
Whole Transcriptome Analysis of Hypothalamus in Mice during Short-Term Starvation
 , ,
, ,
Abstract
:1. Introduction
2. Results
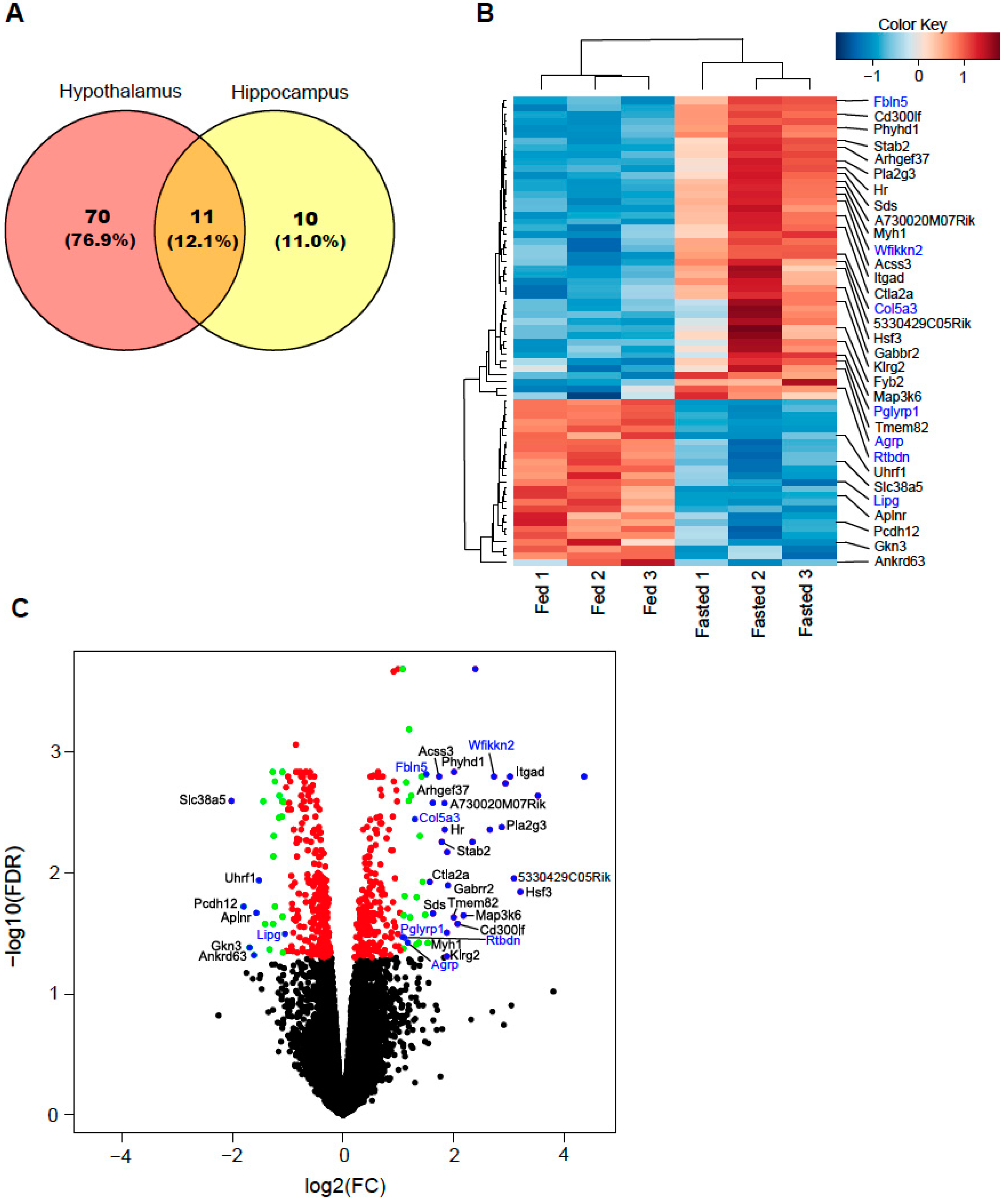
2.1. Differentially Expressed Genes
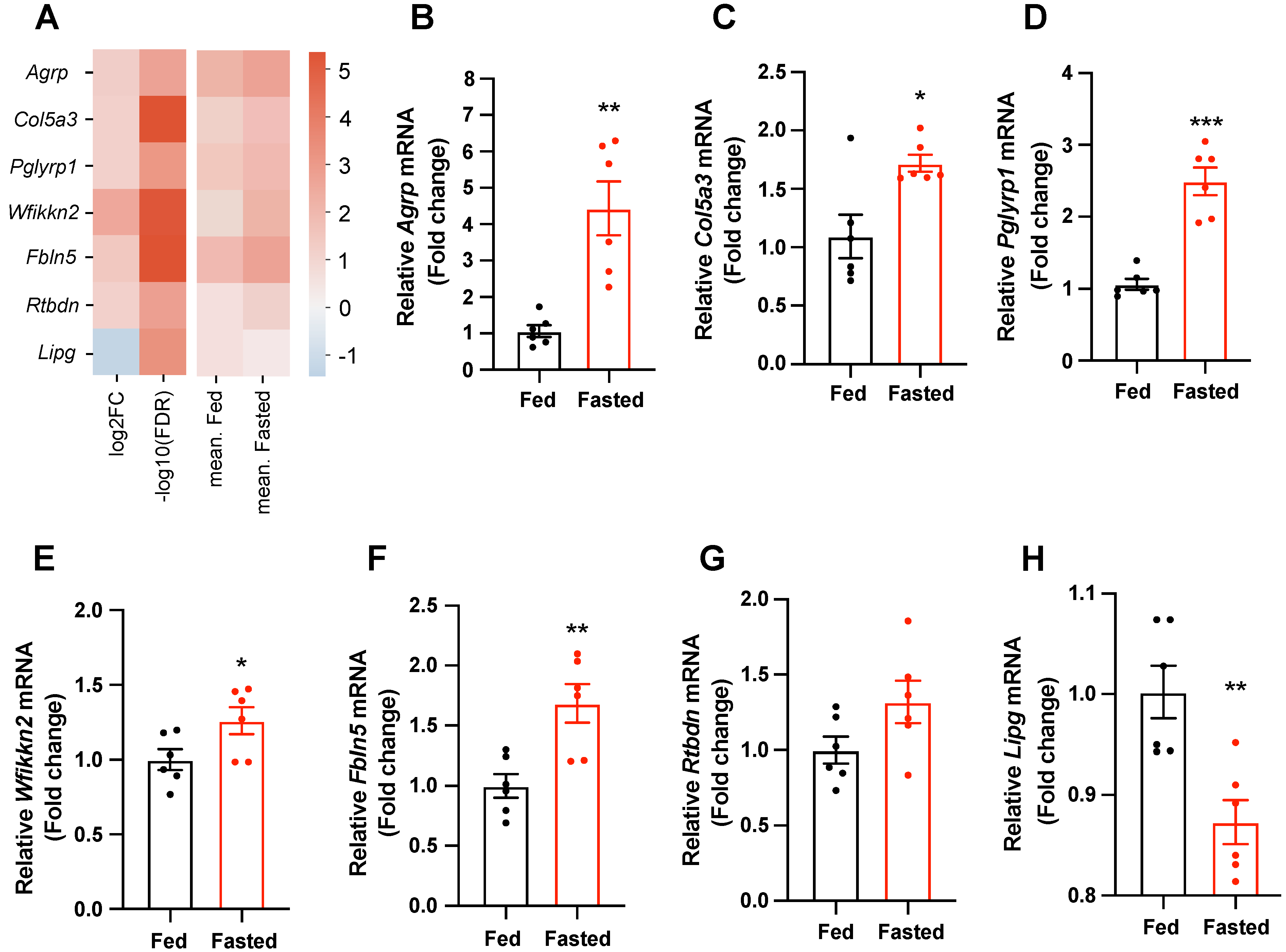
2.2. Secretory-Related Genes Are Altered in the Hypothalamus of Fasted Mice
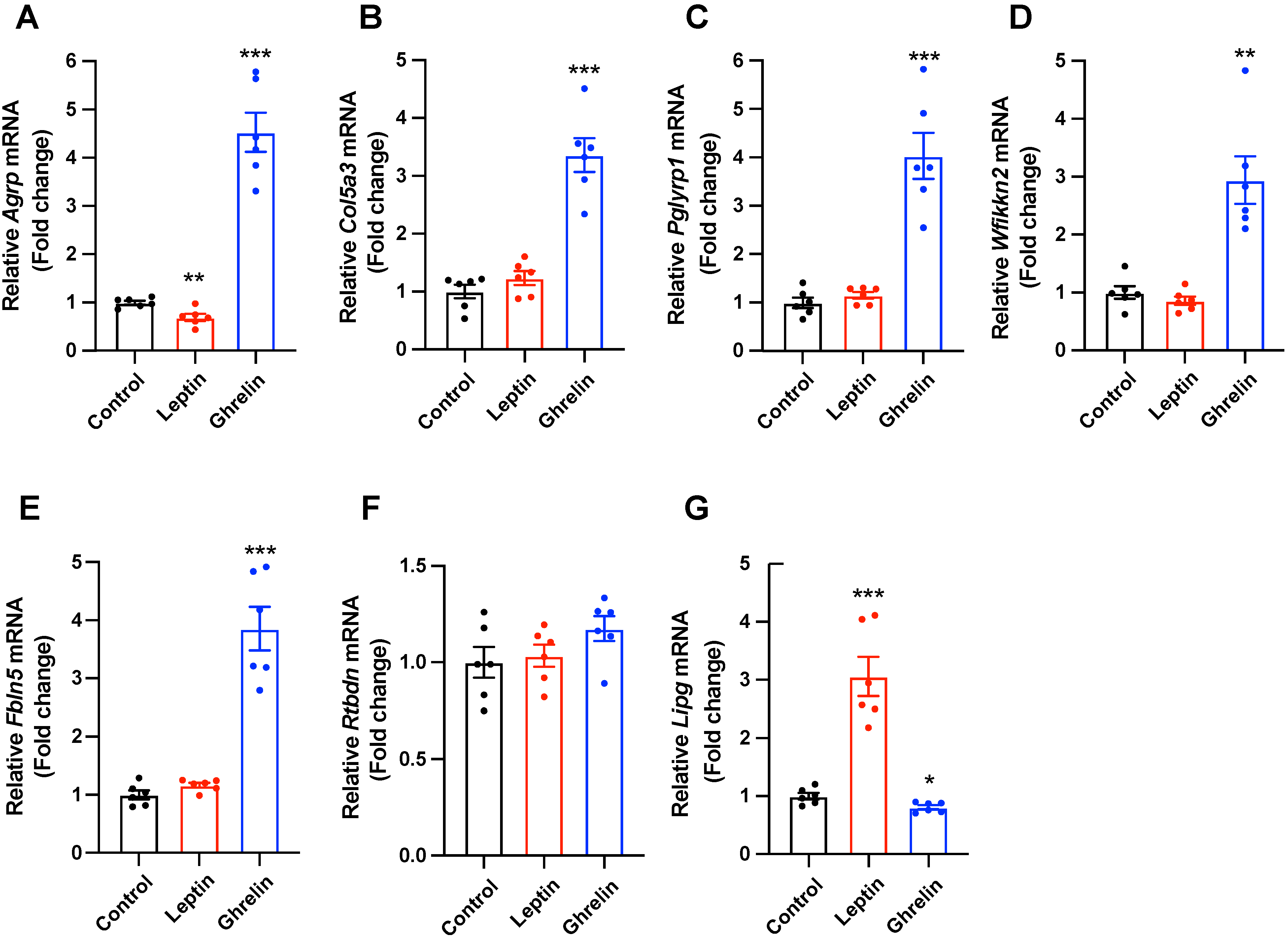
2.3. Selected Secretory-Related Genes Respond to the Metabolic Hormones
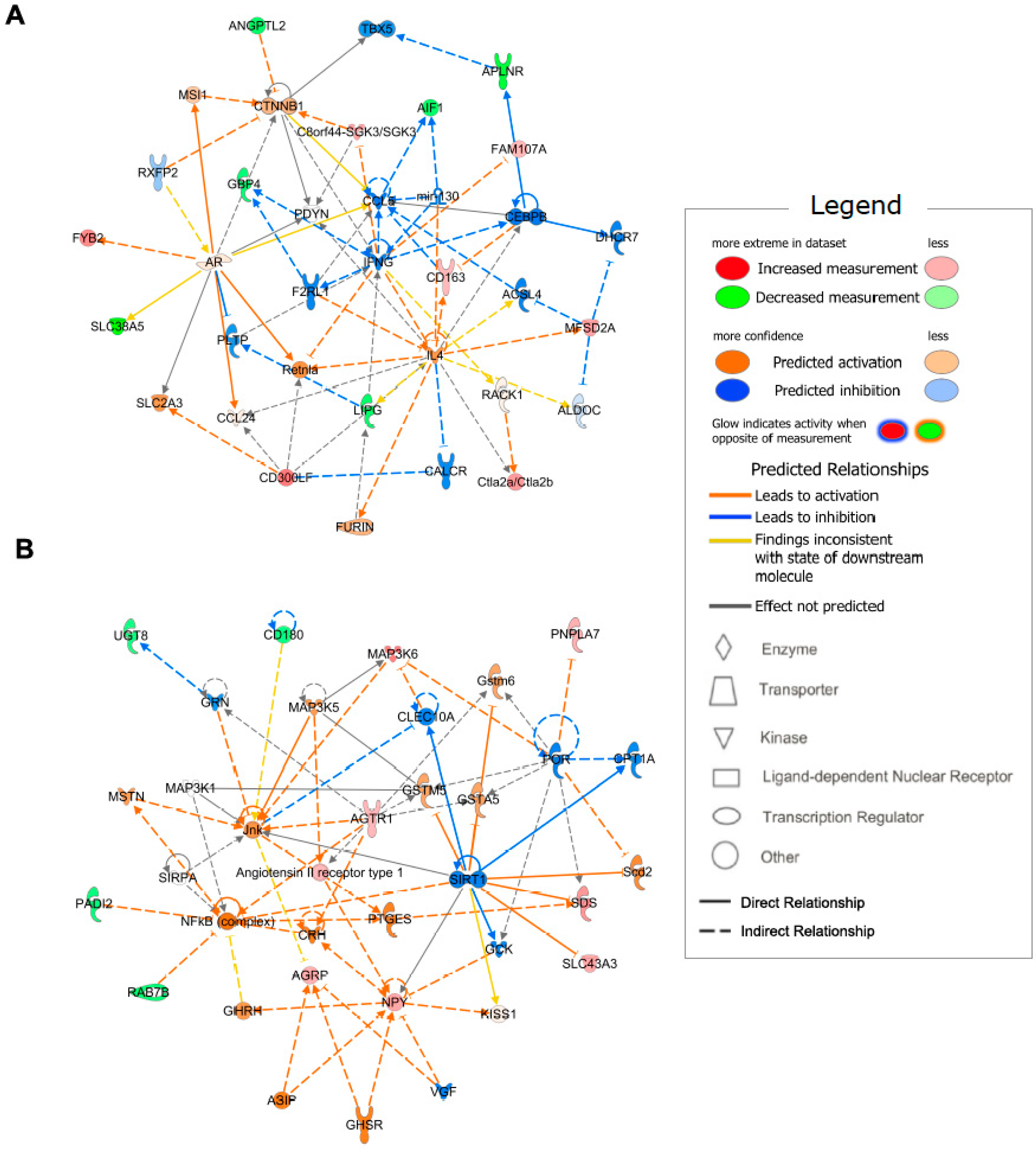
2.4. Enriched Functional Annotation and Canonical Pathway
2.5. Gene Network Identification
3. Discussion
4. Materials and Methods
4.1. Animals
4.2. Culture and Treatment of the Cells
4.3. Quantitative Real-Time Reverse Transcription-Polymerase Chain Reaction
4.4. Whole Transcriptome Sequencing
4.5. Whole Transcriptome Analysis
4.6. Differential Expression Genes Analysis, Secretory Genes Analysis, and Pathway Analysis
4.7. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Xu, Y.; Nedungadi, T.P.; Zhu, L.; Sobhani, N.; Irani, B.G.; Davis, K.E.; Zhang, X.; Zou, F.; Gent, L.M.; Hahner, L.D.; et al. Distinct Hypothalamic Neurons Mediate Estrogenic Effects on Energy Homeostasis and Reproduction. Cell Metab. 2011, 14, 453–465. [Google Scholar] [CrossRef]
- Nestor, C.C.; Qiu, J.; Padilla, S.L.; Zhang, C.; Bosch, M.A.; Fan, W.; Aicher, S.A.; Palmiter, R.D.; Rønnekleiv, O.K.; Kelly, M.J. Optogenetic Stimulation of Arcuate Nucleus Kiss1 Neurons Reveals a Steroid-Dependent Glutamatergic Input to POMC and AgRP Neurons in Male Mice. Mol. Endocrinol. 2016, 30, 630–644. [Google Scholar] [CrossRef]
- Hashikawa, K.; Hashikawa, Y.; Tremblay, R.; Zhang, J.; Feng, J.E.; Sabol, A.; Piper, W.T.; Lee, H.; Rudy, B.; Lin, D. Esr1 + Cells in the Ventromedial Hypothalamus Control Female Aggression. Nat. Neurosci. 2017, 20, 1580–1590. [Google Scholar] [CrossRef]
- Yang, T.; Yang, C.F.; Chizari, M.D.; Maheswaranathan, N.; Burke, K.J.; Borius, M.; Inoue, S.; Chiang, M.C.; Bender, K.J.; Ganguli, S.; et al. Social Control of Hypothalamus-Mediated Male Aggression. Neuron 2017, 95, 955–970. [Google Scholar] [CrossRef]
