
Role of Thylakoid Lipids in Protochlorophyllide Oxidoreductase Activation: Allosteric Mechanism Elucidated by a Computational Study
Abstract
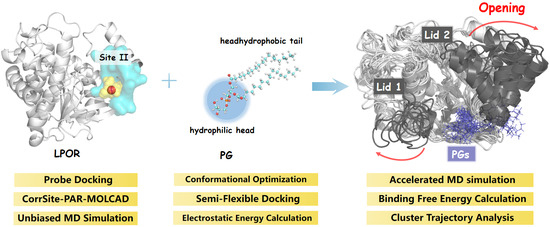
:
1. Introduction
2. Methods
2.1. Structural Preparation
2.2. Exploration of Potential Allosteric Sites
2.3. Molecular Docking
2.4. Molecular Dynamics Simulation
2.4.1. Conventional Molecular Dynamics (cMD) Simulation
2.4.2. Accelerated Molecular Dynamics (aMD) Simulation
2.4.3. Unbiased Molecular Dynamics Simulation
2.5. MM-GBSA Binding Free Energy (BFE) Calculation
2.6. MD Trajectory Analysis
3. Results
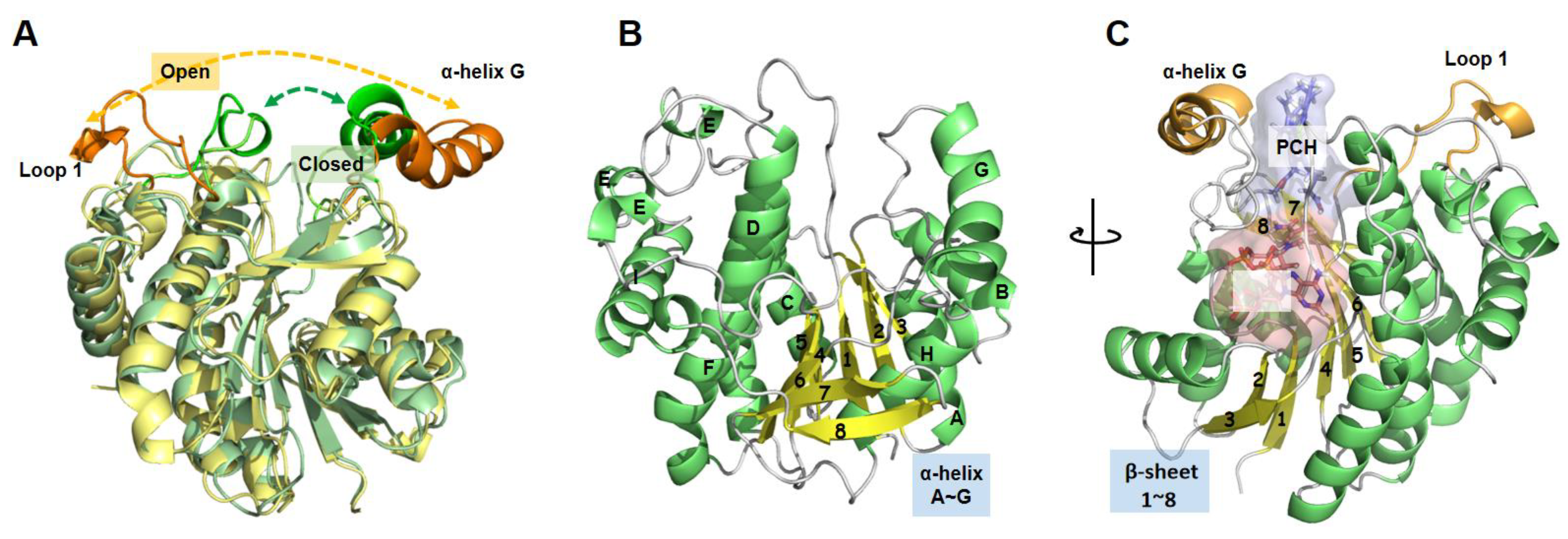
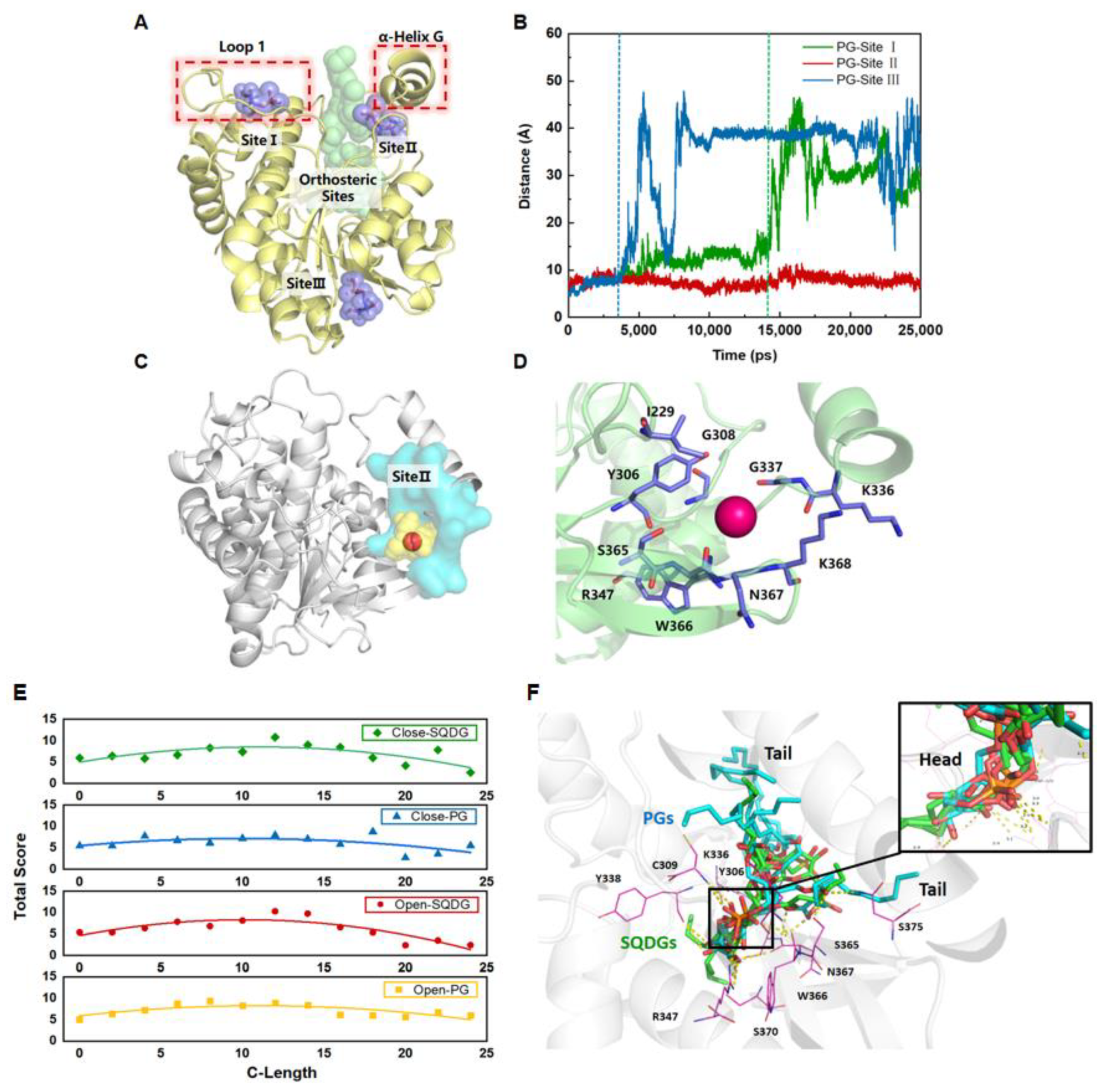
3.1. Exploring the Potential Allosteric Sites in LPOR
3.2. Molecular Docking of PGs and SQDGs to LPOR in Closed and Open States
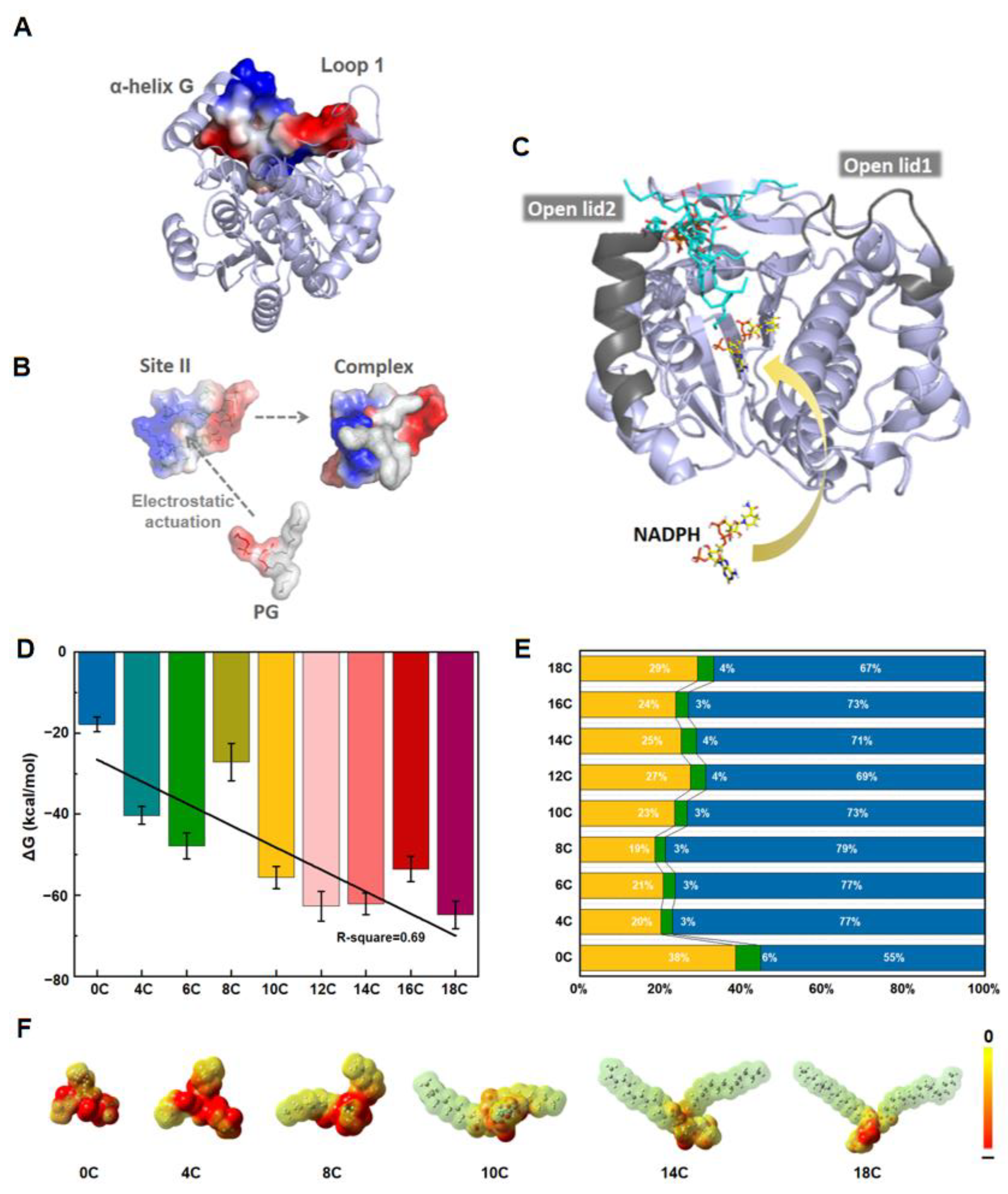
3.3. PGs with LCFA Bind More Favorably at the Site II through Noncovalent Interaction
3.4. The Lid Opening Is Achieved through a Hinge with Salt Bridges
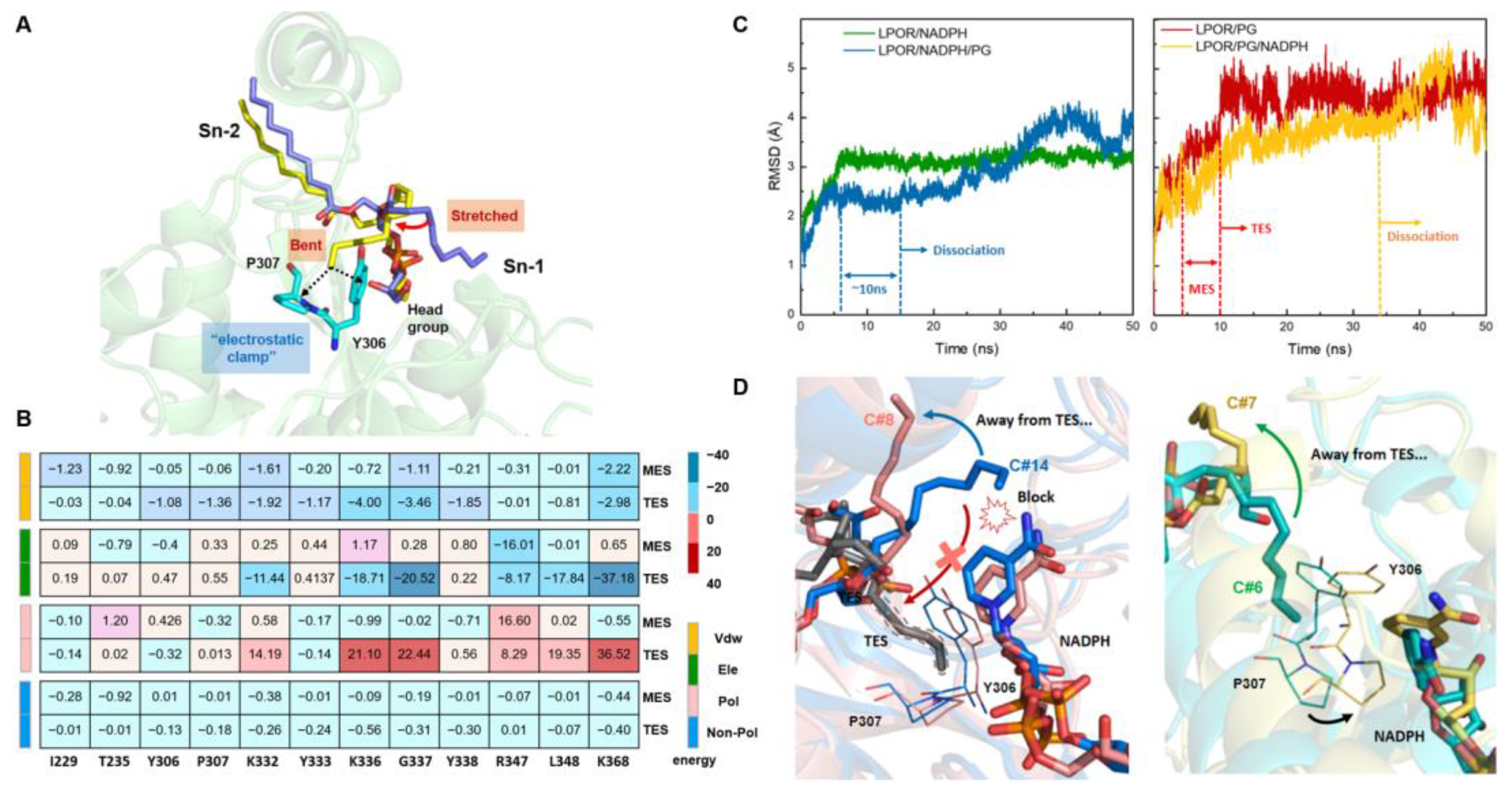
3.5. Dynamic Simulations Reveal Two Modes of Action between PGs and LPOR
3.6. Cofactor Behavior and Bound PG Form a Feedback Loop by Competing for the Electrostatic Clamp
3.7. Comparative Analysis of LPOR/PG Complexes in aMD Simulations with Different Force Fields
4. Conclusion and Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Lichtenthaler, H.K. Chlorophylls and carotenoids: Pigments of photosynthetic biomembranes. Methods Enzymol. 1987, 148, 350–382. [Google Scholar]
- Horton, P.; Ruban, A.V.; Walters, R.G. Regulation of light harvesting in green plants. Annu. Rev. Plant Physiol. Plant Mol. Biol. 1996, 47, 655–684. [Google Scholar] [CrossRef] [PubMed]
- Li, X.-P.; Björkman, O.; Shih, C.; Grossman, A.R.; Rosenquist, M.; Jansson, S.; Niyogi, K.K. A pigment-binding protein essential for regulation of photosynthetic light harvesting. Nature 2000, 403, 391–395. [Google Scholar] [CrossRef]
