
Differential Immunomodulatory Effects of Head and Neck Cancer-Derived Exosomes on B Cells in the Presence of ATP

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Isolation of Tumor-Derived Exosomes (TEX) and Regulatory B Cells (Bregs) from HNSCC
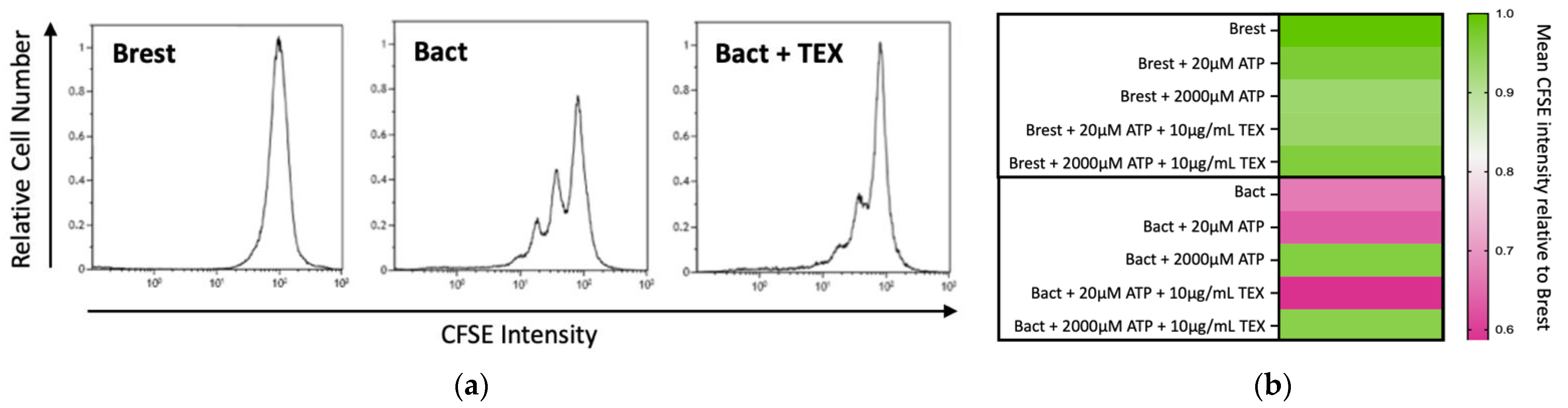
2.2. Effects of TEX and ATP on B-Cell Proliferation
2.2.1. Immunosuppressive Effects of TEX on B-Cell Proliferation
2.2.2. Dose-Dependent Immunomodulatory Effects of ATP and TEX on B Cells
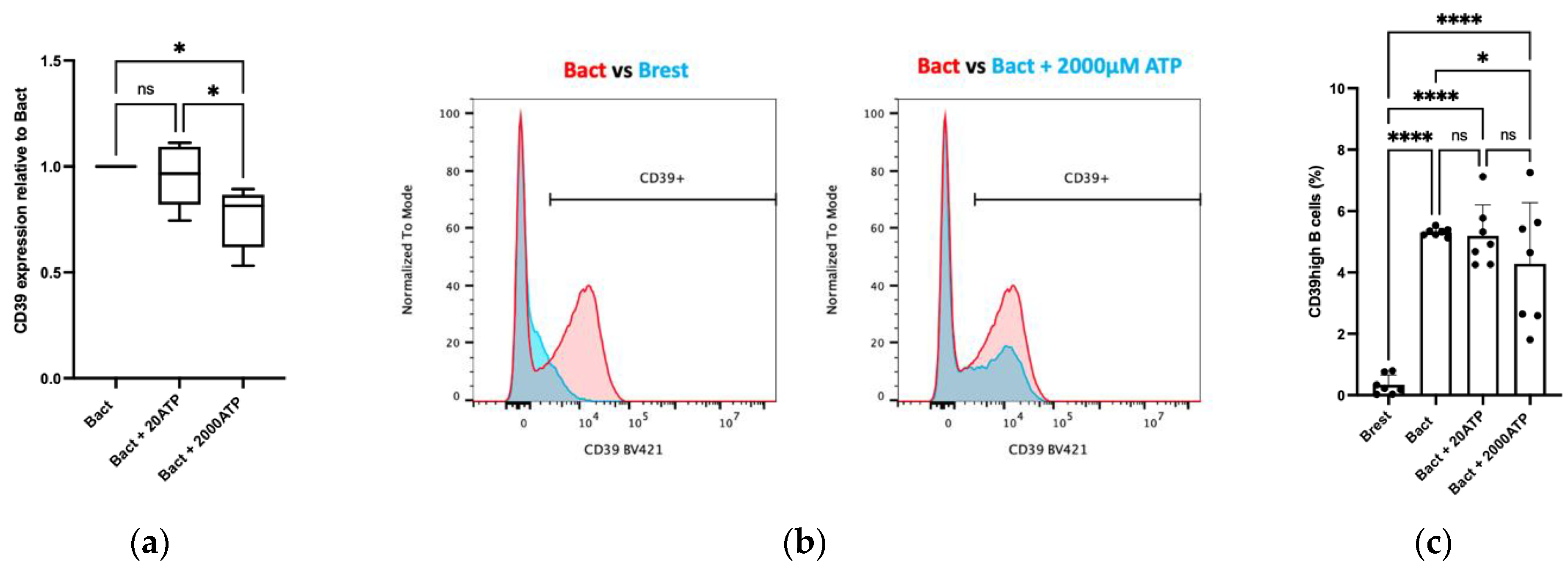
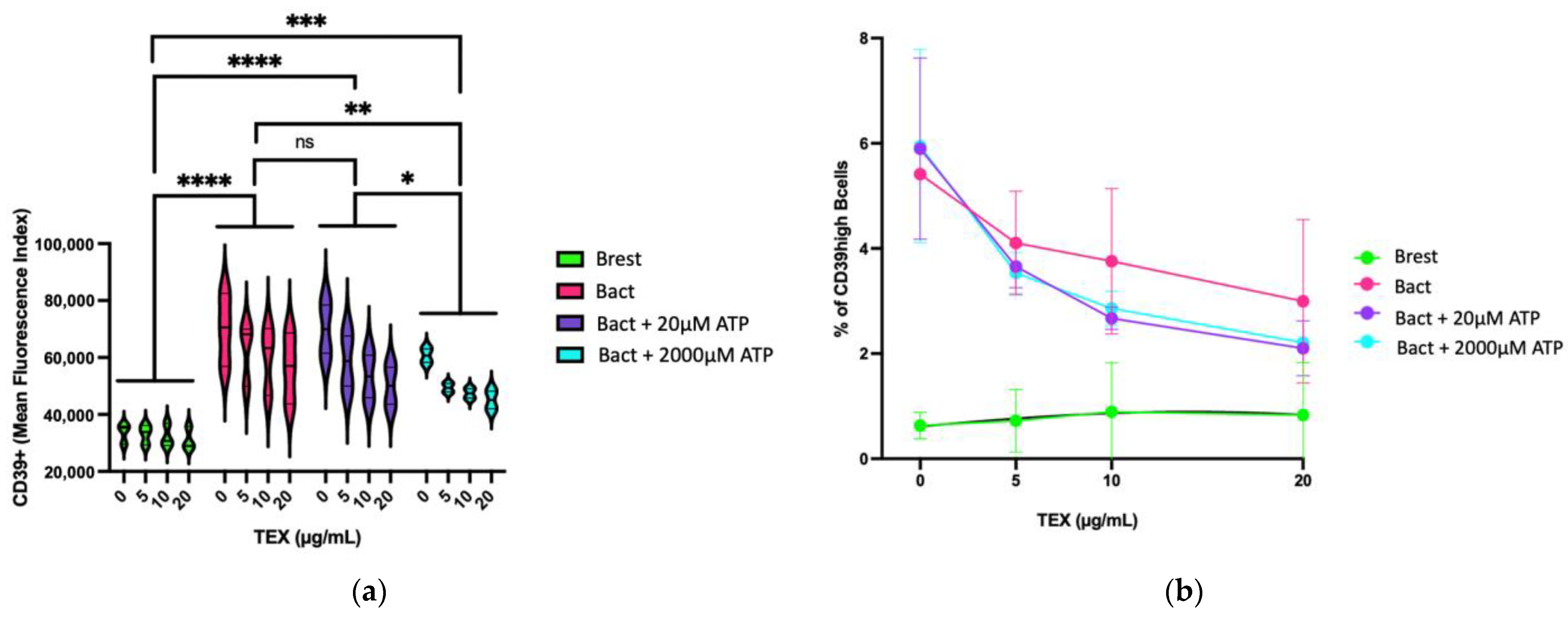
2.3. Effects of ATP and TEX on CD39 Expression on B Cells
2.3.1. Dose-Dependent Effects of ATP on CD39 Expression on B Cells
2.3.2. Dose-Dependent Effect of TEX on CD39 Expression on B Cells in Presence of ATP
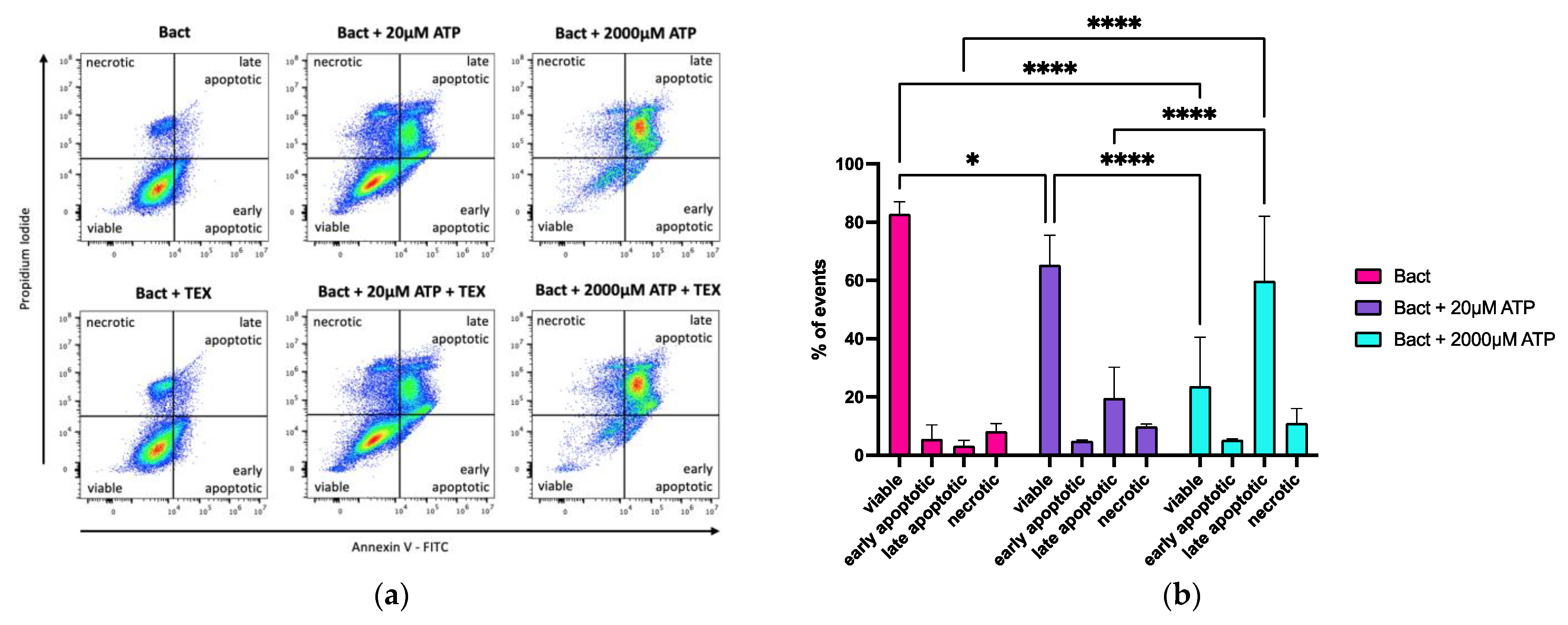
2.4. Effects of ATP and TEX on Cell Death in B cells
2.4.1. Effects of ATP on Annexin V/Propidium Iodide (PI) Positivity in Activated B Cells
2.4.2. Effects of ATP and TEX on Annexin V/Propidium Iodide (PI) Positivity in Activated B Cells
2.5. Effects of ATP and TEX on Apoptosis-Associated Proteins in B Cells
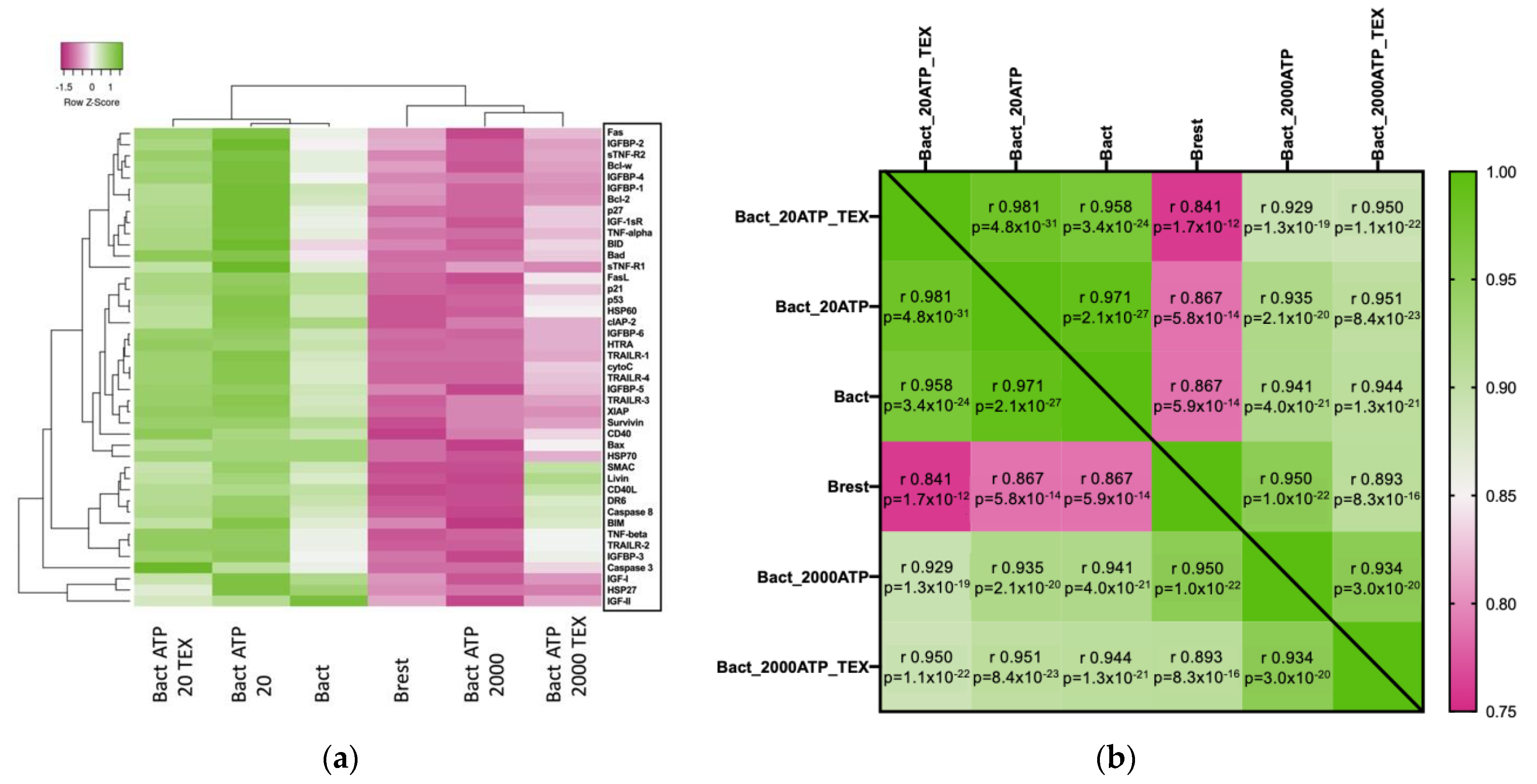
2.5.1. Overall Effects of ATP and TEX on Apoptosis-Associated Proteins in Activated B Cells
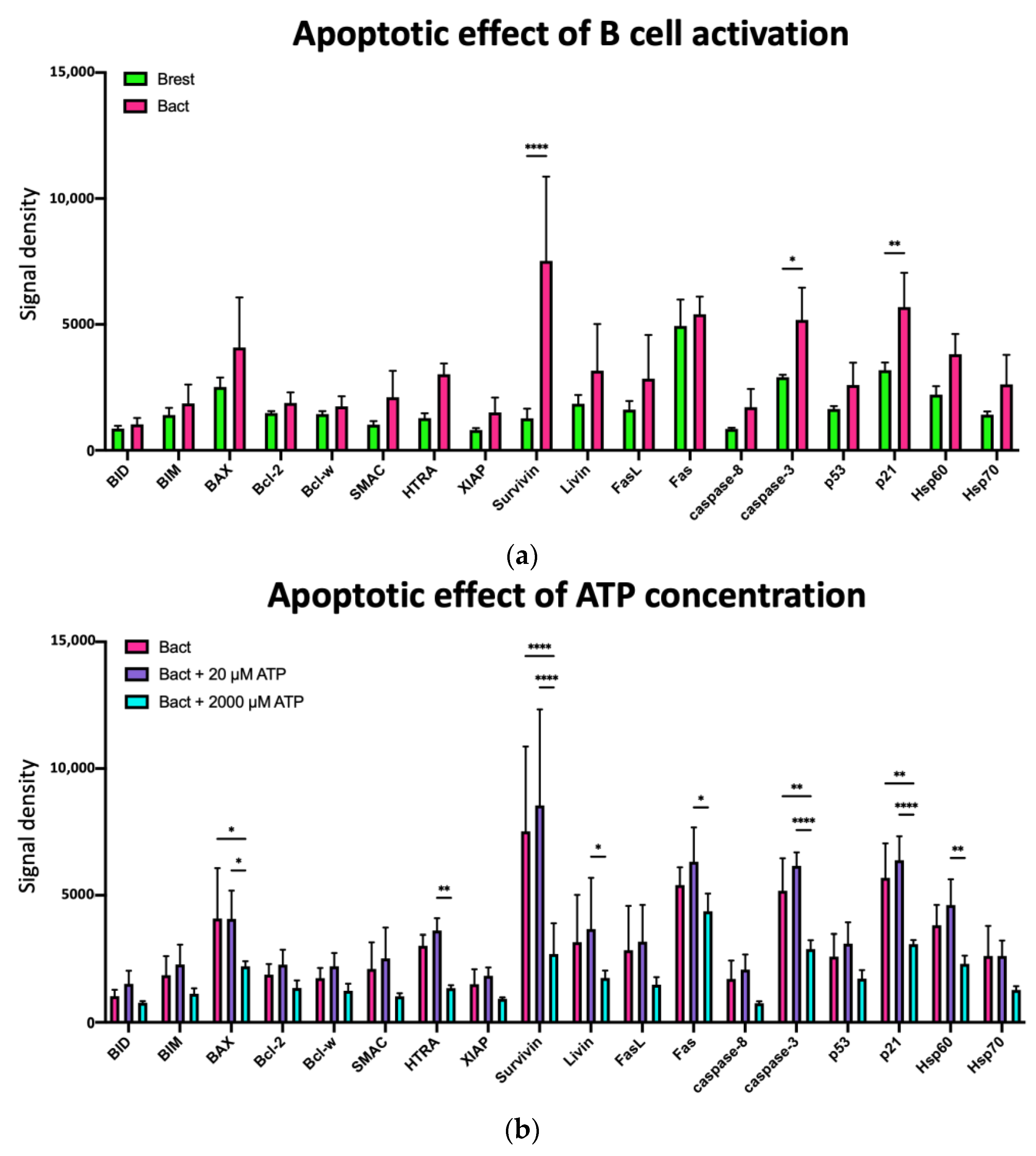
2.5.2. Effects of ATP and TEX on Pro- and Anti-Apoptotic Proteins in Activated B Cells
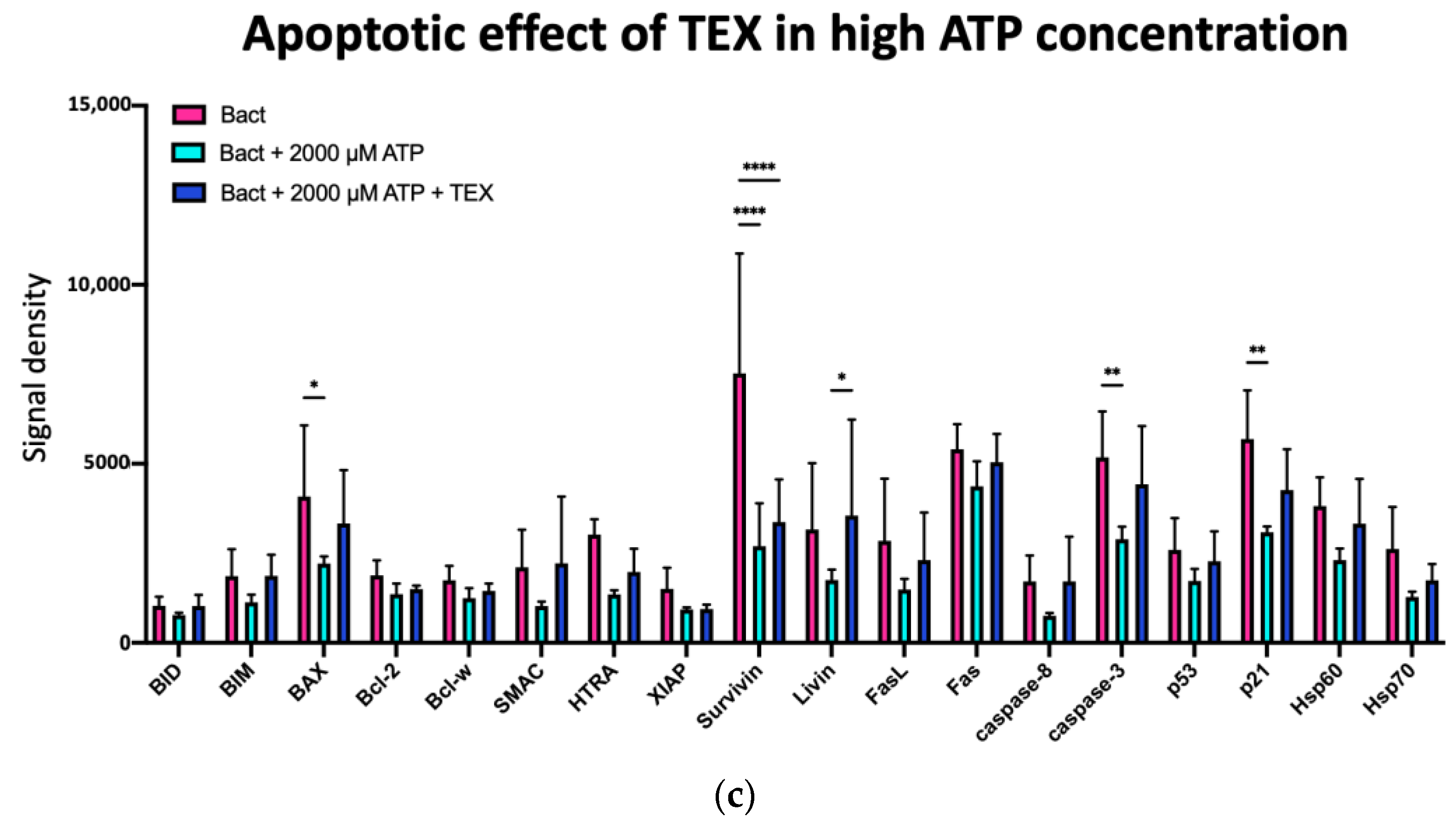
2.5.3. Effects of ATP and TEX on Specific Apoptosis-Associated Proteins in Activated B Cells
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. Isolation of Exosomes
4.3. Quantification and Characterization of Isolated Exosomes
4.4. Peripheral Blood Mononuclear Cells (PBMCs) and Isolation of Peripheral Blood B-Cells
4.5. CFSE-Based Proliferation Assay and Co-Incubation Assays
4.6. Co-Incubation Assays
4.7. Cell Surface Antigen Staining for Flow Cytometry
4.8. Annexin V/Propidium Iodide (PI) Staining Assay
4.9. Apoptotic Protein Profiling
4.10. Western Blotting
4.11. Isolation of Regulatory B Cells (Bregs) from HNSCC Biopsy Samples
4.12. Ethical Approval
4.13. Statistics
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Jemal, A.; Bray, F.; Center, M.M.; Ferlay, J.; Ward, E.; Forman, D. Global cancer statistics. CA Cancer J. Clin. 2011, 61, 69–90. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferris, R.L.; Blumenschein, G., Jr.; Fayette, J.; Guigay, J.; Colevas, A.D.; Licitra, L.; Harrington, K.; Kasper, S.; Vokes, E.E.; Even, C.; et al. Nivolumab for Recurrent Squamous-Cell Carcinoma of the Head and Neck. N. Engl. J. Med. 2016, 375, 1856–1867. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Irani, S. miRNAs Signature in Head and Neck Squamous Cell Carcinoma Metastasis: A Literature Review. J. Dent. 2016, 17, 71–83. [Google Scholar]