- Yu, S.; Qualls-Creekmore, E.; Rezai-Zadeh, K.; Jiang, Y.; Berthoud, H.-R.; Morrison, C.D.; Derbenev, A.V.; Zsombok, A.; Münzberg, H. Glutamatergic Preoptic Area Neurons That Express Leptin Receptors Drive Temperature-Dependent Body Weight Homeostasis. J. Neurosci. 2016, 36, 5034–5046. [Google Scholar] [CrossRef]
- Evans, M.C.; Anderson, G.M. Neuroendocrine Integration of Nutritional Signals on Reproduction. J. Mol. Endocrinol. 2017, 58, R107–R128. [Google Scholar] [CrossRef]
- Clarke, I.J.; Arbabi, L. New Concepts of the Central Control of Reproduction, Integrating Influence of Stress, Metabolic State, and Season. Domest. Anim. Endocrin. 2016, 56, S165–S179. [Google Scholar] [CrossRef]
- Andermann, M.L.; Lowell, B.B. Toward a Wiring Diagram Understanding of Appetite Control. Neuron 2017, 95, 757–778. [Google Scholar] [CrossRef]
- Jais, A.; Brüning, J.C. Hypothalamic Inflammation in Obesity and Metabolic Disease. J. Clin. Investig. 2017, 127, 24–32. [Google Scholar] [CrossRef]
- Timper, K.; Brüning, J.C. Hypothalamic Circuits Regulating Appetite and Energy Homeostasis: Pathways to Obesity. Dis. Model. Mech. 2017, 10, 679–689. [Google Scholar] [CrossRef] [Green Version]
- Miller, G.D. Appetite Regulation: Hormones, Peptides, and Neurotransmitters and Their Role in Obesity. Am. J. Lifestyle Med. 2017, 13, 586–601. [Google Scholar] [CrossRef]
- Guarnieri, D.J.; Brayton, C.E.; Richards, S.M.; Maldonado-Aviles, J.; Trinko, J.R.; Nelson, J.; Taylor, J.R.; Gourley, S.L.; DiLeone, R.J. Gene Profiling Reveals a Role for Stress Hormones in the Molecular and Behavioral Response to Food Restriction. Biol. Psychiat. 2012, 71, 358–365. [Google Scholar] [CrossRef]
- Li, J.-Y.; Kuick, R.; Thompson, R.C.; Misek, D.E.; Lai, Y.-M.; Liu, Y.-Q.; Chai, B.-X.; Hanash, S.M.; Gantz, I. Arcuate Nucleus Transcriptome Profiling Identifies Ankyrin Repeat and Suppressor of Cytokine Signalling Box-Containing Protein 4 as a Gene Regulated by Fasting in Central Nervous System Feeding Circuits. J. Neuroendocrinol. 2005, 17, 394–404. [Google Scholar] [CrossRef]