- Kühlbrandt, W.; Wang, D.N.; Fujiyoshi, Y. Atomic model of plant light-harvesting complex by electron crystallography. Nature 1994, 367, 614–621. [Google Scholar] [CrossRef]
- Escoubas, J.M.; Lomas, M.; LaRoche, J.; Falkowski, P.G. Light intensity regulation of cab gene transcription is signaled by the redox state of the plastoquinone pool. Proc. Natl. Acad. Sci. USA 1995, 92, 10237–10241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Susek, R.E.; Ausubel, F.M.; Chory, J. Signal transduction mutants of arabidopsis uncouple nuclear CAB and RBCS gene expression from chloroplast development. Cell 1993, 74, 787–799. [Google Scholar] [CrossRef] [Green Version]
- Jordan, P.; Fromme, P.; Witt, H.T.; Klukas, O.; Saenger, W.; Krauß, N. Three-dimensional structure of cyanobacterial photosystem I at 2.5 Å resolution. Nature 2001, 411, 909–917. [Google Scholar] [CrossRef] [PubMed]
- Gururani, M.A.; Venkatesh, J.; Tran, L.S.P. Regulation of Photosynthesis during Abiotic Stress-Induced Photoinhibition. Mol. Plant 2015, 8, 1304–1320. [Google Scholar] [CrossRef] [Green Version]
- Meskauskiene, R.; Nater, M.; Goslings, D.; Kessler, F.; Camp, R.O.D.; Apel, K. FLU: A negative regulator of chlorophyll biosynthesis in Arabidopsis thaliana. Proc. Natl. Acad. Sci. USA 2001, 98, 12826–12831. [Google Scholar] [CrossRef] [Green Version]
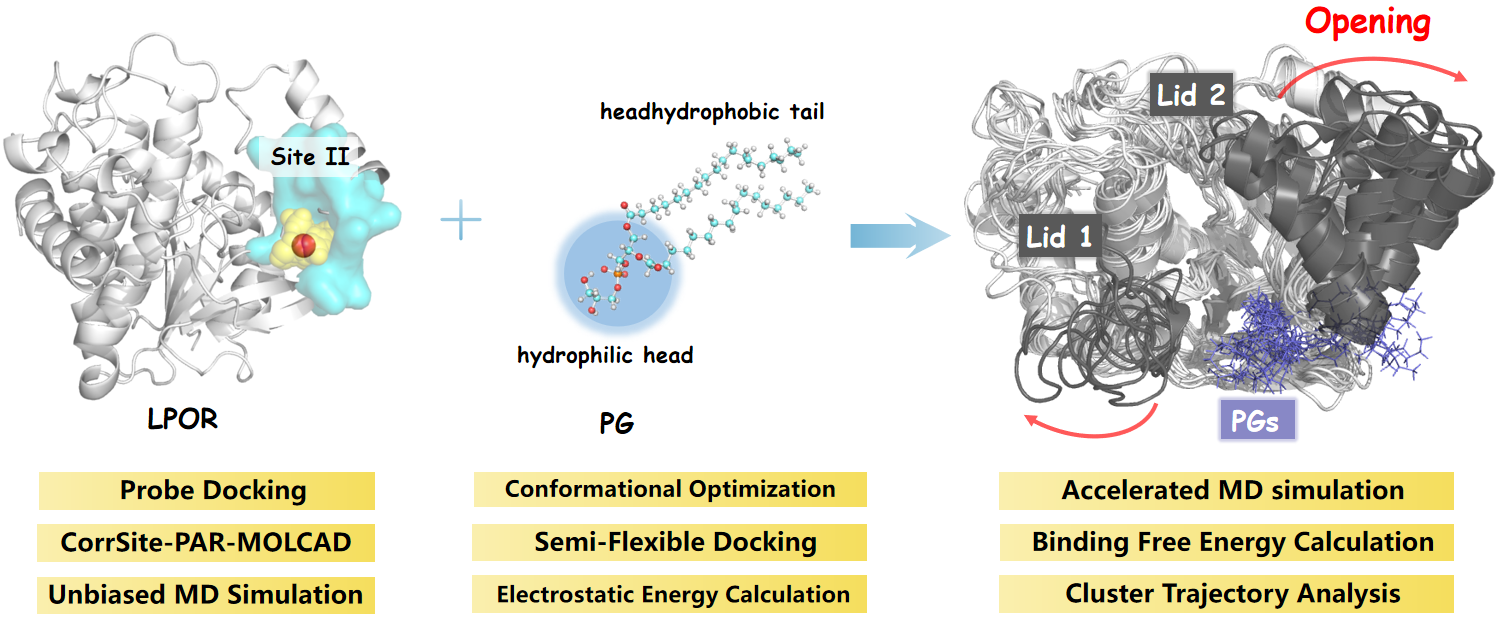
- Gabruk, M.; Mysliwa-Kurdziel, B. Light-Dependent Protochlorophyllide Oxidoreductase: Phylogeny, Regulation, and Catalytic Properties. Biochemistry 2015, 54, 5255–5262. [Google Scholar] [CrossRef]
- Yang, J.; Cheng, Q. Origin and Evolution of the Light-Dependent Protochlorophyllide Oxidoreductase (LPOR) Genes. Plant Biol. 2004, 6, 537–544. [Google Scholar] [CrossRef] [PubMed]