- Luci, G.; Intorre, L.; Ferruzzi, G.; Mani, D.; Giuliotti, L.; Pretti, C.; Tognetti, R.; Bertini, S.; Meucci, V. Determination of ochratoxin A in tissues of wild boar (Sus scrofa L.) by enzymatic digestion (ED) coupled to high-performance liquid chromatography with a fluorescence detector (HPLC-FLD). Mycotoxin Res. 2018, 34, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Meucci, S.; Keilholz, U.; Tinhofer, I.; Ebner, O.A. Mutational load and mutational patterns in relation to age in head and neck cancer. Oncotarget 2016, 7, 69188–69199. [Google Scholar] [CrossRef] [Green Version]
- Kim, R.; Emi, M.; Tanabe, K. Cancer immunoediting from immune surveillance to immune escape. Cancer Immunother. 2007, 121, 9–27. [Google Scholar] [CrossRef]
- Grivennikov, S.I.; Greten, F.R.; Karin, M. Immunity, inflammation, and cancer. Cell 2010, 140, 883–899. [Google Scholar] [CrossRef] [Green Version]
- Gunderson, A.J.; Coussens, L.M. B cells and their mediators as targets for therapy in solid tumors. Exp. Cell Res. 2013, 319, 1644–1649. [Google Scholar] [CrossRef] [Green Version]
- Stagg, J.; Smyth, M.J. Extracellular adenosine triphosphate and adenosine in cancer. Oncogene 2010, 29, 5346–5358. [Google Scholar] [CrossRef] [Green Version]
- Gardani, C.F.F.; Cappellari, A.R.; de Souza, J.B.; da Silva, B.T.; Engroff, P.; Moritz, C.E.J.; Scholl, J.N.; Battastini, A.M.O.; Figueiró, F.; Morrone, F.B. Hydrolysis of ATP, ADP, and AMP is increased in blood plasma of prostate cancer patients. Purinergic Signal. 2019, 15, 95–105. [Google Scholar] [CrossRef]
- Zimmermann, H. Extracellular metabolism of ATP and other nucleotides. Naunyn Schmiedebergs Arch. Pharmacol. 2000, 362, 299–309. [Google Scholar] [CrossRef] [PubMed]
- Sitkovsky, M.; Lukashev, D. Regulation of immune cells by local-tissue oxygen tension: HIF1 alpha and adenosine receptors. Nat. Rev. Immunol. 2005, 5, 712–721. [Google Scholar] [CrossRef] [PubMed]
- Cekic, C.; Linden, J. Purinergic regulation of the immune system. Nat. Rev. Immunol. 2016, 16, 177–192. [Google Scholar] [CrossRef]
- Kepp, O.; Bezu, L.; Yamazaki, T.; Di Virgilio, F.; Smyth, M.J.; Kroemer, G.; Galluzzi, L. ATP and cancer immunosurveillance. EMBO J. 2021, 40, e108130. [Google Scholar] [CrossRef] [PubMed]
- Di Virgilio, F.; Sarti, A.C.; Falzoni, S.; De Marchi, E.; Adinolfi, E. Extracellular ATP and P2 purinergic signalling in the tumour microenvironment. Nat. Rev. Cancer 2018, 18, 601–618. [Google Scholar] [CrossRef] [PubMed]
- Vultaggio-Poma, V.; Sarti, A.C.; Di Virgilio, F. Extracellular ATP: A Feasible Target for Cancer Therapy. Cells 2020, 9, 2496. [Google Scholar] [CrossRef]
