

Copy Number Variations as Determinants of Colorectal Tumor Progression in Liquid Biopsies

Abstract
:1. Introduction
2. CNVs in CRC Disease Progression
2.1. CNVs in Adenoma-Carcinoma
2.2. CNVs in Carcinoma-mCRC

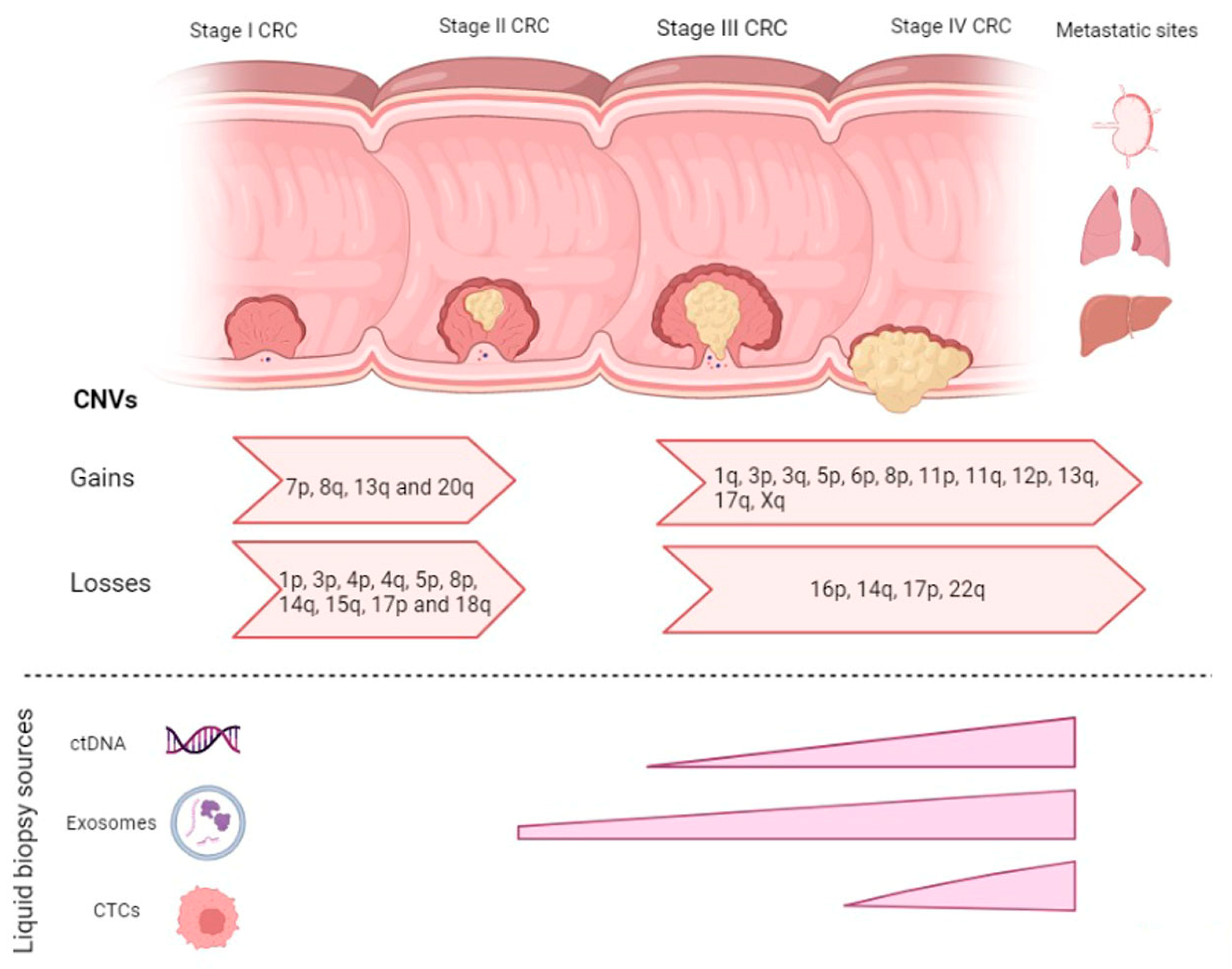{kind=link}
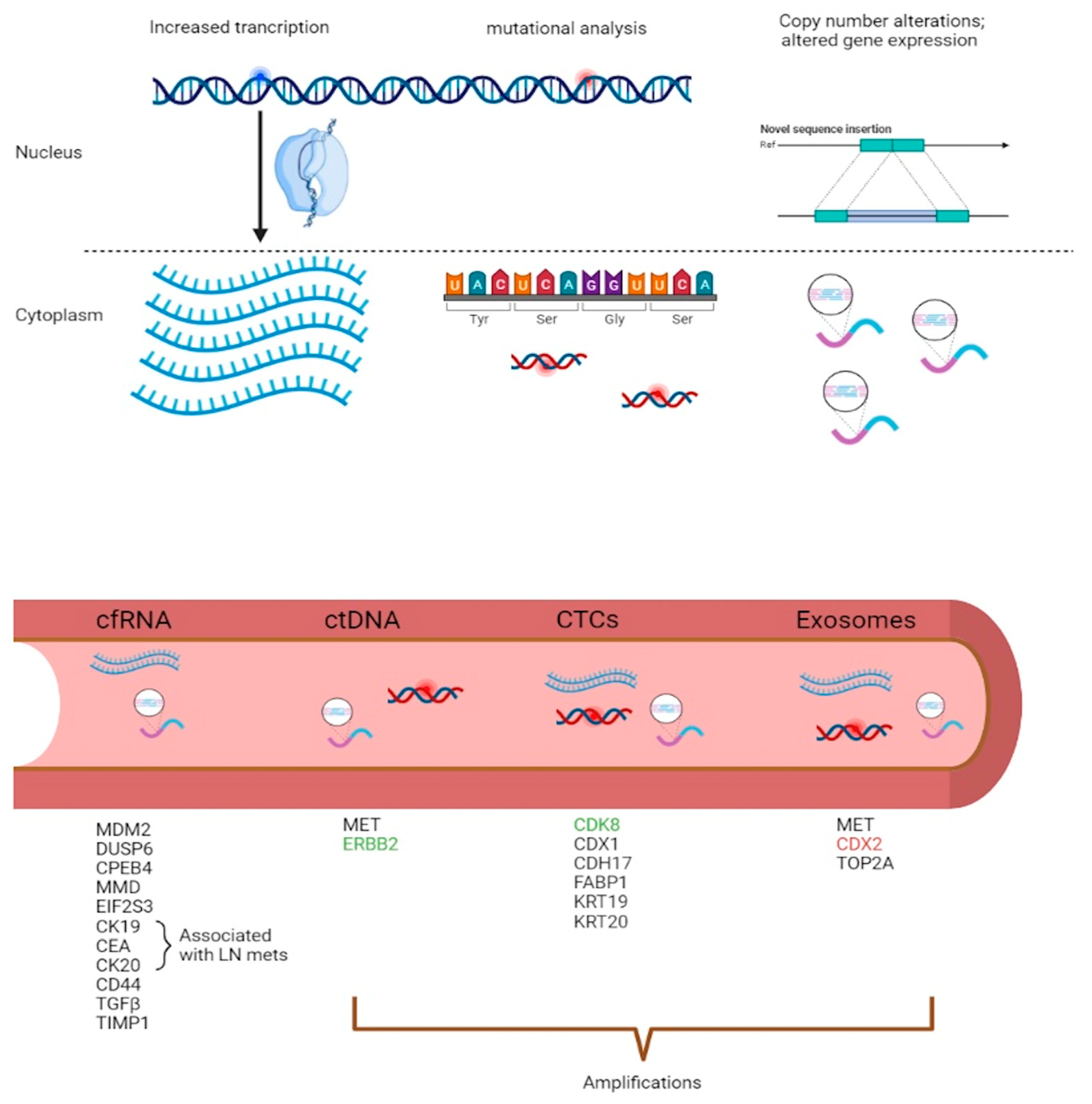{kind=link}
| CNVs | Associated Gene | Reference | |
|---|---|---|---|
| Gain | 7p | EGFR | [11] |
| Gain | 8q | c-MYC | [10,16,17] |
| LYN | [13] | ||
| Gain | 13q | CDX2 | [10,16,17] |
| POLR1D | [13] | ||
| PDX1 | [11] | ||
| Gain | 20q | AURKA | [13,14] |
| TH1L | [14] | ||
| ADRM1 | [14] | ||
| C20orf20 | [14] | ||
| TCFL5 | [14] | ||
| TPX2 | [25] | ||
| PMPEA1 | [13] | ||
| MMP9 | [13] | ||
| MYBL2 | [13,39] | ||
| UBE2C | [13] | ||
| Loss | 1p | [11] | |
| Loss | 3p | FHIT | [30] |
| Loss | 4p | [30] | |
| Loss | 5p | [31] | |
| Loss | 8p | CSMD | [13] |
| DLC1 | [10,16] | ||
| Loss | 14q | [13] | |
| Loss | 15q | [10] | |
| Loss | 17p | TP53 | [10] |
| Loss | 18q | DCC | [12] |
| SMAD2 | [12] | ||
| SMAD4 | [12] | ||
| CCDC68 | [13] | ||
| SERPINB7 | [39] | ||
| CTDP1 | [39] | ||
| Loss | 20p | [13] |
| Copy Number Alterations | Associated Gene | Metastatic Site | Reference |
|---|---|---|---|
| Gain of 1q | TGFB2 | Liver metastasis | [39] |
| Gain of 3p | TGFBR2 CTNNB1 FHIT | Liver metastasis Liver metastasis Liver metastasis | [36] |
| Gain 3q | CBLB KALRN PIK3CA | Liver metastasis Liver metastasis Liver metastasis | [36] |
| Gain 5p | Lung metastasis | [36] | |
| Gain 6p | Liver metastasis | [13,35] | |
| Gain 8p | FGFR1 | Liver metastasis | [36] |
| Gain 11p | Liver metastasis | [39] | |
| Gain 11q | MCAM | Liver metastasis | [39] |
| Gain 12p | Liver metastasis | [39] | |
| Gain 13q | CDK8 | Liver metastasis | [36] |
| Gain 17q | ERBB2 | Liver metastasis | [36] |
| Gain Xq | [35] | ||
| Loss 14q | Liver metastasis | [35,39] | |
| Loss 16p | RBFOX1 | Liver metastasis | [36] |
| Loss 17p | Liver metastasis | [38] | |
| Loss 22q | Liver metastasis | [35,38] |
3. Diagnostic and Predictive Models in Liquid Biopsies
3.1. mRNA Diagnostic Models in CRC
3.2. ctDNA Predictive Models in CRC
3.3. CTCs as Metastatic Markers in CRC
3.4. Exosomal CNVs and Long RNAs as Putative Markers of Disease
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Erratum: Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2020, 70, 313. [CrossRef] [PubMed] [Green Version]
- Vogelstein, B.; Fearon, E.R.; Hamilton, S.R.; Kern, S.E.; Preisinger, A.C.; Leppert, M.; Nakamura, Y.; White, R.; Smits, A.M.; Bos, J.L. Genetic alterations during colorectal-tumor development. N. Engl. J. Med. 1988, 319, 525–532. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malki, A.; ElRuz, R.A.; Gupta, I.; Allouch, A.; Vranic, S.; Al Moustafa, A.E. Molecular Mechanisms of Colon Cancer Progression and Metastasis: Recent Insights and Advancements. Int. J. Mol. Sci. 2020, 22, 130. [Google Scholar] [CrossRef] [PubMed]
- Tauriello, D.V.; Calon, A.; Lonardo, E.; Batlle, E. Determinants of metastatic competency in colorectal cancer. Mol. Oncol. 2017, 11, 97–119. [Google Scholar] [CrossRef] [Green Version]
- Tan, E.S.; Knepper, T.C.; Wang, X.; Permuth, J.B.; Wang, L.; Fleming, J.B.; Xie, H. Copy Number Alterations as Novel Biomarkers and Therapeutic Targets in Colorectal Cancer. Cancers 2022, 14, 2223. [Google Scholar] [CrossRef]
- Ried, T.; Meijer, G.A.; Harrison, D.J.; Grech, G.; Franch-Exposito, S.; Briffa, R.; Carvalho, B.; Camps, J. The landscape of genomic copy number alterations in colorectal cancer and their consequences on gene expression levels and disease outcome. Mol. Aspects Med. 2019, 69, 48–61. [Google Scholar] [CrossRef]
- Fearnhead, N.S.; Britton, M.P.; Bodmer, W.F. The ABC of APC. Hum. Mol. Genet. 2001, 10, 721–733. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Yuan, X.; Shi, H.; Wu, L.; Qian, H.; Xu, W. Exosomes in cancer: Small particle, big player. J. Hematol. Oncol. 2015, 8, 83. [Google Scholar] [CrossRef] [Green Version]