- Scrutton, N.S.; Groot, M.L.; Heyes, D.J. Excited state dynamics and catalytic mechanism of the light-driven enzyme protochlorophyllide oxidoreductase. Phys. Chem. Chem. Phys. 2012, 14, 8818–8824. [Google Scholar] [CrossRef]
- Sanina, N.M.; Goncharova, S.N.; Kostetsky, E.Y. Fatty acid composition of individual polar lipid classes from marine macrophytes. Phytochemistry 2004, 65, 721–730. [Google Scholar] [CrossRef] [PubMed]
- Solymosi, K.; Schoefs, B. Etioplast and etio-chloroplast formation under natural conditions: The dark side of chlorophyll biosynthesis in angiosperms. Photosynth. Res. 2010, 105, 143–166. [Google Scholar] [CrossRef] [PubMed]
- Franck, F.; Sperling, U.; Frick, G.; Pochert, B.; van Cleve, B.; Apel, K.; Armstrong, G.A. Regulation of etioplast pigment-protein complexes, inner membrane architecture, and protochlorophyllide alpha chemical heterogeneity by light-dependent NADPH: Protochlorophyllide oxidoreductases A and B. Plant Physiol. 2000, 124, 1678–1696. [Google Scholar] [CrossRef] [Green Version]
- Grzyb, J.M.; Solymosi, K.; Strzałka, K.; Mysliwa-Kurdziel, B. Visualization and characterization of prolamellar bodies with atomic force microscopy. J. Plant Physiol. 2013, 170, 1217–1227. [Google Scholar] [CrossRef] [PubMed]
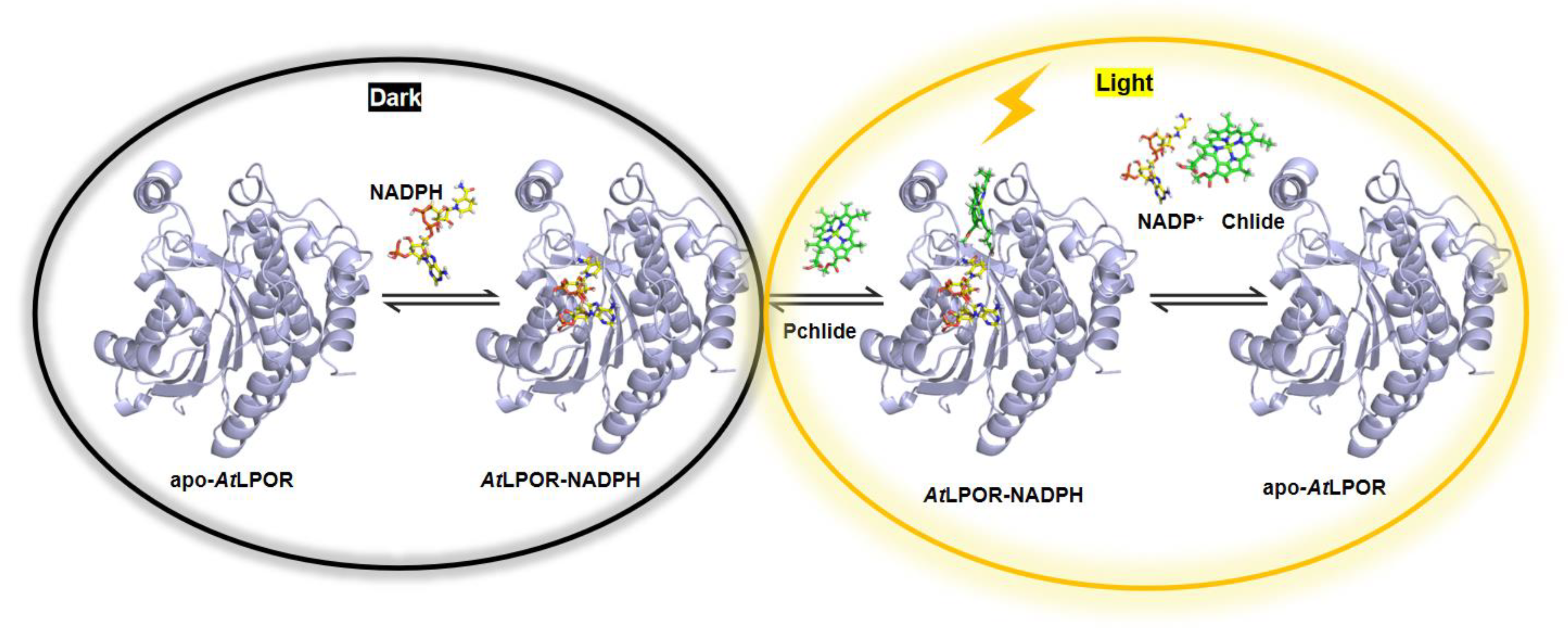
- Kobayashi, K. Role of membrane glycerolipids in photosynthesis, thylakoid biogenesis and chloroplast development. J. Plant Res. 2018, 131, 563. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hölzl, G.; Dörmann, P. Chloroplast Lipids and Their Biosynthesis. Annu. Rev. Plant Biol. 2019, 70, 51–81. [Google Scholar] [CrossRef] [PubMed]
- Demé, B.; Cataye, C.; Block, M.A.; Maréchal, E.; Jouhet, J. Contribution of galactoglycerolipids to the 3-dimensional architecture of thylakoids. FASEB J. 2014, 28, 3373–3383. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kobayashi, K.; Fujii, S.; Sato, M.; Toyooka, K.; Wada, H. Specific role of phosphatidylglycerol and functional overlaps with other thylakoid lipids in Arabidopsis chloroplast biogenesis. Plant Cell Rep. 2014, 34, 631–642. [Google Scholar] [CrossRef]