- Silva, A.C.; Morsch, A.L.; Zanin, R.F.; Correa, M.C.; Arantes, L.C.; Araujo, M.C.; Morsch, V.M.; Schetinger, M.R. Enzymes that hydrolyze adenine nucleotides in chronic renal failure: Relationship between hemostatic defects and renal failure severity. Biochim. Biophys. Acta 2005, 1741, 282–288. [Google Scholar] [CrossRef] [Green Version]
- Fredholm, B.B.; AP, I.J.; Jacobson, K.A.; Linden, J.; Muller, C.E. International Union of Basic and Clinical Pharmacology. LXXXI. Nomenclature and classification of adenosine receptors—an update. Pharmacol. Rev. 2011, 63, 1–34. [Google Scholar] [CrossRef] [Green Version]
- De Andrade Mello, P.; Coutinho-Silva, R.; Savio, L.E.B. Multifaceted Effects of Extracellular Adenosine Triphosphate and Adenosine in the Tumor-Host Interaction and Therapeutic Perspectives. Front. Immunol. 2017, 8, 1526. [Google Scholar] [CrossRef] [Green Version]
- Kasama, H.; Sakamoto, Y.; Kasamatsu, A.; Okamoto, A.; Koyama, T.; Minakawa, Y.; Ogawara, K.; Yokoe, H.; Shiiba, M.; Tanzawa, H.; et al. Adenosine A2b receptor promotes progression of human oral cancer. BMC Cancer 2015, 15, 563. [Google Scholar] [CrossRef] [Green Version]
- Wilkat, M.; Bast, H.; Drees, R.; Dunser, J.; Mahr, A.; Azoitei, N.; Marienfeld, R.; Frank, F.; Brhel, M.; Ushmorov, A.; et al. Adenosine receptor 2B activity promotes autonomous growth, migration as well as vascularization of head and neck squamous cell carcinoma cells. Int. J. Cancer 2020, 147, 202–217. [Google Scholar] [CrossRef]
- Jeske, S.S.; Brand, M.; Ziebart, A.; Laban, S.; Doescher, J.; Greve, J.; Jackson, E.K.; Hoffmann, T.K.; Brunner, C.; Schuler, P.J. Adenosine-producing regulatory B cells in head and neck cancer. Cancer Immunol. Immunother. 2020, 69, 1205–1216. [Google Scholar] [CrossRef] [Green Version]
- Allard, B.; Longhi, M.S.; Robson, S.C.; Stagg, J. The ectonucleotidases CD39 and CD73: Novel checkpoint inhibitor targets. Immunol. Rev. 2017, 276, 121–144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Borsellino, G.; Kleinewietfeld, M.; Di Mitri, D.; Sternjak, A.; Diamantini, A.; Giometto, R.; Hopner, S.; Centonze, D.; Bernardi, G.; Dell’Acqua, M.L.; et al. Expression of ectonucleotidase CD39 by Foxp3+ Treg cells: Hydrolysis of extracellular ATP and immune suppression. Blood 2007, 110, 1225–1232. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Figueiro, F.; Muller, L.; Funk, S.; Jackson, E.K.; Battastini, A.M.; Whiteside, T.L. Phenotypic and functional characteristics of CD39(high) human regulatory B cells (Breg). Oncoimmunology 2016, 5, e1082703. [Google Scholar] [CrossRef] [Green Version]