- Stanczak, A.; Stec, R.; Bodnar, L.; Olszewski, W.; Cichowicz, M.; Kozlowski, W.; Szczylik, C.; Pietrucha, T.; Wieczorek, M.; Lamparska-Przybysz, M. Prognostic significance of Wnt-1, beta-catenin and E-cadherin expression in advanced colorectal carcinoma. Pathol. Oncol. Res. 2011, 17, 955–963. [Google Scholar] [CrossRef] [Green Version]
- Hermsen, M.; Postma, C.; Baak, J.; Weiss, M.; Rapallo, A.; Sciutto, A.; Roemen, G.; Arends, J.W.; Williams, R.; Giaretti, W.; et al. Colorectal adenoma to carcinoma progression follows multiple pathways of chromosomal instability. Gastroenterology 2002, 123, 1109–1119. [Google Scholar] [CrossRef]
- Salari, K.; Spulak, M.E.; Cuff, J.; Forster, A.D.; Giacomini, C.P.; Huang, S.; Ko, M.E.; Lin, A.Y.; van de Rijn, M.; Pollack, J.R. CDX2 is an amplified lineage-survival oncogene in colorectal cancer. Proc. Natl. Acad. Sci. USA 2012, 109, E3196–E3205. [Google Scholar] [CrossRef] [Green Version]
- Shi, Y.; Hata, A.; Lo, R.S.; Massague, J.; Pavletich, N.P. A structural basis for mutational inactivation of the tumour suppressor Smad4. Nature 1997, 388, 87–93. [Google Scholar] [CrossRef] [PubMed]
- Sheffer, M.; Bacolod, M.D.; Zuk, O.; Giardina, S.F.; Pincas, H.; Barany, F.; Paty, P.B.; Gerald, W.L.; Notterman, D.A.; Domany, E. Association of survival and disease progression with chromosomal instability: A genomic exploration of colorectal cancer. Proc. Natl. Acad. Sci. USA 2009, 106, 7131–7136. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carvalho, B.; Diosdado, B.; Terhaar Sive Droste, J.S.; Bolijn, A.S.; Komor, M.A.; de Wit, M.; Bosch, L.J.W.; van Burink, M.; Dekker, E.; Kuipers, E.J.; et al. Evaluation of Cancer-Associated DNA Copy Number Events in Colorectal (Advanced) Adenomas. Cancer Prev. Res. 2018, 11, 403–412. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palin, K.; Pitkanen, E.; Turunen, M.; Sahu, B.; Pihlajamaa, P.; Kivioja, T.; Kaasinen, E.; Valimaki, N.; Hanninen, U.A.; Cajuso, T.; et al. Contribution of allelic imbalance to colorectal cancer. Nat. Commun. 2018, 9, 3664. [Google Scholar] [CrossRef]
- Lee, K.S.; Kwak, Y.; Nam, K.H.; Kim, D.W.; Kang, S.B.; Choe, G.; Kim, W.H.; Lee, H.S. c-MYC Copy-Number Gain Is an Independent Prognostic Factor in Patients with Colorectal Cancer. PLoS ONE 2015, 10, e0139727. [Google Scholar] [CrossRef]
- Cai, Y.; Crowther, J.; Pastor, T.; Abbasi Asbagh, L.; Baietti, M.F.; De Troyer, M.; Vazquez, I.; Talebi, A.; Renzi, F.; Dehairs, J.; et al. Loss of Chromosome 8p Governs Tumor Progression and Drug Response by Altering Lipid Metabolism. Cancer Cell 2016, 29, 751–766. [Google Scholar] [CrossRef]
- Ogino, S.; Nosho, K.; Irahara, N.; Shima, K.; Baba, Y.; Kirkner, G.J.; Meyerhardt, J.A.; Fuchs, C.S. Prognostic significance and molecular associations of 18q loss of heterozygosity: A cohort study of microsatellite stable colorectal cancers. J. Clin. Oncol. 2009, 27, 4591–4598. [Google Scholar] [CrossRef] [Green Version]
- Tanaka, T.; Watanabe, T.; Kazama, Y.; Tanaka, J.; Kanazawa, T.; Kazama, S.; Nagawa, H. Chromosome 18q deletion and Smad4 protein inactivation correlate with liver metastasis: A study matched for T- and N- classification. Br. J. Cancer 2006, 95, 1562–1567. [Google Scholar] [CrossRef] [Green Version]
- Xu, D.; Li, X.F.; Zheng, S.; Jiang, W.Z. Quantitative real-time RT-PCR detection for CEA, CK20 and CK19 mRNA in peripheral blood of colorectal cancer patients. J. Zhejiang Univ. Sci. B 2006, 7, 445–451. [Google Scholar] [CrossRef]
- Nguyen, H.T.; Duong, H.Q. The molecular characteristics of colorectal cancer: Implications for diagnosis and therapy. Oncol. Lett. 2018, 16, 9–18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thiagalingam, S.; Lengauer, C.; Leach, F.S.; Schutte, M.; Hahn, S.A.; Overhauser, J.; Willson, J.K.; Markowitz, S.; Hamilton, S.R.; Kern, S.E.; et al. Evaluation of candidate tumour suppressor genes on chromosome 18 in colorectal cancers. Nat. Genet. 1996, 13, 343–346. [Google Scholar] [CrossRef] [PubMed]
- Postma, C.; Terwischa, S.; Hermsen, M.A.; van der Sijp, J.R.; Meijer, G.A. Gain of chromosome 20q is an indicator of poor prognosis in colorectal cancer. Cell. Oncol. 2007, 29, 73–75. [Google Scholar] [CrossRef] [PubMed]