- Jovanovic, Z.; Tung, Y.C.L.; Lam, B.Y.H.; O’Rahilly, S.; Yeo, G.S.H. Identification of the Global Transcriptomic Response of the Hypothalamic Arcuate Nucleus to Fasting and Leptin. J. Neuroendocrinol. 2010, 22, 915–925. [Google Scholar] [CrossRef]
- Gley, K.; Murani, E.; Trakooljul, N.; Zebunke, M.; Puppe, B.; Wimmers, K.; Ponsuksili, S. Transcriptome Profiles of Hypothalamus and Adrenal Gland Linked to Haplotype Related to Coping Behavior in Pigs. Sci. Rep. 2019, 9, 13038. [Google Scholar] [CrossRef]
- Wang, C.; Ma, W. Hypothalamic and Pituitary Transcriptome Profiling Using RNA-Sequencing in High-Yielding and Low-Yielding Laying Hens. Sci. Rep. 2019, 9, 10285. [Google Scholar] [CrossRef]
- Gao, G.; Li, Q.; Zhao, X.; Ding, N.; Han, Q.; Su, J.; Wang, Q. Transcriptome Profiling of the Hypothalamus during Prelaying and Laying Periods in Sichuan White Geese (Anser Cygnoides): Transcriptome Profiling of Geese Hypothalamus. Anim. Sci. J. 2015, 86, 800–805. [Google Scholar] [CrossRef]
- Li, Z.; Liu, X.; Zhang, P.; Han, R.; Sun, G.; Jiang, R.; Wang, Y.; Liu, X.; Li, W.; Kang, X.; et al. Comparative Transcriptome Analysis of Hypothalamus-Regulated Feed Intake Induced by Exogenous Visfatin in Chicks. BMC Genom. 2018, 19, 249. [Google Scholar] [CrossRef]
- Meinken, J.; Walker, G.; Cooper, C.R.; Min, X.J. MetazSecKB: The Human and Animal Secretome and Subcellular Proteome Knowledgebase. Database 2015, 2015, bav077. [Google Scholar] [CrossRef] [PubMed]
- Acakpo-Satchivi, L.J.R.; Edelmann, W.; Sartorius, C.; Lu, B.D.; Wahr, P.A.; Watkins, S.C.; Metzger, J.M.; Leinwand, L.; Kucherlapati, R. Growth and Muscle Defects in Mice Lacking Adult Myosin Heavy Chain Genes. J. Cell Biol. 1997, 139, 1219–1229. [Google Scholar] [CrossRef] [PubMed]
- Sandu, C.; Rexhepaj, R.; Grahammer, F.; McCormick, J.A.; Henke, G.; Palmada, M.; Nammi, S.; Lang, U.; Metzger, M.; Just, L.; et al. Decreased Intestinal Glucose Transport in the Sgk3-Knockout Mouse. Pflügers Arch. 2005, 451, 437–444. [Google Scholar] [CrossRef] [PubMed]