- White, N.; Burnstock, G. P2 receptors and cancer. Trends Pharmacol. Sci. 2006, 27, 211–217. [Google Scholar] [CrossRef]
- Schetinger, M.R.; Morsch, V.M.; Bonan, C.D.; Wyse, A.T. NTPDase and 5′-nucleotidase activities in physiological and disease conditions: New perspectives for human health. Biofactors 2007, 31, 77–98. [Google Scholar] [CrossRef]
- Braganhol, E.; Zanin, R.F.; Bernardi, A.; Bergamin, L.S.; Cappellari, A.R.; Campesato, L.F.; Morrone, F.B.; Campos, M.M.; Calixto, J.B.; Edelweiss, M.I.; et al. Overexpression of NTPDase2 in gliomas promotes systemic inflammation and pulmonary injury. Purinergic Signal. 2012, 8, 235–243. [Google Scholar] [CrossRef] [Green Version]
- Burnstock, G.; Di Virgilio, F. Purinergic signalling and cancer. Purinergic Signal. 2013, 9, 491–540. [Google Scholar] [CrossRef]
- Lal, H.; Kumar, L.; Kohli, G.S.; Sharma, A.; Goel, H. Serum enzymes in head and neck cancer. IV: 5-nucleotidase. J. Laryngol. Otol. 1989, 103, 200–202. [Google Scholar] [CrossRef]
- Whiteside, T.L. Exosomes and tumor-mediated immune suppression. J. Clin. Investig. 2016, 126, 1216–1223. [Google Scholar] [CrossRef]
- Whiteside, T.L. Immune modulation of T-cell and NK (natural killer) cell activities by TEXs (tumour-derived exosomes). Biochem. Soc. Trans. 2013, 41, 245–251. [Google Scholar] [CrossRef]
- Thery, C.; Zitvogel, L.; Amigorena, S. Exosomes: Composition, biogenesis and function. Nat. Rev. Immunol. 2002, 2, 569–579. [Google Scholar] [CrossRef]
- Raposo, G.; Stoorvogel, W. Extracellular vesicles: Exosomes, microvesicles, and friends. J. Cell Biol. 2013, 200, 373–383. [Google Scholar] [CrossRef] [Green Version]
- Whiteside, T.L. Tumor-Derived Exosomes and Their Role in Cancer Progression. Adv Clin Chem 2016, 74, 103–141. [Google Scholar] [CrossRef] [Green Version]
- Clayton, A.; Al-Taei, S.; Webber, J.; Mason, M.D.; Tabi, Z. Cancer exosomes express CD39 and CD73, which suppress T cells through adenosine production. J. Immunol. 2011, 187, 676–683. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Gu, Y.; Cao, X. The exosomes in tumor immunity. Oncoimmunology 2015, 4, e1027472. [Google Scholar] [CrossRef] [Green Version]
- Whiteside, T.L. The effect of tumor-derived exosomes on immune regulation and cancer immunotherapy. Future Oncol. 2017, 13, 2583–2592. [Google Scholar] [CrossRef]
- Atay, S.; Godwin, A.K. Tumor-derived exosomes: A message delivery system for tumor progression. Commun. Integr. Biol. 2014, 7, e28231. [Google Scholar] [CrossRef]
- Zech, D.; Rana, S.; Buchler, M.W.; Zoller, M. Tumor-exosomes and leukocyte activation: An ambivalent crosstalk. Cell Commun. Signal 2012, 10, 37. [Google Scholar] [CrossRef] [Green Version]