- Schaller, S.; Latowski, D.; Jemioła-Rzemińska, M.; Dawood, A.; Wilhelm, C.; Strzałka, K.; Goss, R. Regulation of LHCII aggregation by different thylakoid membrane lipids. Biochim. Biophys. Acta 2011, 1807, 326–335. [Google Scholar] [CrossRef] [PubMed]
- Selstam, E.; Schelin, J.; Brain, T.; Williams, W.P. The effects of low pH on the properties of protochlorophyllide oxidoreductase and the organization of prolamellar bodies of maize (Zea mays). JBIC J. Biol. Inorg. Chem. 2002, 269, 2336–2346. [Google Scholar] [CrossRef] [PubMed]
- Selstam, E.; Schelin, J.; Williams, W.P.; Brain, A.P.R. Structural organisation of prolamellar bodies (PLB) isolated from Zea mays. Parallel TEM, SAXS and absorption spectra measurements on samples subjected to freeze-thaw, reduced pH and high-salt perturbation. Biochim. Biophys. Acta Biomembr. 2007, 1768, 2235–2245. [Google Scholar] [CrossRef] [Green Version]
- Di Baccio, D.; Quartacci, M.F.; Vecchia, F.D.; La Rocca, N.; Rascio, N.; Navari-Izzo, F. Bleaching herbicide effects on plastids of dark-grown plants: Lipid composition of etioplasts in amitrole and norflurazon-treated barley leaves. J. Exp. Bot. 2002, 53, 1857–1865. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kopečná, J.; Pilný, J.; Krynická, V.; Tomčala, A.; Kis, M.; Gombos, Z.; Komenda, J.; Sobotka, R. Lack of Phosphatidylglycerol Inhibits Chlorophyll Biosynthesis at Multiple Sites and Limits Chlorophyllide Reutilization in Synechocystis sp. Strain PCC 6803. Plant Physiol. 2015, 169, 1307–1317. [Google Scholar] [CrossRef] [Green Version]
