garcia-Muñoz, N.; et al. Evaluation of Humoral and Cellular Immune Responses to the SARS-CoV-2 Vaccine in Patients With Common Variable Immunodeficiency Phenotype and Patient Receiving B-Cell Depletion Therapy. Front. Immunol. 2022, 13, 895209. [Google Scholar] [CrossRef] [PubMed]
- Quinti, I.; Locatelli, F.; Carsetti, R. The Immune Response to SARS-CoV-2 Vaccination: Insights Learned From Adult Patients With Common Variable Immune Deficiency. Front. Immunol. 2021, 12, 815404. [Google Scholar] [CrossRef] [PubMed]
- Sallusto, F.; Lanzavecchia, A.; Araki, K.; Ahmed, R. From vaccines to memory and back. Immunity 2010, 33, 451–463. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akondy, R.S.; Johnson, P.L.; Nakaya, H.I.; Edupuganti, S.; Mulligan, M.J.; Lawson, B.; Miller, J.D.; Pulendran, B.; Antia, R.; Ahmed, R. Initial viral load determines the magnitude of the human CD8 T cell response to yellow fever vaccination. Proc. Natl. Acad. Sci. USA 2015, 112, 3050–3055. [Google Scholar] [CrossRef] [Green Version]
- Akondy, R.S.; Fitch, M.; Edupuganti, S.; Yang, S.; Kissick, H.T.; Li, K.W.; Youngblood, B.A.; Abdelsamed, H.A.; McGuire, D.J.; Cohen, K.W.; et al. Origin and differentiation of human memory CD8 T cells after vaccination. Nature 2017, 552, 362–367. [Google Scholar] [CrossRef]
- Woodland, D.L. Jump-starting the immune system: Prime-boosting comes of age. Trends Immunol. 2004, 25, 98–104. [Google Scholar] [CrossRef]
- Estcourt, M.J.; Ramsay, A.J.; Brooks, A.; Thomson, S.A.; Medveckzy, C.J.; Ramshaw, I.A. Prime-boost immunization generates a high frequency, high-avidity CD8(+) cytotoxic T lymphocyte population. Int. Immunol. 2002, 14, 31–37. [Google Scholar] [CrossRef] [Green Version]
- Yewdell, J.W.; Bennink, J.R. Immunodominance in major histocompatibility complex class I-restricted T lymphocyte responses. Annu. Rev. Immunol. 1999, 17, 51–88. [Google Scholar] [CrossRef]
- Walsh, S.R.; Moodie, Z.; Fiore-Gartland, A.J.; Morgan, C.; Wilck, M.B.; Hammer, S.M.; Buchbinder, S.P.; Kalams, S.A.; Goepfert, P.A.; Mulligan, M.J.; et al. Vaccination With Heterologous HIV-1 Envelope Sequences and Heterologous Adenovirus Vectors Increases T-Cell Responses to Conserved Regions: HVTN 083. J. Infect. Dis. 2016, 213, 541–550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kardani, K.; Bolhassani, A.; Shahbazi, S. Prime-boost vaccine strategy against viral infections: Mechanisms and benefits. Vaccine 2016, 34, 413–423. [Google Scholar] [CrossRef] [PubMed]
- Koutsakos, M.; Illing, P.T.; Nguyen, T.H.O.; Mifsud, N.A.; Crawford, J.C.; Rizzetto, S.; Eltahla, A.A.; Clemens, E.B.; Sant, S.; Chua, B.Y.; et al. Human CD8+ T cell cross-reactivity across influenza A, B and C viruses. Nat. Immunol. 2019, 20, 613–625. [Google Scholar] [CrossRef] [PubMed]
- Lipsitch, M.; Grad, Y.H.; Sette, A.; Crotty, S. Cross-reactive memory T cells and herd immunity to SARS-CoV-2. Nat. Rev. Immunol. 2020, 20, 709–713. [Google Scholar] [CrossRef]
- Mallajosyula, V.; Ganjavi, C.; Chakraborty, S.; McSween, A.M.; Pavlovitch-Bedzyk, A.J.; Wilhelmy, J.; Nau, A.; Manohar, M.; Nadeau, K.C.; Davis, M.M. CD8+ T cells specific for conserved coronavirus epitopes correlate with milder disease in COVID-19 patients. Sci. Immunol. 2021, 6, eabg5669. [Google Scholar] [CrossRef]
- Slütter, B.; Pewe, L.L.; Lauer, P.; Harty, J.T. Cutting edge: Rapid boosting of cross-reactive memory CD8 T cells broadens the protective capacity of the Flumist vaccine. J. Immunol. 2013, 190, 3854–3858. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tarke, A.; Coelho, C.H.; Zhang, Z.; Dan, J.M.; Yu, E.D.; Methot, N.; Bloom, N.I.; Goodwin, B.; Phillips, E.; Mallal, S.; et al. SARS-CoV-2 vaccination induces immunological T cell memory able to cross-recognize variants from Alpha to Omicron. Cell 2022, 185, 847–859.e11. [Google Scholar] [CrossRef]
- Hovav, A.H.; Panas, M.W.; Osuna, C.E.; Cayabyab, M.J.; Autissier, P.; Letvin, N.L. The impact of a boosting immunogen on the differentiation of secondary memory CD8+ T cells. J. Virol. 2007, 81, 12793–12802. [Google Scholar] [CrossRef] [Green Version]
- Matzinger, P.; (National Institutes of Health, Bethesda, MD, USA). Personal communication, 1993.