- Xu, Z.; Zeng, S.; Gong, Z.; Yan, Y. Exosome-based immunotherapy: A promising approach for cancer treatment. Mol. Cancer 2020, 19, 160. [Google Scholar] [CrossRef]
- Whiteside, T.L. The role of regulatory T cells in cancer immunology. Immunotargets Ther. 2015, 4, 159–171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beccard, I.J.; Hofmann, L.; Schroeder, J.C.; Ludwig, S.; Laban, S.; Brunner, C.; Lotfi, R.; Hoffmann, T.K.; Jackson, E.K.; Schuler, P.J.; et al. Immune Suppressive Effects of Plasma-Derived Exosome Populations in Head and Neck Cancer. Cancers 2020, 12, 1997. [Google Scholar] [CrossRef]
- Lykken, J.M.; Candando, K.M.; Tedder, T.F. Regulatory B10 cell development and function. Int. Immunol. 2015, 27, 471–477. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosser, E.C.; Mauri, C. Regulatory B cells: Origin, phenotype, and function. Immunity 2015, 42, 607–612. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lechner, A.; Schlosser, H.A.; Thelen, M.; Wennhold, K.; Rothschild, S.I.; Gilles, R.; Quaas, A.; Siefer, O.G.; Huebbers, C.U.; Cukuroglu, E.; et al. Tumor-associated B cells and humoral immune response in head and neck squamous cell carcinoma. Oncoimmunology 2019, 8, 1535293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saze, Z.; Schuler, P.J.; Hong, C.S.; Cheng, D.; Jackson, E.K.; Whiteside, T.L. Adenosine production by human B cells and B cell-mediated suppression of activated T cells. Blood 2013, 122, 9–18. [Google Scholar] [CrossRef] [Green Version]
- Schroeder, J.C.; Puntigam, L.; Hofmann, L.; Jeske, S.S.; Beccard, I.J.; Doescher, J.; Laban, S.; Hoffmann, T.K.; Brunner, C.; Theodoraki, M.N.; et al. Circulating Exosomes Inhibit B Cell Proliferation and Activity. Cancers 2020, 12, 2110. [Google Scholar] [CrossRef]
- Hasan, M.M.; Thompson-Snipes, L.; Klintmalm, G.; Demetris, A.J.; O’Leary, J.; Oh, S.; Joo, H. CD24(hi)CD38(hi) and CD24(hi)CD27(+) Human Regulatory B Cells Display Common and Distinct Functional Characteristics. J. Immunol. 2019, 203, 2110–2120. [Google Scholar] [CrossRef]
- Blair, P.A.; Norena, L.Y.; Flores-Borja, F.; Rawlings, D.J.; Isenberg, D.A.; Ehrenstein, M.R.; Mauri, C. CD19(+)CD24(hi)CD38(hi) B cells exhibit regulatory capacity in healthy individuals but are functionally impaired in systemic Lupus Erythematosus patients. Immunity 2010, 32, 129–140. [Google Scholar] [CrossRef] [Green Version]
- Adinolfi, E.; Giuliani, A.L.; De Marchi, E.; Pegoraro, A.; Orioli, E.; Di Virgilio, F. The P2X7 receptor: A main player in inflammation. Biochem. Pharmacol. 2018, 151, 234–244. [Google Scholar] [CrossRef] [PubMed]