- Sillars-Hardebol, A.H.; Carvalho, B.; Belien, J.A.; de Wit, M.; Delis-van Diemen, P.M.; Tijssen, M.; van de Wiel, M.A.; Ponten, F.; Fijneman, R.J.; Meijer, G.A. BCL2L1 has a functional role in colorectal cancer and its protein expression is associated with chromosome 20q gain. J. Pathol. 2012, 226, 442–450. [Google Scholar] [CrossRef] [PubMed]
- Sillars-Hardebol, A.H.; Carvalho, B.; Tijssen, M.; Belien, J.A.; de Wit, M.; Delis-van Diemen, P.M.; Ponten, F.; van de Wiel, M.A.; Fijneman, R.J.; Meijer, G.A. TPX2 and AURKA promote 20q amplicon-driven colorectal adenoma to carcinoma progression. Gut 2012, 61, 1568–1575. [Google Scholar] [CrossRef] [Green Version]
- Cammareri, P.; Scopelliti, A.; Todaro, M.; Eterno, V.; Francescangeli, F.; Moyer, M.P.; Agrusa, A.; Dieli, F.; Zeuner, A.; Stassi, G. Aurora-a is essential for the tumorigenic capacity and chemoresistance of colorectal cancer stem cells. Cancer Res. 2010, 70, 4655–4665. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Liang, L.; Fang, J.Y.; Xu, J. Somatic gene copy number alterations in colorectal cancer: New quest for cancer drivers and biomarkers. Oncogene 2016, 35, 2011–2019. [Google Scholar] [CrossRef]
- Jung, P.; Horst, D.; Kirchner, T.; Klauschen, F.; Neumann, J. AURKA is a prognostic biomarker for good overall survival in stage II colorectal cancer patients. Pathol. Res. Pract. 2022, 235, 153936. [Google Scholar] [CrossRef]
- Yin, X.L.; Chen, S.; Gu, J.X. Identification of TH1 as an interaction partner of A-Rafkinase. Mol. Cell. Biochem. 2002, 231, 69–74. [Google Scholar] [CrossRef]
- Xie, T.; D’Ario, G.; Lamb, J.R.; Martin, E.; Wang, K.; Tejpar, S.; Delorenzi, M.; Bosman, F.T.; Roth, A.D.; Yan, P.; et al. A comprehensive characterization of genome-wide copy number aberrations in colorectal cancer reveals novel oncogenes and patterns ofalterations. PLoS ONE 2012, 7, e42001. [Google Scholar] [CrossRef]
- Kasem, K.; Gopalan, V.; Salajegheh, A.; Lu, C.T.; Smith, R.A.; Lam, A.K. JK1 (FAM134B) gene and colorectal cancer: A pilot study on the gene copy number alterations and correlations with clinicopathological parameters. Exp. Mol. Pathol. 2014, 97, 31–36. [Google Scholar] [CrossRef] [PubMed]
- Fares, J.; Fares, M.Y.; Khachfe, H.H.; Salhab, H.A.; Fares, Y. Molecular principles of metastasis: A hallmark of cancer revisited. Signal. Transduct. Target Ther. 2020, 5, 28. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zacksenhaus, E.; Egan, S.E. Progression to Metastasis of Solid Cancer. Cancers 2021, 13, 717. [Google Scholar] [CrossRef] [PubMed]
- Riihimaki, M.; Hemminki, A.; Sundquist, J.; Hemminki, K. Patterns of metastasis in colon and rectal cancer. Sci. Rep. 2016, 6, 29765. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tanami, H.; Tsuda, H.; Okabe, S.; Iwai, T.; Sugihara, K.; Imoto, I.; Inazawa, J. Involvement of cyclin D3 in liver metastasis of colorectal cancer, revealed by genome-wide copy-number analysis. Lab. Investig. 2005, 85, 1118–1129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kawamata, F.; Patch, A.M.; Nones, K.; Bond, C.; McKeone, D.; Pearson, S.A.; Homma, S.; Liu, C.; Fennell, L.; Dumenil, T.; et al. Copy number profiles of paired primary and metastaticcolorectal cancers. Oncotarget 2017, 9, 3394–3405. [Google Scholar] [CrossRef] [Green Version]
- Al-Mulla, F.; Keith, W.N.; Pickford, I.R.; Going, J.J.; Birnie, G.D. Comparative genomic hybridization analysis of primary colorectal carcinomas and their synchronous metastases. Genes Chromosomes Cancer 1999, 24, 306–314. [Google Scholar] [CrossRef]
- Gonzalez-Gonzalez, M.; Munoz-Bellvis, L.; Mackintosh, C.; Fontanillo, C.; Gutierrez, M.L.; Abad, M.M.; Bengoechea, O.; Teodosio, C.; Fonseca, E.; Fuentes, M.; et al. Prognostic Impact of del(17p) and del(22q) as assessed by interphase FISH in sporadic colorectal carcinomas. PLoS ONE 2012, 7, e42683. [Google Scholar] [CrossRef] [Green Version]