- Pace, L.; Tempez, A.; Arnold-Schrauf, C.; Lemaitre, F.; Bousso, P.; Fetler, L.; Sparwasser, T.; Amigorena, S. Regulatory T cells increase the avidity of primary CD8+ T cell responses and promote memory. Science 2012, 338, 532–536. [Google Scholar] [CrossRef] [Green Version]
- Aviles, J.; Bello, A.; Wong, G.; Fausther-Bovendo, H.; Qiu, X.; Kobinger, G. Optimization of Prime-Boost Vaccination Strategies Against Mouse-Adapted Ebolavirus in a Short-Term Protection Study. J. Infect. Dis. 2015, 212 (Suppl. 2), S389–S397. [Google Scholar] [CrossRef]
- Sedegah, M.; Jones, T.R.; Kaur, M.; Hedstrom, R.; Hobart, P.; Tine, J.A.; Hoffman, S.L. Boosting with recombinant vaccinia increases immunogenicity and protective efficacy of malaria DNA vaccine. Proc. Natl. Acad. Sci. USA 1998, 95, 7648–7653. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schneider, J.; Gilbert, S.C.; Hannan, C.M.; Degano, P.; Prieur, E.; Sheu, E.G.; Plebanski, M.; Hill, A.V. Induction of CD8+ T cells using heterologous prime-boost immunisation strategies. Immunol. Rev. 1999, 170, 29–38. Available online: https://0-www-ncbi-nlm-nih-gov.brum.beds.ac.uk/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=10566139 (accessed on 30 September 2022). [CrossRef] [PubMed]
- Barros-Martins, J.; Hammerschmidt, S.I.; Cossmann, A.; Odak, I.; Stankov, M.V.; Morillas Ramos, G.; Dopfer-Jablonka, A.; Heidemann, A.; Ritter, C.; Friedrichsen, M.; et al. Immune responses against SARS-CoV-2 variants after heterologous and homologous ChAdOx1 nCoV-19/BNT162b2 vaccination. Nat. Med. 2021, 27, 1525–1529. [Google Scholar] [CrossRef] [PubMed]
- Fuchs, E.J.; Matzinger, P. B cells turn off virgin but not memory T cells. Science 1992, 258, 1156–1159. [Google Scholar] [CrossRef] [Green Version]
- Brandes, M.; Willimann, K.; Moser, B. Professional antigen-presentation function by human gammadelta T Cells. Science 2005, 309, 264–268. [Google Scholar] [CrossRef]
- Schwarz, K.; Meijerink, E.; Speiser, D.E.; Tissot, A.C.; Cielens, I.; Renhof, R.; Dishlers, A.; Pumpens, P.; Bachmann, M.F. Efficient homologous prime-boost strategies for T cell vaccination based on virus-like particles. Eur. J. Immunol. 2005, 35, 816–821. [Google Scholar] [CrossRef]
- MacKerracher, A.; Sommershof, A.; Groettrup, M. PLGA particle vaccination elicits resident memory CD8 T cells protecting from tumors and infection. Eur. J. Pharm. Sci. 2022, 175, 106209. [Google Scholar] [CrossRef]
- Çuburu, N.; Khan, S.; Thompson, C.D.; Kim, R.; Vellinga, J.; Zahn, R.; Lowy, D.R.; Scheper, G.; Schiller, J.T. Adenovirus vector-based prime-boost vaccination via heterologous routes induces cervicovaginal CD8+ T cell responses against HPV16 oncoproteins. Int. J. Cancer 2018, 142, 1467–1479. [Google Scholar] [CrossRef] [Green Version]
- Olsen, T.M.; Stone, B.C.; Chuenchob, V.; Murphy, S.C. Prime-and-Trap Malaria Vaccination To Generate Protective CD8+ Liver-Resident Memory T Cells. J. Immunol. 2018, 201, 1984–1993. [Google Scholar] [CrossRef] [Green Version]
- Waltz, E. China and India approve nasal COVID vaccines—Are they a game changer. Nature 2022, 609, 450. [Google Scholar] [CrossRef]