- Pupovac, A.; Geraghty, N.J.; Watson, D.; Sluyter, R. Activation of the P2X7 receptor induces the rapid shedding of CD23 from human and murine B cells. Immunol. Cell Biol. 2015, 93, 77–85. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Babicki, S.; Arndt, D.; Marcu, A.; Liang, Y.; Grant, J.R.; Maciejewski, A.; Wishart, D.S. Heatmapper: Web-enabled heat mapping for all. Nucleic Acids Res. 2016, 44, W147–W153. [Google Scholar] [CrossRef] [PubMed]
- Ichim, G.; Tait, S.W. A fate worse than death: Apoptosis as an oncogenic process. Nat. Rev. Cancer 2016, 16, 539–548. [Google Scholar] [CrossRef] [Green Version]
- Haupt, S.; Berger, M.; Goldberg, Z.; Haupt, Y. Apoptosis—the p53 network. J. Cell Sci. 2003, 116, 4077–4085. [Google Scholar] [CrossRef] [Green Version]
- Lanneau, D.; Brunet, M.; Frisan, E.; Solary, E.; Fontenay, M.; Garrido, C. Heat shock proteins: Essential proteins for apoptosis regulation. J. Cell Mol. Med. 2008, 12, 743–761. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghosh, J.C.; Dohi, T.; Kang, B.H.; Altieri, D.C. Hsp60 regulation of tumor cell apoptosis. J. Biol. Chem. 2008, 283, 5188–5194. [Google Scholar] [CrossRef] [Green Version]
- Kao, T.Y.; Chiu, Y.C.; Fang, W.C.; Cheng, C.W.; Kuo, C.Y.; Juan, H.F.; Wu, S.H.; Lee, A.Y. Mitochondrial Lon regulates apoptosis through the association with Hsp60-mtHsp70 complex. Cell Death Dis. 2015, 6, e1642. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hofmann, L.; Ludwig, S.; Vahl, J.M.; Brunner, C.; Hoffmann, T.K.; Theodoraki, M.N. The Emerging Role of Exosomes in Diagnosis, Prognosis, and Therapy in Head and Neck Cancer. Int. J. Mol. Sci. 2020, 21, 4072. [Google Scholar] [CrossRef]
- Brunner, M.; Maier, K.; Rummele, P.; Jacobsen, A.; Merkel, S.; Benard, A.; Krautz, C.; Kersting, S.; Grutzmann, R.; Weber, G.F. Upregulation of CD20 Positive B-Cells and B-Cell Aggregates in the Tumor Infiltration Zone is Associated with Better Survival of Patients with Pancreatic Ductal Adenocarcinoma. Int. J. Mol. Sci. 2020, 21, 1779. [Google Scholar] [CrossRef] [Green Version]
- Savio, L.E.B.; de Andrade Mello, P.; da Silva, C.G.; Coutinho-Silva, R. The P2X7 Receptor in Inflammatory Diseases: Angel or Demon? Front. Pharmacol. 2018, 9, 52. [Google Scholar] [CrossRef] [PubMed]
- Gu, B.J.; Zhang, W.Y.; Bendall, L.J.; Chessell, I.P.; Buell, G.N.; Wiley, J.S. Expression of P2X(7) purinoceptors on human lymphocytes and monocytes: Evidence for nonfunctional P2X(7) receptors. Am. J. Physiol. Cell Physiol. 2000, 279, C1189–C1197. [Google Scholar] [CrossRef] [PubMed]
- Bian, S.; Sun, X.; Bai, A.; Zhang, C.; Li, L.; Enjyoji, K.; Junger, W.G.; Robson, S.C.; Wu, Y. P2X7 integrates PI3K/AKT and AMPK-PRAS40-mTOR signaling pathways to mediate tumor cell death. PLoS ONE 2013, 8, e60184. [Google Scholar] [CrossRef] [PubMed]