- Diep, C.B.; Kleivi, K.; Ribeiro, F.R.; Teixeira, M.R.; Lindgjaerde, O.C.; Lothe, R.A. The order of genetic events associated with colorectal cancer progression inferred from meta-analysis of copy number changes. Genes Chromosomes Cancer 2006, 45, 31–41. [Google Scholar] [CrossRef]
- Mamlouk, S.; Childs, L.H.; Aust, D.; Heim, D.; Melching, F.; Oliveira, C.; Wolf, T.; Durek, P.; Schumacher, D.; Blaker, H.; et al. DNA copy number changes define spatial patterns of heterogeneity in colorectal cancer. Nat. Commun. 2017, 8, 14093. [Google Scholar] [CrossRef]
- Saha, S.; Bardelli, A.; Buckhaults, P.; Velculescu, V.E.; Rago, C.; St Croix, B.; Romans, K.E.; Choti, M.A.; Lengauer, C.; Kinzler, K.W.; et al. A phosphatase associated with metastasis of colorectal cancer. Science 2001, 294, 1343–1346. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Knosel, T.; Schluns, K.; Dietel, M.; Petersen, I. Chromosomal alterations in lung metastases of colorectal carcinomas: Associations with tissue specific tumor dissemination. Clin. Exp. Metastasis 2005, 22, 533–538. [Google Scholar] [CrossRef] [PubMed]
- Tohme, S.; Simmons, R.L.; Tsung, A. Surgery for Cancer: A Trigger for Metastases. Cancer Res. 2017, 77, 1548–1552. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ribatti, D.; Mangialardi, G.; Vacca, A. Stephen Paget and the ’seed and soil’ theory of metastatic dissemination. Clin. Exp. Med. 2006, 6, 145–149. [Google Scholar] [CrossRef] [PubMed]
- De Luca, A.; Pignata, S.; Casamassimi, A.; D’Antonio, A.; Gridelli, C.; Rossi, A.; Cremona, F.; Parisi, V.; De Matteis, A.; Normanno, N. Detection of circulating tumor cells in carcinoma patients by a novel epidermal growth factor receptor reverse transcription-PCR assay. Clin. Cancer Res. 2000, 6, 1439–1444. [Google Scholar] [PubMed]
- Wang, W.; Li, Y.; Zhang, X.; Jing, J.; Zhao, X.; Wang, Y.; Han, C. Evaluating the significance of expression of CEA mRNA and levels of CEA and its related proteins in colorectal cancer patients. J. Surg. Oncol. 2014, 109, 440–444. [Google Scholar] [CrossRef]
- Yeh, C.S.; Wang, J.Y.; Wu, C.H.; Chong, I.W.; Chung, F.Y.; Wang, Y.H.; Yu, Y.P.; Lin, S.R. Molecular detection of circulating cancer cells in the peripheral blood of patients with colorectal cancer by using membrane array with a multiple mRNA marker panel. Int. J. Oncol. 2006, 28, 411–420. [Google Scholar] [CrossRef]
- Chang, Y.; Huang, C.; Yao, C.; Su, S.; Terng, H.; Chou, H.; Chou, Y.; Chen, K.; Shih, Y.; Lu, C.; et al. Gene expression profile of peripheral blood in colorectal cancer. World J. Gastroenterol. 2014, 20, 14463–14471. [Google Scholar] [CrossRef]
- Galamb, O.; Sipos, F.; Solymosi, N.; Spisak, S.; Krenacs, T.; Toth, K.; Tulassay, Z.; Molnar, B. Diagnostic mRNA expression patterns of inflamed, benign, and malignant colorectal biopsy specimen and their correlation with peripheral blood results. Cancer Epidemiol. Biomarkers Prev. 2008, 17, 2835–2845. [Google Scholar] [CrossRef] [Green Version]
- Garcia-Pardo, M.; Makarem, M.; Li, J.J.N.; Kelly, D.; Leighl, N.B. Integrating circulating-free DNA (cfDNA) analysis into clinical practice: Opportunities and challenges. Br. J. Cancer 2022, 127, 592–602. [Google Scholar] [CrossRef]
- Pascual, J.; Attard, G.; Bidard, F.; Curigliano, G.; De Mattos-Arruda, L.; Diehn, M.; Italiano, A.; Lindberg, J.; Merker, J.D.; Montagut, C.; et al. ESMO recommendations on the use of circulating tumour DNA assays for patients with cancer: A report from the ESMO Precision Medicine Working Group. Ann. Oncol. 2022, 33, 750–768. [Google Scholar] [CrossRef] [PubMed]
- Tie, J.; Wang, Y.; Tomasetti, C.; Li, L.; Springer, S.; Kinde, I.; Silliman, N.; Tacey, M.; Wong, H.; Christie, M.; et al. Circulating tumor DNA analysis detects minimal residual disease and predicts recurrence in patients with stage II colon cancer. Sci. Transl. Med. 2016, 8, 346ra92. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reinert, T.; Henriksen, T.V.; Christensen, E.; Sharma, S.; Salari, R.; Sethi, H.; Knudsen, M.; Nordentoft, I.; Wu, H.; Tin, A.S.; et al. Analysis of Plasma Cell-Free DNA by Ultradeep Sequencing in Patients With Stages I to III Colorectal Cancer. JAMA Oncol. 2019, 5, 1124–1131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tie, J.; Cohen, J.D.; Wang, Y.; Christie, M.; Simons, K.; Lee, M.; Wong, R.; Kosmider, S.; Ananda, S.; McKendrick, J.; et al. Circulating Tumor DNA Analyses as Markers of Recurrence Risk and Benefit of Adjuvant Therapy for Stage III Colon Cancer. JAMA Oncol. 2019, 5, 1710–1717. [Google Scholar] [CrossRef]