- Schaller-Laudel, S.; Latowski, D.; Jemiola-Rzeminska, M.; Strzalka, K.; Daum, S.; Bacia, K.; Wilhelm, C.; Goss, R. Influence of thylakoid membrane lipids on the structure of aggregated light-harvesting complexes of the diatom Thalassiosira pseudonana and the green alga Mantoniella squamata. Physiol. Plant. 2017, 160, 339–358. [Google Scholar] [CrossRef]
- Gabruk, M.; Mysliwa-Kurdziel, B.; Kruk, J. MGDG, PG and SQDG regulate the activity of light-dependent protochlorophyllide oxidoreductase. Biochem. J. 2017, 474, 1307–1320. [Google Scholar] [CrossRef]
- Nguyen, H.C.; Melo, A.A.; Kruk, J.; Frost, A.; Gabruk, M. Photocatalytic LPOR forms helical lattices that shape membranes for chlorophyll synthesis. Nat. Plants 2021, 7, 437–444. [Google Scholar] [CrossRef]
- Altschul, S. Hot papers-Bioinformatics-Gapped BLAST and PSI-BLAST: A new generation of protein database search programs by S.F. Altschul, T.L. Madden, A.A. Schaffer, J.H. Zhang, Z. Zhang, W. Miller, D.J. Lipman—Comments. Scientist 1999, 13, 15. [Google Scholar]
- Waterhouse, A.; Bertoni, M.; Bienert, S.; Studer, G.; Tauriello, G.; Gumienny, R.; Heer, F.T.; De Beer, T.A.P.; Rempfer, C.; Bordoli, L.; et al. SWISS-MODEL: Homology modelling of protein structures and complexes. Nucleic Acids Res. 2018, 46, W296–W303. [Google Scholar] [CrossRef] [Green Version]
- Laskowski, R.A.; Chistyakov, V.V.; Thornton, J.M. PDBsum more: New summaries and analyses of the known 3D structures of proteins and nucleic acids. Nucleic Acids Res. 2005, 33, D266–D268. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Colovos, C.; Yeates, T.O. Verification of protein structures: Patterns of nonbonded atomic interactions. Protein Sci. 1993, 2, 1511–1519. [Google Scholar] [CrossRef] [PubMed]
- Eisenberg, D.; Luthy, R.; Bowie, J.U. VERIFY3D: Assessment of protein models with three-dimensional profiles. Method Enzym. 1997, 277, 396–404. [Google Scholar]