- Muller, L.; Hong, C.S.; Stolz, D.B.; Watkins, S.C.; Whiteside, T.L. Isolation of biologically-active exosomes from human plasma. J. Immunol. Methods 2014, 411, 55–65. [Google Scholar] [CrossRef] [Green Version]
- Ludwig, N.; Azambuja, J.H.; Rao, A.; Gillespie, D.G.; Jackson, E.K.; Whiteside, T.L. Adenosine receptors regulate exosome production. Purinergic Signal. 2020, 16, 231–240. [Google Scholar] [CrossRef]
- Bidula, S.; Dhuna, K.; Helliwell, R.; Stokes, L. Positive allosteric modulation of P2X7 promotes apoptotic cell death over lytic cell death responses in macrophages. Cell Death Dis. 2019, 10, 882. [Google Scholar] [CrossRef] [Green Version]
- Bradbury, D.A.; Simmons, T.D.; Slater, K.J.; Crouch, S.P. Measurement of the ADP:ATP ratio in human leukaemic cell lines can be used as an indicator of cell viability, necrosis and apoptosis. J. Immunol. Methods 2000, 240, 79–92. [Google Scholar] [CrossRef]
- Kasof, G.M.; Gomes, B.C. Livin, a novel inhibitor of apoptosis protein family member. J. Biol. Chem. 2001, 276, 3238–3246. [Google Scholar] [CrossRef] [Green Version]
- Murgia, M.; Pizzo, P.; Steinberg, T.H.; Di Virgilio, F. Characterization of the cytotoxic effect of extracellular ATP in J774 mouse macrophages. Biochem. J. 1992, 288, 897–901. [Google Scholar] [CrossRef] [Green Version]
- Wheatley, S.P.; Altieri, D.C. Survivin at a glance. J. Cell Sci. 2019, 132. [Google Scholar] [CrossRef] [Green Version]
- Aswad, F.; Kawamura, H.; Dennert, G. High sensitivity of CD4+CD25+ regulatory T cells to extracellular metabolites nicotinamide adenine dinucleotide and ATP: A role for P2X7 receptors. J. Immunol. 2005, 175, 3075–3083. [Google Scholar] [CrossRef]
- Hong, C.S.; Sharma, P.; Yerneni, S.S.; Simms, P.; Jackson, E.K.; Whiteside, T.L.; Boyiadzis, M. Circulating exosomes carrying an immunosuppressive cargo interfere with cellular immunotherapy in acute myeloid leukemia. Sci. Rep. 2017, 7, 14684. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vecchio, E.; Caiazza, C.; Mimmi, S.; Avagliano, A.; Iaccino, E.; Brusco, T.; Nistico, N.; Maisano, D.; Aloisio, A.; Quinto, I.; et al. Metabolites Profiling of Melanoma Interstitial Fluids Reveals Uridine Diphosphate as Potent Immune Modulator Capable of Limiting Tumor Growth. Front. Cell Dev. Biol. 2021, 9, 730726. [Google Scholar] [CrossRef]
- Whiteside, T.L. Induced regulatory T cells in inhibitory microenvironments created by cancer. Expert Opin. Biol. Ther. 2014, 14, 1411–1425. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoffmann, T.K.; Dworacki, G.; Tsukihiro, T.; Meidenbauer, N.; Gooding, W.; Johnson, J.T.; Whiteside, T.L. Spontaneous apoptosis of circulating T lymphocytes in patients with head and neck cancer and its clinical importance. Clin. Cancer Res. 2002, 8, 2553–2562. [Google Scholar]