- Dotan, E.; Cohen, S.J. Challenges in the management of stage II colon cancer. Semin. Oncol. 2011, 38, 511–520. [Google Scholar] [CrossRef] [Green Version]
- Chakrabarti, S.; Peterson, C.Y.; Sriram, D.; Mahipal, A. Early stage colon cancer: Current treatment standards, evolving paradigms, and future directions. World J. Gastrointest. Oncol. 2020, 12, 808–832. [Google Scholar] [CrossRef]
- Siravegna, G.; Marsoni, S.; Siena, S.; Bardelli, A. Integrating liquid biopsies into the management of cancer. Nat. Rev. Clin. Oncol. 2017, 14, 531–548. [Google Scholar] [CrossRef]
- Wan, J.C.M.; Massie, C.; Garcia-Corbacho, J.; Mouliere, F.; Brenton, J.D.; Caldas, C.; Pacey, S.; Baird, R.; Rosenfeld, N. Liquid biopsies come of age: Towards implementation of circulating tumour DNA. Nat. Rev. Cancer 2017, 17, 223–238. [Google Scholar] [CrossRef]
- Molparia, B.; Oliveira, G.; Wagner, J.L.; Spencer, E.G.; Torkamani, A. A feasibility study of colorectal cancer diagnosis via circulating tumor DNA derived CNV detection. PLoS ONE 2018, 13, e0196826. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Dittmar, R.L.; Xia, S.; Zhang, H.; Du, M.; Huang, C.; Druliner, B.R.; Boardman, L.; Wang, L. Cell-free DNA copy number variations in plasma from colorectal cancer patients. Mol. Oncol. 2017, 11, 1099–1111. [Google Scholar] [CrossRef]
- Takegawa, N.; Yonesaka, K.; Sakai, K.; Ueda, H.; Watanabe, S.; Nonagase, Y.; Okuno, T.; Takeda, M.; Maenishi, O.; Tsurutani, J.; et al. HER2 genomic amplification in circulating tumor DNA from patients withcetuximab-resistant colorectal cancer. Oncotarget 2016, 7, 3453–3460. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakamura, Y.; Okamoto, W.; Kato, T.; Esaki, T.; Kato, K.; Komatsu, Y.; Yuki, S.; Masuishi, T.; Nishina, T.; Ebi, H.; et al. Circulating tumor DNA-guided treatment with pertuzumab plus trastuzumab for HER2-amplified metastatic colorectal cancer: A phase 2 trial. Nat. Med. 2021, 27, 1899–1903. [Google Scholar] [CrossRef] [PubMed]
- Wei, Q.; Zhang, Y.; Gao, J.; Li, J.; Li, J.; Li, Y.; Zhou, J.; Lu, M.; Gong, J.; Zhang, X.; et al. Clinicopathologic Characteristics of HER2-positive Metastatic Colorectal Cancer and Detection of HER2 in Plasma Circulating Tumor DNA. Clin. Colorectal Cancer 2019, 18, 175–182. [Google Scholar] [CrossRef] [PubMed]
- Mohamed, A.A.; Lau, D.K.; Chau, I. HER2 targeted therapy in colorectal cancer: Newhorizons. Cancer Treat. Rev. 2022, 105, 102363. [Google Scholar] [CrossRef]
- Jia, J.; Morse, M.A.; Nagy, R.J.; Lanman, R.B.; Strickler, J.H. Cell-Free DNA Profiling to Discover Mechanisms of Exceptional Response to Cabozantinib Plus Panitumumab in a Patient With Treatment Refractory Metastatic Colorectal Cancer. Front. Oncol. 2018, 8, 305. [Google Scholar] [CrossRef] [Green Version]
- Jenkins, S.; Yang, J.C.; Ramalingam, S.S.; Yu, K.; Patel, S.; Weston, S.; Hodge, R.; Cantarini, M.; Janne, P.A.; Mitsudomi, T.; et al. Plasma ctDNA Analysis for Detection of the EGFR T790M Mutation in Patients with Advanced Non-Small Cell Lung Cancer. J. Thorac. Oncol. 2017, 12, 1061–1070. [Google Scholar] [CrossRef] [Green Version]
- Sacher, A.G.; Paweletz, C.; Dahlberg, S.E.; Alden, R.S.; O’Connell, A.; Feeney, N.; Mach, S.L.; Janne, P.A.; Oxnard, G.R. Prospective Validation of Rapid Plasma Genotyping for the Detection of EGFR and KRAS Mutations in Advanced Lung Cancer. JAMA Oncol. 2016, 2, 1014–1022. [Google Scholar] [CrossRef] [Green Version]
- Leighl, N.B.; Page, R.D.; Raymond, V.M.; Daniel, D.B.; Divers, S.G.; Reckamp, K.L.; Villalona-Calero, M.A.; Dix, D.; Odegaard, J.I.; Lanman, R.B.; et al. Clinical Utility of Comprehensive Cell-free DNA Analysis to Identify Genomic Biomarkers in Patients with Newly Diagnosed Metastatic Non-small Cell Lung Cancer. Clin. Cancer Res. 2019, 25, 4691–4700. [Google Scholar] [CrossRef] [Green Version]