- Pontius, J.; Richelle, J.; Wodak, S. Deviations from Standard Atomic Volumes as a Quality Measure for Protein Crystal Structures. J. Mol. Biol. 1996, 264, 121–136. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Panjkovich, A.; Daura, X. PARS: A web server for the prediction of Protein Allosteric and Regulatory Sites. Bioinformatics 2014, 30, 1314–1315. [Google Scholar] [CrossRef] [Green Version]
- Panjkovich, A.; Daura, X. Exploiting protein flexibility to predict the location of allosteric sites. BMC Bioinform. 2012, 13, 273. [Google Scholar] [CrossRef] [Green Version]
- Xu, Y.; Wang, S.; Hu, Q.; Gao, S.; Ma, X.; Zhang, W.; Shen, Y.; Chen, F.; Lai, L.; Pei, J. CavityPlus: A web server for protein cavity detection with pharmacophore modelling, allosteric site identification and covalent ligand binding ability prediction. Nucleic Acids Res. 2019, 46, W374–W379. [Google Scholar] [CrossRef]
- Yuan, Y.; Pei, J.; Lai, L. Binding Site Detection and Druggability Prediction of Protein Targets for Structure- Based Drug Design. Curr. Pharm. Des. 2013, 19, 2326–2333. [Google Scholar] [CrossRef]
- Ma, X.; Meng, H.; Lai, L. Motions of Allosteric and Orthosteric Ligand-Binding Sites in Proteins are Highly Correlated. J. Chem. Inf. Model. 2016, 56, 1725–1733. [Google Scholar] [CrossRef]
- Haliloglu, T.; Bahar, I.; Erman, B. Gaussian Dynamics of Folded Proteins. Phys. Rev. Lett. 1997, 79, 3090–3093. [Google Scholar] [CrossRef]
- Zhang, W.; Pei, J.; Lai, L. Statistical Analysis and Prediction of Covalent Ligand Targeted Cysteine Residues. J. Chem. Inf. Model. 2017, 57, 1453–1460. [Google Scholar] [CrossRef] [PubMed]
- Patel, B.; Patel, A.; Patel, A.; Bhatt, H. CoMFA, CoMSIA, molecular docking and MOLCAD studies of pyrimidinone derivatives to design novel and selective tankyrase inhibitors. J. Mol. Struct. 2020, 1221. [Google Scholar] [CrossRef]
- Sybyl Version 7.3; Tripos Inc.: St. Louis, MO, USA, 2006; Available online: www.tripos.com (accessed on 10 March 2008).