- Reichert, T.E.; Strauss, L.; Wagner, E.M.; Gooding, W.; Whiteside, T.L. Signaling abnormalities, apoptosis, and reduced proliferation of circulating and tumor-infiltrating lymphocytes in patients with oral carcinoma. Clin. Cancer Res. 2002, 8, 3137–3145. [Google Scholar] [PubMed]
- Verdi, J.; Ketabchi, N.; Noorbakhsh, N.; Saleh, M.; Ebrahimi-Barough, S.; Seyhoun, I.; Kavianpour, M. Development and Clinical Application of Tumor-derived Exosomes in Patients with Cancer. Curr. Stem Cell Res. Ther. 2022, 17, 91–102. [Google Scholar] [CrossRef] [PubMed]
- Kahlert, C.; Kalluri, R. Exosomes in tumor microenvironment influence cancer progression and metastasis. J. Mol. Med. 2013, 91, 431–437. [Google Scholar] [CrossRef] [Green Version]
- Zhao, R.; Chen, X.; Song, H.; Bie, Q.; Zhang, B. Dual Role of MSC-Derived Exosomes in Tumor Development. Stem Cells Int. 2020, 2020, 8844730. [Google Scholar] [CrossRef]
- Chen, W.; Jiang, J.; Xia, W.; Huang, J. Tumor-Related Exosomes Contribute to Tumor-Promoting Microenvironment: An Immunological Perspective. J. Immunol. Res. 2017, 2017, 1073947. [Google Scholar] [CrossRef]
- Boyiadzis, M.; Whiteside, T.L. The emerging roles of tumor-derived exosomes in hematological malignancies. Leukemia 2017, 31, 1259–1268. [Google Scholar] [CrossRef] [PubMed]
- Baig, M.S.; Roy, A.; Rajpoot, S.; Liu, D.; Savai, R.; Banerjee, S.; Kawada, M.; Faisal, S.M.; Saluja, R.; Saqib, U.; et al. Tumor-derived exosomes in the regulation of macrophage polarization. Inflamm. Res. 2020, 69, 435–451. [Google Scholar] [CrossRef] [PubMed]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Coray, M.; Göldi, V.; Schmid, L.; Benecke, L.; Figueiró, F.; Muller, L. Differential Immunomodulatory Effects of Head and Neck Cancer-Derived Exosomes on B Cells in the Presence of ATP. Int. J. Mol. Sci. 2022, 23, 14446. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms232214446
Coray M, Göldi V, Schmid L, Benecke L, Figueiró F, Muller L. Differential Immunomodulatory Effects of Head and Neck Cancer-Derived Exosomes on B Cells in the Presence of ATP. International Journal of Molecular Sciences. 2022; 23(22):14446. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms232214446
Chicago/Turabian StyleCoray, Mali, Veronika Göldi, Lukas Schmid, Laura Benecke, Fabrício Figueiró, and Laurent Muller. 2022. "Differential Immunomodulatory Effects of Head and Neck Cancer-Derived Exosomes on B Cells in the Presence of ATP" International Journal of Molecular Sciences 23, no. 22: 14446. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms232214446