- Markus, H.; Chandrananda, D.; Moore, E.; Mouliere, F.; Morris, J.; Brenton, J.D.; Smith, C.G.; Rosenfeld, N. Refined characterization of circulating tumor DNA through biological feature integration. Sci. Rep. 2022, 12, 1928. [Google Scholar] [CrossRef]
- Kang, Q.; Henry, N.L.; Paoletti, C.; Jiang, H.; Vats, P.; Chinnaiyan, A.M.; Hayes, D.F.; Merajver, S.D.; Rae, J.M.; Tewari, M. Comparative analysis of circulating tumor DNA stability In K(3)EDTA, Streck, and CellSave blood collection tubes. Clin. Biochem. 2016, 49, 1354–1360. [Google Scholar] [CrossRef]
- (Parpart-Li, S.; Bartlett, B.; Popoli, M.; Adleff, V.; Tucker, L.; Steinberg, R.; Georgiadis, A.; Phallen, J.; Brahmer, J.; Azad, N.; et al. The Effect of Preservative and Temperature on the Analysis of Circulating Tumor DNA. Clin. Cancer Res. 2017, 23, 2471–2477. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tivey, A.; Church, M.; Rothwell, D.; Dive, C.; Cook, N. Circulating tumour DNA - looking beyond the blood. Nat. Rev. Clin. Oncol. 2022, 19, 600–612. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Lian, J.; Chen, Y.; Zhao, X.; Du, C.; Xu, Y.; Hu, H.; Rao, H.; Hong, X. Circulating Tumor Cells (CTCs): A Unique Model of Cancer Metastases and Non-invasive Biomarkers of Therapeutic Response. Front. Genet. 2021, 12, 734595. [Google Scholar] [CrossRef] [PubMed]
- Gasch, C.; Bauernhofer, T.; Pichler, M.; Langer-Freitag, S.; Reeh, M.; Seifert, A.M.; Mauermann, O.; Izbicki, J.R.; Pantel, K.; Riethdorf, S. Heterogeneity of epidermal growth factor receptor status and mutations of KRAS/PIK3CA in circulating tumor cells of patients with colorectal cancer. Clin. Chem. 2013, 59, 252–260. [Google Scholar] [CrossRef] [Green Version]
- Krebs, M.G.; Metcalf, R.L.; Carter, L.; Brady, G.; Blackhall, F.H.; Dive, C. Molecular analysis of circulating tumour cells-biology and biomarkers. Nat. Rev. Clin. Oncol. 2014, 11, 129–144. [Google Scholar] [CrossRef]
- Heitzer, E.; Auer, M.; Gasch, C.; Pichler, M.; Ulz, P.; Hoffmann, E.M.; Lax, S.; Waldispuehl-Geigl, J.; Mauermann, O.; Lackner, C.; et al. Complex tumor genomes inferred from single circulating tumor cells by array-CGH and next-generation sequencing. Cancer Res. 2013, 73, 2965–2975. [Google Scholar] [CrossRef] [Green Version]
- Firestein, R.; Bass, A.J.; Kim, S.Y.; Dunn, I.F.; Silver, S.J.; Guney, I.; Freed, E.; Ligon, A.H.; Vena, N.; Ogino, S.; et al. CDK8 is a colorectal cancer oncogene that regulates beta-catenin activity. Nature 2008, 455, 547–551. [Google Scholar] [CrossRef] [Green Version]
- Dickson, M.A.; Shah, M.A.; Rathkopf, D.; Tse, A.; Carvajal, R.D.; Wu, N.; Lefkowitz, R.A.; Gonen, M.; Cane, L.M.; Dials, H.J.; et al. A phase I clinical trial of FOLFIRI in combination with the pan-cyclin-dependent kinase (CDK) inhibitor flavopiridol. Cancer Chemother. Pharmacol. 2010, 66, 1113–1121. [Google Scholar] [CrossRef] [Green Version]
- Ramaswamy, B.; Phelps, M.A.; Baiocchi, R.; Bekaii-Saab, T.; Ni, W.; Lai, J.; Wolfson, A.; Lustberg, M.E.; Wei, L.; Wilkins, D.; et al. A dose-finding, pharmacokinetic and pharmacodynamic study of a novel schedule of flavopiridol in patients with advanced solid tumors. Investig. New Drugs 2012, 30, 629–638. [Google Scholar] [CrossRef] [Green Version]
- Mostert, B.; Sieuwerts, A.M.; Bolt-de Vries, J.; Kraan, J.; Lalmahomed, Z.; van Galen, A.; van der Spoel, P.; de Weerd, V.; Ramirez-Moreno, R.; Smid, M.; et al. mRNA expression profiles in circulating tumor cells of metastatic colorectal cancer patients. Mol. Oncol. 2015, 9, 920–932. [Google Scholar] [CrossRef]