- Geerling, J.J.; Wang, Y.; Havekes, L.M.; Romijn, J.A.; Rensen, P.C.N. Acute Central Neuropeptide Y Administration Increases Food Intake but Does Not Affect Hepatic Very Low-Density Lipoprotein (Vldl) Production in Mice. PLoS ONE 2013, 8, e55217. [Google Scholar] [CrossRef] [PubMed]
- Hohmann, J.G.; Teklemichael, D.N.; Weinshenker, D.; Wynick, D.; Clifton, D.K.; Steiner, R.A. Obesity and Endocrine Dysfunction in Mice with Deletions of Both Neuropeptide Y and Galanin. Mol. Cell Biol. 2004, 24, 2978–2985. [Google Scholar] [CrossRef] [PubMed]
- Redmann, S.M.; Argyropoulos, G. AgRP-Deficiency Could Lead to Increased Lifespan. Biochem. Biophys. Res. Commun. 2006, 351, 860–864. [Google Scholar] [CrossRef]
- Benoit, S.C.; Tracy, A.L. Behavioral Controls of Food Intake. Peptides 2007, 29, 139–147. [Google Scholar] [CrossRef] [PubMed]
- Mena, J.D.; Sadeghian, K.; Baldo, B.A. Induction of Hyperphagia and Carbohydrate Intake by μ-Opioid Receptor Stimulation in Circumscribed Regions of Frontal Cortex. J. Neurosci. Off. J. Soc. Neurosci. 2011, 31, 3249–3260. [Google Scholar] [CrossRef]
- González-García, I.; Fernø, J.; Diéguez, C.; Nogueiras, R.; López, M. Hypothalamic Lipids: Key Regulators of Whole Body Energy Balance. Neuroendocrinology 2017, 104, 398–411. [Google Scholar] [CrossRef]
- Song, N.; Jeong, D.Y.; Tu, T.H.; Park, B.S.; Yang, H.R.; Kim, Y.J.; Kim, J.K.; Park, J.T.; Yeh, J.-Y.; Yang, S.; et al. Adiponectin Controls Nutrient Availability in Hypothalamic Astrocytes. Int. J. Mol. Sci. 2021, 22, 1587. [Google Scholar] [CrossRef]
- Wang, Y.; Wu, R. The Effect of Fasting on Human Metabolism and Psychological Health. Dis. Markers 2022, 2022, 5653739. [Google Scholar] [CrossRef]
- Roh, E.; Kim, M.-S. Brain Regulation of Energy Metabolism. Endocrinol. Metab. 2016, 31, 519–524. [Google Scholar] [CrossRef] [Green Version]