- Manak, M.; Jirkovsky, L.; Kolingerova, I. Interactive Analysis of Connolly Surfaces for Various Probes. Comput. Graph. Forum 2016, 36, 160–172. [Google Scholar] [CrossRef]
- Perrot, G.; Cheng, B.; Gibson, K.; Vilá, J.; Palmer, K.; Nayeem, A.; Maigret, B.; Scheraga, H. MSEED: A program for the rapid analytical determination of accessible surface areas and their derivatives. J. Comput. Chem. 1992, 13, 1–11. [Google Scholar] [CrossRef]
- Ohlrogge, J.; Thrower, N.; Mhaske, V.; Stymne, S.; Baxter, M.; Yang, W.; Liu, J.; Shaw, K.; Shorrosh, B.; Zhang, M.; et al. PlantFAdb: A resource for exploring hundreds of plant fatty acid structures synthesized by thousands of plants and their phylogenetic relationships. Plant J. 2018, 96, 1299–1308. [Google Scholar] [CrossRef] [Green Version]
- Jain, A.N. Surflex-Dock 2.1: Robust performance from ligand energetic modeling, ring flexibility, and knowledge-based search. J. Comput. Aided Mol. Des. 2007, 21, 281–306. [Google Scholar] [CrossRef] [Green Version]
- Claußen, H.; Buning, C.; Rarey, M.; Lengauer, T. FlexE: Efficient molecular docking considering protein structure variations. J. Mol. Biol. 2001, 308, 377–395. [Google Scholar] [CrossRef] [Green Version]
- Matthey, T.; Ko, A.; Izaguirre, J.A. PROTOMOL: A molecular dynamics research framework for algorithmic development. Lect. Notes Comput. Sc. 2003, 2659, 50–59. [Google Scholar]
- Wang, J.; Wang, W.; Kollman, P.A.; Case, D.A. Automatic atom type and bond type perception in molecular mechanical calculations. J. Mol. Graph. Model. 2006, 25, 247–260. [Google Scholar] [CrossRef]
- Madej, B.D.; Skjevik, A.; Teigen, K.; Walker, R.C. AMBER-Lipid 11: A new modular lipid force field for molecular dynamics. Abstr. Pap. Am. Chem. S. 2012, 243, 1155. [Google Scholar]
- Dickson, C.J.; Walker, R.C.; Gould, I.R. Lipid21: Complex Lipid Membrane Simulations with AMBER. J. Chem. Theory Comput. 2022, 18, 1726–1736. [Google Scholar] [CrossRef] [PubMed]
- Anandakrishnan, R.; Aguilar, B.; Onufriev, A.V. H++ 3.0: Automating pK prediction and the preparation of biomolecular structures for atomistic molecular modeling and simulations. Nucleic Acids Res. 2012, 40, W537–W541. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maier, J.A.; Martinez, C.; Kasavajhala, K.; Wickstrom, L.; Hauser, K.E.; Simmerling, C. ff14SB: Improving the accuracy of protein side chain and backbone parameters from ff99SB. J. Chem. Theory Comput. 2015, 11, 3696–3713. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.M.; Wang, W.; Kollman, P.A. Antechamber: An accessory software package for molecular mechanical calculations. Abstr. Pap. Am. Chem. S. 2001, 222, U403. [Google Scholar]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16 A.03; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Wang, J.M.; Cieplak, P.; Kollman, P.A. How well does a restrained electrostatic potential (RESP) model perform in calculating conformational energies of organic and biological molecules? J. Comput. Chem. 2000, 21, 1049–1074. [Google Scholar] [CrossRef]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar] [CrossRef]
- Kini, R.M.; Evans, H.J. Molecular Modeling of Proteins: A Strategy for Energy Minimization by Molecular Mechanics in the AMBER Force Field. J. Biomol. Struct. Dyn. 1991, 9, 475–488. [Google Scholar] [CrossRef]
- Darden, T.; York, D.; Pedersen, L. Particle Mesh Ewald—An N. Log(N) Method for Ewald Sums in Large Systems. J. Chem. Phys. 1993, 98, 10089–10092. [Google Scholar]
- Ryckaert, J.-P.; Ciccotti, G.; Berendsen, H.J.C. Numerical integration of the cartesian equations of motion of a system with constraints: Molecular dynamics of n-alkanes. J. Comput. Phys. 1977, 23, 327–341. [Google Scholar] [CrossRef] [Green Version]
- Berendsen, H.J.C.; Postma, J.P.M.; Van Gunsteren, W.F.; DiNola, A.; Haak, J.R. Molecular dynamics with coupling to an external bath. J. Chem. Phys. 1984, 81, 3684–3690. [Google Scholar] [CrossRef] [Green Version]
- Hamelberg, D.; De Oliveira, C.A.F.; McCammon, J.A. Sampling of slow diffusive conformational transitions with accelerated molecular dynamics. J. Chem. Phys. 2007, 127, 155102. [Google Scholar] [CrossRef] [PubMed]
- Salomon-Ferrer, R.; Götz, A.W.; Poole, D.; Le Grand, S.; Walker, R.C. Routine Microsecond Molecular Dynamics Simulations with AMBER on GPUs. 2. Explicit Solvent Particle Mesh Ewald. J. Chem. Theory Comput. 2013, 9, 3878–3888. [Google Scholar] [CrossRef] [PubMed]
- Knapp, B.; Ospina-Forero, L.; Deane, C.M. Avoiding False Positive Conclusions in Molecular Simulation: The Importance of Replicas. J. Chem. Theory Comput. 2018, 14, 6127–6138. [Google Scholar] [CrossRef] [PubMed]
- Liu, K.; Kokubo, H. Exploring the Stability of Ligand Binding Modes to Proteins by Molecular Dynamics Simulations: A Cross-docking Study. J. Chem. Inf. Model. 2017, 57, 2514–2522. [Google Scholar] [CrossRef]