- de Wit, S.; Manicone, M.; Rossi, E.; Lampignano, R.; Yang, L.; Zill, B.; Rengel-Puertas, A.; Ouhlen, M.; Crespo, M.; Berghuis, A.M.S.; et al. EpCAM(high) and EpCAM(low) circulating tumor cells in metastatic prostate and breast cancer patients. Oncotarget 2018, 9, 35705–35716. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Wit, S.; Rossi, E.; Weber, S.; Tamminga, M.; Manicone, M.; Swennenhuis, J.F.; Groothuis-Oudshoorn, C.G.M.; Vidotto, R.; Facchinetti, A.; Zeune, L.L.; et al. Single tube liquid biopsy for advanced non-small cell lung cancer. Int. J. Cancer 2019, 144, 3127–3137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Wit, S.; van Dalum, G.; Lenferink, A.T.M.; Tibbe, A.G.J.; Hiltermann, T.J.N.; Groen, H.J.M.; van Rijn, C.J.M.; Terstappen, L.W.M.M. The detection of EpCAM(+) and EpCAM(-) circulating tumor cells. Sci. Rep. 2015, 5, 12270. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Polyak, K.; Weinberg, R.A. Transitions between epithelial and mesenchymal states: Acquisition of malignant and stem cell traits. Nat. Rev. Cancer 2009, 9, 265–273. [Google Scholar] [CrossRef]
- Sieuwerts, A.M.; Kraan, J.; Bolt, J.; van der Spoel, P.; Elstrodt, F.; Schutte, M.; Martens, J.W.M.; Gratama, J.; Sleijfer, S.; Foekens, J.A. Anti-epithelial cell adhesion molecule antibodies and the detection of circulating normal-like breast tumor cells. J. Natl. Cancer Inst. 2009, 101, 61–66. [Google Scholar] [CrossRef]
- Huang, L.R.; Cox, E.C.; Austin, R.H.; Sturm, J.C. Continuous particle separation through deterministic lateral displacement. Science 2004, 304, 987–990. [Google Scholar] [CrossRef]
- Di Carlo, D.; Irimia, D.; Tompkins, R.G.; Toner, M. Continuous inertial focusing, ordering, and separation of particles in microchannels. Proc. Natl. Acad. Sci. USA 2007, 104, 18892–18897. [Google Scholar] [CrossRef] [Green Version]
- Karabacak, N.M.; Spuhler, P.S.; Fachin, F.; Lim, E.J.; Pai, V.; Ozkumur, E.; Martel, J.M.; Kojic, N.; Smith, K.; Chen, P.; et al. Microfluidic, marker-free isolation of circulating tumor cells from blood samples. Nat. Protoc. 2014, 9, 694–710. [Google Scholar] [CrossRef] [Green Version]
- Harb, W.; Fan, A.; Tran, T.; Danila, D.C.; Keys, D.; Schwartz, M.; Ionescu-Zanetti, C. Mutational Analysis of Circulating Tumor Cells Using a Novel Microfluidic Collection Device and qPCR Assay. Transl. Oncol. 2013, 6, 528–538. [Google Scholar] [CrossRef] [Green Version]
- Pantel, K.; Brakenhoff, R.H.; Brandt, B. Detection, clinical relevance and specific biological properties of disseminating tumour cells. Nat. Rev. Cancer. 2008, 8, 329–340. [Google Scholar] [CrossRef]
- Thery, C.; Zitvogel, L.; Amigorena, S. Exosomes: Composition, biogenesis and function. Nat. Rev. Immunol. 2002, 2, 569–579. [Google Scholar] [CrossRef] [PubMed]
- Yu, W.; Hurley, J.; Roberts, D.; Chakrabortty, S.K.; Enderle, D.; Noerholm, M.; Breakefield, X.O.; Skog, J.K. Exosome-based liquid biopsies in cancer: Opportunities and challenges. Ann. Oncol. 2021, 32, 466–477. [Google Scholar] [CrossRef] [PubMed]
- Gurung, S.; Perocheau, D.; Touramanidou, L.; Baruteau, J. The exosome journey: From biogenesis to uptake and intracellular signalling. Cell. Commun. Signal. 2021, 19, 47. [Google Scholar] [CrossRef] [PubMed]
- Becker, A.; Thakur, B.K.; Weiss, J.M.; Kim, H.S.; Peinado, H.; Lyden, D. Extracellular Vesicles in Cancer: Cell-to-Cell Mediators of Metastasis. Cancer Cell 2016, 30, 836–848. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Valadi, H.; Ekstrom, K.; Bossios, A.; Sjostrand, M.; Lee, J.J.; Lotvall, J.O. Exosome-mediated transfer of mRNAs and microRNAs is a novel mechanism of genetic exchange between cells. Nat. Cell Biol. 2007, 9, 654–659. [Google Scholar] [CrossRef] [Green Version]
- Hagiwara, S.; Kantharidis, P.; Cooper, M.E. MicroRNA as biomarkers and regulator of cardiovascular development and disease. Curr. Pharm. Des. 2014, 20, 2347–2370. [Google Scholar] [CrossRef]
- Wang, J.; Yue, B.L.; Huang, Y.Z.; Lan, X.Y.; Liu, W.J.; Chen, H. Exosomal RNAs: Novel Potential Biomarkers for Diseases-A Review. Int. J. Mol. Sci. 2022, 23, 2461. [Google Scholar] [CrossRef]
- Xu, H.; Dong, X.; Chen, Y.; Wang, X. Serum exosomal hnRNPH1 mRNA as a novel marker for hepatocellular carcinoma. Clin. Chem. Lab. Med. 2018, 56, 479–484. [Google Scholar] [CrossRef]