- Tran, L.T.; Park, S.; Kim, S.K.; Lee, J.S.; Kim, K.W.; Kwon, O. Hypothalamic Control of Energy Expenditure and Thermogenesis. Exp. Mol. Med. 2022, 54, 358–369. [Google Scholar] [CrossRef]
- Kim, Y.J.; Kang, D.; Yang, H.R.; Park, B.S.; Tu, T.H.; Jeong, B.; Lee, B.J.; Kim, J.K.; Kim, J.G. Metabolic Profiling of the Hypothalamus of Mice during Short-Term Food Deprivation. Metabolites 2022, 12, 407. [Google Scholar] [CrossRef]
- Park, B.S.; Kim, Y.J.; Jeong, D.Y.; Kim, Y.T.; Kim, J.K.; Lee, B.J.; Kim, J.G. Enhanced Lipid Utilization Is Coupled to the Sickness Responses Triggered by Lipopolysaccharide. Biochem. Biophys. Res. Commun. 2021, 558, 44–50. [Google Scholar] [CrossRef] [PubMed]
- Zaghlool, S.B.; Sharma, S.; Molnar, M.; Matías-García, P.R.; Elhadad, M.A.; Waldenberger, M.; Peters, A.; Rathmann, W.; Graumann, J.; Gieger, C.; et al. Revealing the Role of the Human Blood Plasma Proteome in Obesity Using Genetic Drivers. Nat. Commun. 2021, 12, 1279. [Google Scholar] [CrossRef] [PubMed]
- González, O.; Tobia, C.; Ebersole, J.; Novak, M. Caloric Restriction and Chronic Inflammatory Diseases. Oral Dis. 2012, 18, 16–31. [Google Scholar] [CrossRef]
- Martin, M. Cutadapt Removes Adapter Sequences from High-Throughput Sequencing Reads. EMBnet. J. 2011, 17, 10–12. [Google Scholar] [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A Flexible Trimmer for Illumina Sequence Data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast Universal RNA-Seq Aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef] [PubMed]
- Liao, Y.; Smyth, G.K.; Shi, W. featureCounts: An Efficient General Purpose Program for Assigning Sequence Reads to Genomic Features. Bioinformatics 2014, 30, 923–930. [Google Scholar] [CrossRef]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. edgeR: A Bioconductor Package for Differential Expression Analysis of Digital Gene Expression Data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef] [Green Version]
- Durinck, S.; Spellman, P.T.; Birney, E.; Huber, W. Mapping Identifiers for the Integration of Genomic Datasets with the R/Bioconductor Package biomaRt. Nat. Protoc. 2009, 4, 1184–1191. [Google Scholar] [CrossRef] [PubMed]