- Zhang, L.; Wang, W.; Wang, S. Effect of vaccine administration modality on immunogenicity and efficacy. Expert Rev. Vaccines 2015, 14, 1509–1523. [Google Scholar] [CrossRef] [Green Version]
- Cromer, D.; Steain, M.; Reynaldi, A.; Schlub, T.E.; Wheatley, A.K.; Juno, J.A.; Kent, S.J.; Triccas, J.A.; Khoury, D.S.; Davenport, M.P. Neutralising antibody titres as predictors of protection against SARS-CoV-2 variants and the impact of boosting: A meta-analysis. Lancet Microbe 2022, 3, e52–e61. [Google Scholar] [CrossRef]
- Liu, J.; Chandrashekar, A.; Sellers, D.; Barrett, J.; Jacob-Dolan, C.; Lifton, M.; McMahan, K.; Sciacca, M.; VanWyk, H.; Wu, C.; et al. Vaccines elicit highly conserved cellular immunity to SARS-CoV-2 Omicron. Nature 2022, 603, 493–496. [Google Scholar] [CrossRef] [PubMed]
- Crotty, S. Hybrid immunity. Science 2021, 372, 1392–1393. [Google Scholar] [CrossRef]
- Goldberg, Y.; Mandel, M.; Bar-On, Y.M.; Bodenheimer, O.; Freedman, L.S.; Ash, N.; Alroy-Preis, S.; Huppert, A.; Milo, R. Protection and Waning of Natural and Hybrid Immunity to SARS-CoV-2. N. Engl. J. Med. 2022, 386, 2201–2212. [Google Scholar] [CrossRef]
- Lim, J.M.E.; Tan, A.T.; Le Bert, N.; Hang, S.K.; Low, J.G.H.; Bertoletti, A. SARS-CoV-2 breakthrough infection in vaccinees induces virus-specific nasal-resident CD8+ and CD4+ T cells of broad specificity. J. Exp. Med. 2022, 219, e20220780. [Google Scholar] [CrossRef]
- Rollier, C.S.; Hill, A.V.; Reyes-Sandoval, A. Influence of adenovirus and MVA vaccines on the breadth and hierarchy of T cell responses. Vaccine 2016, 34, 4470–4474. [Google Scholar] [CrossRef]
- Atmar, R.L.; Lyke, K.E.; Deming, M.E.; Jackson, L.A.; Branche, A.R.; El Sahly, H.M.; Rostad, C.A.; Martin, J.M.; Johnston, C.; Rupp, R.E.; et al. Homologous and Heterologous Covid-19 Booster Vaccinations. N. Engl. J. Med. 2022, 386, 1046–1057. [Google Scholar] [CrossRef]
- Munro, A.P.S.; Janani, L.; Cornelius, V.; Aley, P.K.; Babbage, G.; Baxter, D.; Bula, M.; Cathie, K.; Chatterjee, K.; Dodd, K.; et al. Safety and immunogenicity of seven COVID-19 vaccines as a third dose (booster) following two doses of ChAdOx1 nCov-19 or BNT162b2 in the UK (COV-BOOST): A blinded, multicentre, randomised, controlled, phase 2 trial. Lancet 2021, 398, 2258–2276. [Google Scholar] [CrossRef]
- Vesin, B.; Lopez, J.; Noirat, A.; Authié, P.; Fert, I.; Le Chevalier, F.; Moncoq, F.; Nemirov, K.; Blanc, C.; Planchais, C.; et al. An intranasal lentiviral booster reinforces the waning mRNA vaccine-induced SARS-CoV-2 immunity that it targets to lung mucosa. Mol. Ther. 2022, 30, 2984–2997. [Google Scholar] [CrossRef]
- Van Doremalen, N.; Purushotham, J.N.; Schulz, J.E.; Holbrook, M.G.; Bushmaker, T.; Carmody, A.; Port, J.R.; Yinda, C.K.; Okumura, A.; Saturday, G.; et al. Intranasal ChAdOx1 nCoV-19/AZD1222 vaccination reduces viral shedding after SARS-CoV-2 D614G challenge in preclinical models. Sci. Transl. Med. 2021, 13, eabh0755. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues, C.M.C.; Plotkin, S.A. The influence of interval between doses on response to vaccines. Vaccine 2021, 39, 7123–7127. [Google Scholar] [CrossRef] [PubMed]