- Skog, J.; Wurdinger, T.; van Rijn, S.; Meijer, D.H.; Gainche, L.; Sena-Esteves, M.; Curry, W.T., Jr.; Carter, B.S.; Krichevsky, A.M.; Breakefield, X.O. Glioblastoma microvesicles transport RNA and proteins that promote tumour growth and provide diagnostic biomarkers. Nat. Cell Biol. 2008, 10, 1470–1476. [Google Scholar] [CrossRef]
- Hong, B.S.; Cho, J.H.; Kim, H.; Choi, E.J.; Rho, S.; Kim, J.; Kim, J.H.; Choi, D.S.; Kim, Y.K.; Hwang, D.; et al. Colorectal cancer cell-derived microvesicles are enriched in cell cycle-related mRNAs that promote proliferation of endothelial cells. BMC Genom. 2009, 10, 556. [Google Scholar] [CrossRef]
- Dong, L.; Lin, W.; Qi, P.; Xu, M.D.; Wu, X.; Ni, S.; Huang, D.; Weng, W.W.; Tan, C.; Sheng, W.; et al. Circulating Long RNAs in Serum Extracellular Vesicles: Their Characterization and Potential Application as Biomarkers for Diagnosis of Colorectal Cancer. Cancer Epidemiol. Biomarkers Prev. 2016, 25, 1158–1166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baldacchino, S.; Grech, G. Somatic copy number aberrations in metastatic patients: The promise of liquid biopsies. Semin. Cancer Biol. 2020, 60, 302–310. [Google Scholar] [CrossRef]
- Valencia, K.; Montuenga, L.M. Exosomes in Liquid Biopsy: The Nanometric World in the Pursuit of Precision Oncology. Cancers 2021, 13, 2147. [Google Scholar] [CrossRef] [PubMed]
- Whiteside, T.L. Tumor-Derived Exosomes and Their Role in Cancer Progression. Adv. Clin. Chem. 2016, 74, 103–141. [Google Scholar]
- He, M.; Crow, J.; Roth, M.; Zeng, Y.; Godwin, A.K. Integrated immunoisolation and protein analysis of circulating exosomes using microfluidic technology. Lab Chip 2014, 14, 3773–3780. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Logozzi, M.; Angelini, D.F.; Giuliani, A.; Mizzoni, D.; Di Raimo, R.; Maggi, M.; Gentilucci, A.; Marzio, V.; Salciccia, S.; Borsellino, G.; et al. Increased Plasmatic Levels of PSA-Expressing Exosomes Distinguish Prostate Cancer Patients from Benign Prostatic Hyperplasia: A Prospective Study. Cancers 2019, 11, 1449. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Melo, S.A.; Luecke, L.B.; Kahlert, C.; Fernandez, A.F.; Gammon, S.T.; Kaye, J.; LeBleu, V.S.; Mittendorf, E.A.; Weitz, J.; Rahbari, N.; et al. Glypican-1 identifies cancer exosomes and detects early pancreatic cancer. Nature 2015, 523, 177–182. [Google Scholar] [CrossRef] [Green Version]
- Im, H.; Shao, H.; Park, Y.I.; Peterson, V.M.; Castro, C.M.; Weissleder, R.; Lee, H. Label-free detection and molecular profiling of exosomes with a nano-plasmonic sensor. Nat. Biotechnol. 2014, 32, 490–495. [Google Scholar] [CrossRef]


| ctDNA | CTC | Extracellular Vesicles | |
|---|---|---|---|
| Advantages | |||
| Detection of tumor heterogeneity | √ | √ | √ |
| Offers RNA- and DNA-based measurements | √ | √ | |
| Use of standardized methodology exemplified by CE-IVD platform for enumeration and capturing and ctDNA mutation and methylation assays | √ | √ | |
| Can monitor disease progression and relapse | √ | √ | |
| Disadvantages | |||
| Present in low numbers—large amount of sample required | √ | √ | |
| Patient-derived material is specific to a disease stage exemplified by CTC in metastatic disease | √ | ||
| Extensive analytic and clinical validity required | √ | √ |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Debattista, J.; Grech, L.; Scerri, C.; Grech, G. Copy Number Variations as Determinants of Colorectal Tumor Progression in Liquid Biopsies. Int. J. Mol. Sci. 2023, 24, 1738. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms24021738
Debattista J, Grech L, Scerri C, Grech G. Copy Number Variations as Determinants of Colorectal Tumor Progression in Liquid Biopsies. International Journal of Molecular Sciences. 2023; 24(2):1738. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms24021738
Chicago/Turabian StyleDebattista, Jessica, Laura Grech, Christian Scerri, and Godfrey Grech. 2023. "Copy Number Variations as Determinants of Colorectal Tumor Progression in Liquid Biopsies" International Journal of Molecular Sciences 24, no. 2: 1738. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms24021738