- Waskom, M. Seaborn: Statistical Data Visualization. J. Open Source Softw. 2021, 6, 3021. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Description | log2FC | p-Value | FDR |
|---|---|---|---|---|
| Phyhd1 | phytanoyl-CoA dioxygenase domain containing 1 | 2.01 | 1.10 × 10−6 | 1.47 × 10−3 |
| Fbln5 | fibulin 5 | 1.51 | 1.70 × 10−6 | 1.54 × 10−3 |
| Acss3 | acyl-CoA synthetase short chain family member 3 | 1.74 | 2.81 × 10−6 | 1.61 × 10−3 |
| Wfikkn2 | WAP, follistatin/kazal, immunoglobulin, kunitz and netrin domain containing 2 | 2.73 | 2.43 × 10−6 | 1.61 × 10−3 |
| Col5a3 | collagen type V alpha 3 chain | 1.14 | 4.44 × 10−6 | 1.80 × 10−3 |
| Itgad | integrin subunit alpha D | 2.94 | 4.65 × 10−6 | 1.83 × 10−3 |
| Slc38a5 | solute carrier family 38 member 5 | −2.02 | 9.71 × 10−6 | 2.56 × 10−3 |
| Arhgef37 | Rho guanine nucleotide exchange factor 37 | 1.63 | 1.14 × 10−5 | 2.65 × 10−3 |
| A730020M07Rik | RIKEN cDNA A730020M07 gene | 1.84 | 1.16 × 10−5 | 2.67 × 10−3 |
| Pla2g3 | phospholipase A2 group III | 2.87 | 2.41 × 10−5 | 4.21 × 10−3 |
| Hr | HR lysine demethylase and nuclear receptor corepressor | 1.84 | 2.87 × 10−5 | 4.41 × 10−3 |
| Stab2 | stabilin 2 | 1.79 | 4.20 × 10−5 | 5.57 × 10−3 |
| 5330429C05Rik | RIKEN cDNA 5330429C05 gene | 3.09 | 1.15 × 10−4 | 1.12 × 10−2 |
| Uhrf1 | ubiquitin like with PHD and ring finger domains 1 | −1.52 | 1.22 × 10−4 | 1.16 × 10−2 |
| Ctla2a | cytotoxic T lymphocyte-associated protein 2 alpha | 1.57 | 1.30 × 10−4 | 1.19 × 10−2 |
| Gabrr2 | gamma-aminobutyric acid type A receptor subunit rho2 | 1.90 | 1.50 × 10−4 | 1.28 × 10−2 |
| Hsf3 | heat shock transcription factor 3 | 3.21 | 1.84 × 10−4 | 1.44 × 10−2 |
| Pcdh12 | protocadherin 12 | −1.80 | 3.05 × 10−4 | 1.91 × 10−2 |
| Aplnr | apelin receptor | −1.57 | 3.91 × 10−4 | 2.15 × 10−2 |
| Sds | serine dehydratase | 1.63 | 4.00 × 10−4 | 2.17 × 10−2 |
| Tmem82 | transmembrane protein 82 | 2.18 | 4.23 × 10−4 | 2.25 × 10−2 |