- Castiglione, F.; Mantile, F.; De Berardinis, P.; Prisco, A. How the interval between prime and boost injection affects the immune response in a computational model of the immune system. Comput. Math. Methods Med. 2012, 2012, 842329. [Google Scholar] [CrossRef] [Green Version]
- Khan, S.H.; Martin, M.D.; Starbeck-Miller, G.R.; Xue, H.H.; Harty, J.T.; Badovinac, V.P. The Timing of Stimulation and IL-2 Signaling Regulate Secondary CD8 T Cell Responses. PLoS Pathog. 2015, 11, e1005199. [Google Scholar] [CrossRef]
- Knudsen, M.L.; Ljungberg, K.; Kakoulidou, M.; Kostic, L.; Hallengärd, D.; García-Arriaza, J.; Merits, A.; Esteban, M.; Liljeström, P. Kinetic and phenotypic analysis of CD8+ T cell responses after priming with alphavirus replicons and homologous or heterologous booster immunizations. J. Virol. 2014, 88, 12438–12451. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thompson, E.A.; Beura, L.K.; Nelson, C.E.; Anderson, K.G.; Vezys, V. Shortened Intervals during Heterologous Boosting Preserve Memory CD8 T Cell Function but Compromise Longevity. J. Immunol. 2016, 196, 3054–3063. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Masopust, D.; Ha, S.J.; Vezys, V.; Ahmed, R. Stimulation history dictates memory CD8 T cell phenotype: Implications for prime-boost vaccination. J. Immunol. 2006, 177, 831–839. Available online: http://0-www-ncbi-nlm-nih-gov.brum.beds.ac.uk/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=16818737 (accessed on 30 September 2022). [CrossRef] [Green Version]
- Prlic, M.; Sacks, J.A.; Bevan, M.J. Dissociating markers of senescence and protective ability in memory T cells. PLoS ONE 2012, 7, e32576. [Google Scholar] [CrossRef] [Green Version]
- Natalini, A.; Simonetti, S.; Favaretto, G.; Lucantonio, L.; Peruzzi, G.; Muñoz-Ruiz, M.; Kelly, G.; Contino, A.M.; Sbrocchi, R.; Battella, S.; et al. Improved memory CD8 T cell response to delayed vaccine boost is associated with a distinct molecular signature. bioRxiv 2022. [Google Scholar] [CrossRef]
- Di Rosa, F. Two Niches in the Bone Marrow: A Hypothesis on Life-long T Cell Memory. Trends Immunol. 2016, 37, 503–512. [Google Scholar] [CrossRef]
- Alvarez-Dominguez, J.R.; Melton, D.A. Cell maturation: Hallmarks, triggers, and manipulation. Cell 2022, 185, 235–249. [Google Scholar] [CrossRef] [PubMed]
- Jung, S.; Jung, J.H.; Noh, J.Y.; Kim, W.J.; Yoon, S.Y.; Jung, J.; Kim, E.S.; Kim, H.B.; Cheong, H.J.; Kim, W.J.; et al. The generation of stem cell-like memory cells early after BNT162b2 vaccination is associated with durability of memory CD8+ T cell responses. Cell Rep. 2022, 40, 111138. [Google Scholar] [CrossRef] [PubMed]
- Payne, R.P.; Longet, S.; Austin, J.A.; Skelly, D.T.; Dejnirattisai, W.; Adele, S.; Meardon, N.; Faustini, S.; Al-Taei, S.; Moore, S.C.; et al. Immunogenicity of standard and extended dosing intervals of BNT162b2 mRNA vaccine. Cell 2021, 184, 5699–5714.e11. [Google Scholar] [CrossRef] [PubMed]