- Chodera, J.; Swope, W.C.; Pitera, J.W.; Dill, K.A. Long-Time Protein Folding Dynamics from Short-Time Molecular Dynamics Simulations. Multiscale Model. Simul. 2006, 5, 1214–1226. [Google Scholar] [CrossRef] [Green Version]
- Huang, S.-Y.; Zou, X.-W.; Tan, Z.-J.; Jin, Z.-Z. Short-time critical dynamics in two-dimensional vapor–liquid transition. Phys. Lett. A 2002, 297, 105–109. [Google Scholar] [CrossRef]
- Harada, R.; Shigeta, Y. An assessment of optimal time scale of conformational resampling for parallel cascade selection molecular dynamics. Mol. Simul. 2018, 44, 206–212. [Google Scholar] [CrossRef]
- McAllister, R.G.; Konermann, L. Challenges in the Interpretation of Protein H/D Exchange Data: A Molecular Dynamics Simulation Perspective. Biochemistry 2015, 54, 2683–2692. [Google Scholar] [CrossRef] [PubMed]
- Homeyer, N.; Gohlke, H. Free Energy Calculations by the Molecular Mechanics Poisson−Boltzmann Surface Area Method. Mol. Inform. 2012, 31, 114–122. [Google Scholar] [CrossRef] [PubMed]
- Onufriev, A.; Case, D.A.; Bashford, D. Effective Born radii in the generalized Born approximation: The importance of being perfect. J. Comput. Chem. 2002, 23, 1297–1304. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef] [PubMed]
- Khan, K.; Rehman, S.U.; Aziz, K.; Fong, S.; Sarasvady, S. DBSCAN: Past, Present and Future. In Proceedings of the 2014 Fifth International Conference on the Applications of Digital Information and Web Technologies (Icadiwt), Chennai, India, 17–19 February 2014; pp. 232–238. [Google Scholar]
- Schoier, G.; Borruso, G. A clustering method for large spatial databases. Comput. Sci. Its Appl. 2004, 3044 Pt 2, 1089–1095. [Google Scholar]
- DeLano, W.L.; Lam, J.W. PyMOL: A communications tool for computational models. Abstr. Pap. Am. Chem. S. 2005, 230, U1371–U1372. [Google Scholar]
- Felline, A.; Seeber, M.; Fanelli, F. webPSN v2.0: A webserver to infer fingerprints of structural communication in biomacromolecules. Nucleic Acids Res. 2020, 48, W94–W103. [Google Scholar] [CrossRef]
- Kavanagh, K.; Jornvall, H.; Persson, B.; Oppermann, U. The SDR superfamily: Functional and structural diversity within a family of metabolic and regulatory enzymes. Cell Mol. Life Sci. 2008, 65, 3895–3906. [Google Scholar] [CrossRef] [Green Version]
- Zhang, S.; Heyes, D.J.; Feng, L.; Sun, W.; Johannissen, L.O.; Liu, H.; Levy, C.W.; Li, X.; Yang, J.; Yu, X.; et al. Structural basis for enzymatic photocatalysis in chlorophyll biosynthesis. Nature 2019, 574, 722–725. [Google Scholar] [CrossRef]
- Dong, C.-S.; Zhang, W.-L.; Wang, Q.; Li, Y.-S.; Wang, X.; Zhang, M.; Liu, L. Crystal structures of cyanobacterial light-dependent protochlorophyllide oxidoreductase. Proc. Natl. Acad. Sci. USA 2020, 117, 8455–8461. [Google Scholar] [CrossRef]
- Zhang, S.; Godwin, A.R.F.; Taylor, A.; Hardman, S.J.O.; Jowitt, T.A.; Johannissen, L.O.; Hay, S.; Baldock, C.; Heyes, D.J.; Scrutton, N.S. Dual role of the active site ‘lid’ regions of protochlorophyllide oxidoreductase in photocatalysis and plant development. FEBS J. 2020, 288, 175–189. [Google Scholar] [CrossRef]
- Arêas, J.A.G.; Gröbner, G.J.; Pellacani, L.B.; Glaubitz, C.; Watts, A. Use of solid-state2H NMR for studying protein-lipid interactions at emulsion interfaces. Org. Magn. Reson. 1997, 35, S119–S124. [Google Scholar] [CrossRef]
- Schnell, J.R.; Chou, J.J. Structure and mechanism of the M2 proton channel of influenza A virus. Nature 2008, 451, 591–595. [Google Scholar] [CrossRef] [Green Version]
- Bhattacharya, A.A.; Grüne, T.; Curry, S. Crystallographic analysis reveals common modes of binding of medium and long-chain fatty acids to human serum albumin. J. Mol. Biol. 2000, 303, 721–732. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Binding Site | Residence Time (ps) | |||||
|---|---|---|---|---|---|---|
| PG | SQDG | |||||
| 1 | 2 | 3 | 1 | 2 | 3 | |
| I | 12,301 | 10,546 | 13,147 | 10,980 | 14,011 | 13,753 |
| II | >25,000 | >25,000 | >25,000 | >25,000 | >25,000 | >25,000 |
| III | 2230 | 2317 | 3793 | 1224 | 1329 | 2957 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, R.; Wang, L.; Meng, Y.; Li, F.; Nie, H.; Lu, H. Role of Thylakoid Lipids in Protochlorophyllide Oxidoreductase Activation: Allosteric Mechanism Elucidated by a Computational Study. Int. J. Mol. Sci. 2023, 24, 307. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms24010307
Liu R, Wang L, Meng Y, Li F, Nie H, Lu H. Role of Thylakoid Lipids in Protochlorophyllide Oxidoreductase Activation: Allosteric Mechanism Elucidated by a Computational Study. International Journal of Molecular Sciences. 2023; 24(1):307. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms24010307
Chicago/Turabian StyleLiu, Ruiyuan, Leng Wang, Yue Meng, Fang Li, Haiyu Nie, and Huizhe Lu. 2023. "Role of Thylakoid Lipids in Protochlorophyllide Oxidoreductase Activation: Allosteric Mechanism Elucidated by a Computational Study" International Journal of Molecular Sciences 24, no. 1: 307. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms24010307