| Map3k6 | mitogen-activated protein kinase kinase kinase 6 | 2.01 | 4.63 × 10−4 | 2.33 × 10−2 |
| Cd300lf | CD300 molecule like family member f | 2.07 | 5.76 × 10−4 | 2.63 × 10−2 |
| Lipg | lipase G, endothelial type | −1.26 | 5.80 × 10−4 | 2.64 × 10−2 |
| Pglyrp1 | peptidoglycan recognition protein 1 | 1.08 | 8.66 × 10−4 | 3.39 × 10−2 |
| Rtbdn | retbindin | 1.17 | 1.02 × 10−3 | 3.76 × 10−2 |
| Myh1 | myosin heavy chain 1 | 1.54 | 1.05 × 10−3 | 3.79 × 10−2 |
| Gkn3 | gastrokine 3 | −1.69 | 1.21 × 10−3 | 4.14 × 10−2 |
| Ankrd63 | ankyrin repeat domain 63 | −1.61 | 1.62 × 10−3 | 4.78 × 10−2 |
| Klrg2 | killer cell lectin like receptor G2 | 1.88 | 1.67 × 10−3 | 4.89 × 10−2 |
| Fyb2 | FYN binding protein 2 | 1.83 | 1.73 × 10−3 | 5.01 × 10−2 |
| Agrp | agouti related neuropeptide | 1.24 | 1.98 × 10−3 | 5.38 × 10−2 |
| Functional Annotation | p-Value | Genes |
|---|---|---|
| Ingestion by mice | 2.03 × 10−4 | AGRP, AGTR1, APLNR, C8orf44-SGK3/SGK3, MYH1, NPY |
| Quantity of lysophosphatidylcholine | 1.75 × 10−3 | MFSD2A, PLA2G3 |
| Hyperphagia | 2.07 × 10−3 | AGRP, AGTR1, NPY |
| Formation of actin bundles | 2.14 × 10−3 | AIF1, FAM107A |
| Appetite | 2.56 × 10−3 | AGRP, NPY |
| Function of vascular endothelial cells | 2.79 × 10−3 | LIPG, MFSD2A |
| Abnormal skeletal muscle fiber type ratio | 3.03 × 10−3 | MYH1, WFIKKN2 |
| Cutis laxa | 3.28 × 10−3 | FBLN5 |
| Transmembrane potential of mitochondrial outer membrane | 3.28 × 10−3 | HR |
| Demyelination of cervical spinal cord | 3.28 × 10−3 | CD300LF |
| Organization of elastic fibers | 3.28 × 10−3 | FBLN5 |
| Activation of BMMC cells | 3.28 × 10−3 | CD300LF |
| Internalization of phospholipid | 3.28 × 10−3 | MFSD2A |
| Elastinopathy | 3.28 × 10−3 | FBLN5 |
| Transport of lysophosphatidic acid | 3.28 × 10−3 | MFSD2A |
| Hydrolysis of 1-oleoyl lysophosphatidylcholine | 3.28 × 10−3 | PNPLA7 |
| Development of infundibulum of hair follicle | 3.28 × 10−3 | HR |
| Infection by Norovirus | 3.28 × 10−3 | CD300LF |
| Obesity of epididymal fat | 3.28 × 10−3 | AGTR1 |