- Shaw, R.H.; Liu, X.; Stuart, A.S.V.; Greenland, M.; Aley, P.K.; Andrews, N.J.; Cameron, J.C.; Charlton, S.; Clutterbuck, E.A.; Collins, A.M.; et al. Effect of priming interval on reactogenicity, peak immunological response, and waning after homologous and heterologous COVID-19 vaccine schedules: Exploratory analyses of Com-COV, a randomised control trial. Lancet Respir. Med. 2022, 10, 1049–1060. [Google Scholar] [CrossRef]
- Parry, H.; Bruton, R.; Stephens, C.; Bentley, C.; Brown, K.; Amirthalingam, G.; Hallis, B.; Otter, A.; Zuo, J.; Moss, P. Extended interval BNT162b2 vaccination enhances peak antibody generation. NPJ Vaccines 2022, 7, 14. [Google Scholar] [CrossRef]
- Sidney, J.; Peters, B.; Sette, A. Epitope prediction and identification- adaptive T cell responses in humans. Semin. Immunol. 2020, 50, 101418. [Google Scholar] [CrossRef]
- Tarke, A.; Sidney, J.; Kidd, C.K.; Dan, J.M.; Ramirez, S.I.; Yu, E.D.; Mateus, J.; da Silva Antunes, R.; Moore, E.; Rubiro, P.; et al. Comprehensive analysis of T cell immunodominance and immunoprevalence of SARS-CoV-2 epitopes in COVID-19 cases. Cell Rep. Med. 2021, 2, 100204. [Google Scholar] [CrossRef]
- Schwarz, M.; Torre, D.; Lozano-Ojalvo, D.; Tan, A.T.; Tabaglio, T.; Mzoughi, S.; Sanchez-Tarjuelo, R.; Le Bert, N.; Lim, J.M.E.; Hatem, S.; et al. Rapid, scalable assessment of SARS-CoV-2 cellular immunity by whole-blood PCR. Nat. Biotechnol. 2022, 40, 1680–1689. [Google Scholar] [CrossRef]
- Castro, J.T.; Azevedo, P.; Fumagalli, M.J.; Hojo-Souza, N.S.; Salazar, N.; Almeida, G.G.; Oliveira, L.I.; Faustino, L.; Antonelli, L.R.; Marçal, T.G.; et al. Promotion of neutralizing antibody-independent immunity to wild-type and SARS-CoV-2 variants of concern using an RBD-Nucleocapsid fusion protein. Nat. Commun. 2022, 13, 4831. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| T Cell IFN-γ ELISPOT | Anti-Spike IgG | |||
|---|---|---|---|---|
| Short Interval | Long Interval | Short Interval | Long Interval | |
| ChAd/ChAd | ++ | + | + | ++ |
| BNT/BNT | +++ | ++ | ++++ | +++++ |
| ChAd/BNT | +++++ | ++++ | ++++ | ++++ |
| BNT/ChAd | ++++ | + | +++ | ++++ |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Natalini, A.; Simonetti, S.; Sher, C.; D’Oro, U.; Hayday, A.C.; Di Rosa, F. Durable CD8 T Cell Memory against SARS-CoV-2 by Prime/Boost and Multi-Dose Vaccination: Considerations on Inter-Dose Time Intervals. Int. J. Mol. Sci. 2022, 23, 14367. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms232214367
Natalini A, Simonetti S, Sher C, D’Oro U, Hayday AC, Di Rosa F. Durable CD8 T Cell Memory against SARS-CoV-2 by Prime/Boost and Multi-Dose Vaccination: Considerations on Inter-Dose Time Intervals. International Journal of Molecular Sciences. 2022; 23(22):14367. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms232214367
Chicago/Turabian StyleNatalini, Ambra, Sonia Simonetti, Carmel Sher, Ugo D’Oro, Adrian C. Hayday, and Francesca Di Rosa. 2022. "Durable CD8 T Cell Memory against SARS-CoV-2 by Prime/Boost and Multi-Dose Vaccination: Considerations on Inter-Dose Time Intervals" International Journal of Molecular Sciences 23, no. 22: 14367. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms232214367