| Synthesis of neointima | 3.28 × 10−3 | AGTR1 |
| Ingenuity Canonical Pathways | −log(p-Value) | Ratio | Molecules |
|---|---|---|---|
| Apelin Cardiac Fibroblast Signaling Pathway | 2.59 | 0.087 | AGTR1, APLNR |
| Apelin Liver Signaling Pathway | 2.49 | 0.077 | APLNR, COL5A3 |
| Thiosulfate Disproportionation III (Rhodanese) | 2.01 | 0.333 | TST |
| L-serine Degradation | 2.01 | 0.333 | SDS |
| Acetate Conversion to Acetyl-CoA | 1.88 | 0.250 | ACSS3 |
| Triacylglycerol Degradation | 1.87 | 0.037 | LIPG, PNPLA7 |
| Protein Citrullination | 1.79 | 0.200 | PADI2 |
| Phospholipases | 1.75 | 0.032 | LIPG, PLA2G3 |
| Gustation Pathway | 1.63 | 0.016 | GABRR2, LIPG, PKD2L1 |
| Hepatic Fibrosis/Hepatic Stellate Cell Activation | 1.63 | 0.016 | AGTR1, COL5A3, MYH1 |
| Leptin Signaling in Obesity | 1.60 | 0.027 | AGRP, NPY |
| VDR/RXR Activation | 1.57 | 0.026 | HR, SERPINB1 |
| RHOGDI Signaling | 1.51 | 0.014 | CDH19, ITGAD, MYH1 |
| Glycine Betaine Degradation | 1.49 | 0.100 | SDS |
| Ketogenesis | 1.45 | 0.091 | HMGCS2 |
| Mevalonate Pathway I | 1.38 | 0.077 | HMGCS2 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Oh, E.-Y.; Park, B.S.; Yang, H.R.; Lee, H.G.; Tu, T.H.; Yang, S.; Han, M.-R.; Kim, J.G. Whole Transcriptome Analysis of Hypothalamus in Mice during Short-Term Starvation. Int. J. Mol. Sci. 2023, 24, 3204. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms24043204
Oh E-Y, Park BS, Yang HR, Lee HG, Tu TH, Yang S, Han M-R, Kim JG. Whole Transcriptome Analysis of Hypothalamus in Mice during Short-Term Starvation. International Journal of Molecular Sciences. 2023; 24(4):3204. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms24043204
Chicago/Turabian StyleOh, Eun-Young, Byong Seo Park, Hye Rim Yang, Ho Gyun Lee, Thai Hien Tu, Sunggu Yang, Mi-Ryung Han, and Jae Geun Kim. 2023. "Whole Transcriptome Analysis of Hypothalamus in Mice during Short-Term Starvation" International Journal of Molecular Sciences 24, no. 4: 3204. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms24043204