
Multi-Omics Profiling of Hypertrophic Cardiomyopathy Reveals Altered Mechanisms in Mitochondrial Dynamics and Excitation–Contraction Coupling

 , , , , ,
, , , , ,
Abstract
:1. Introduction
2. Results
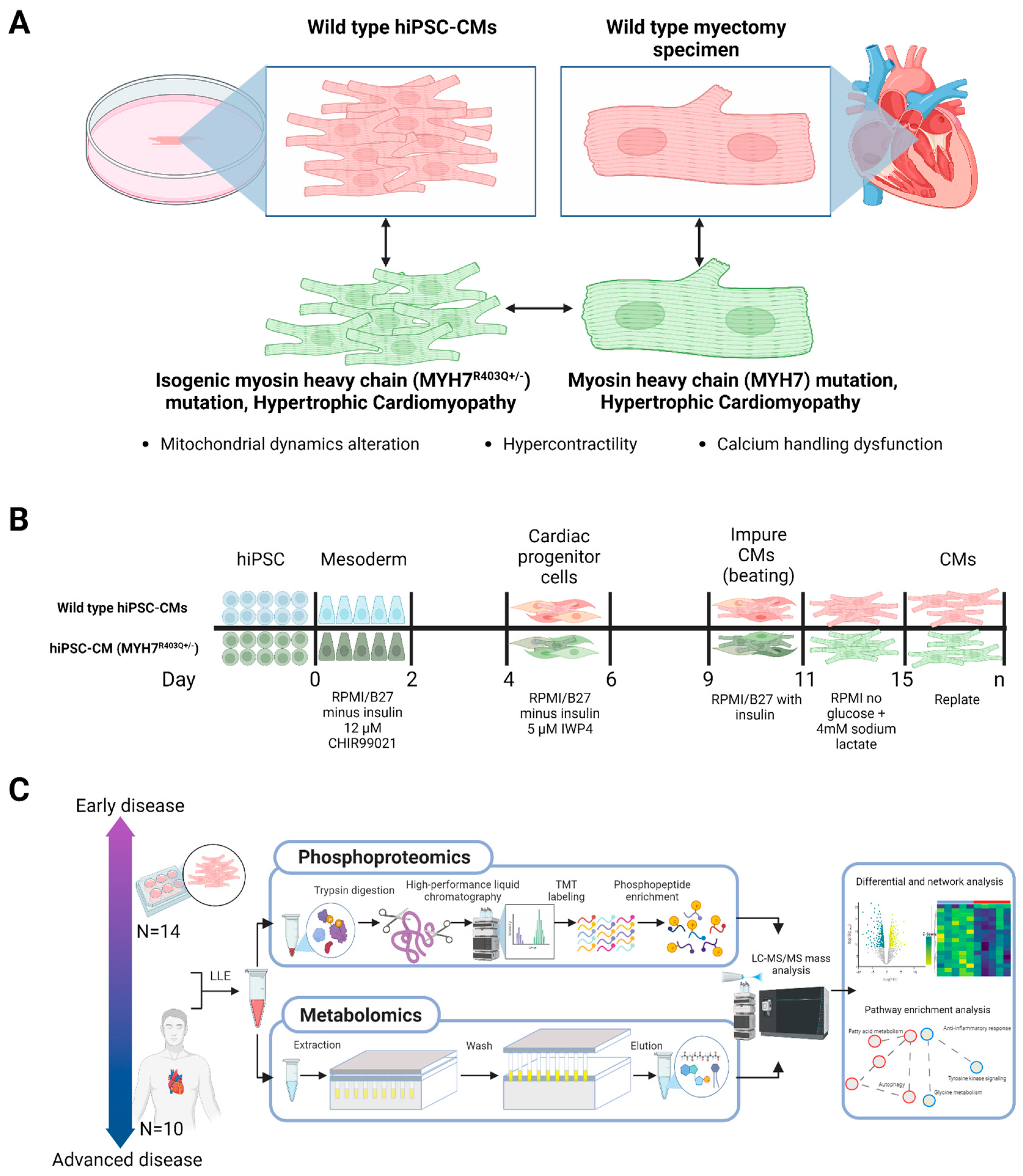
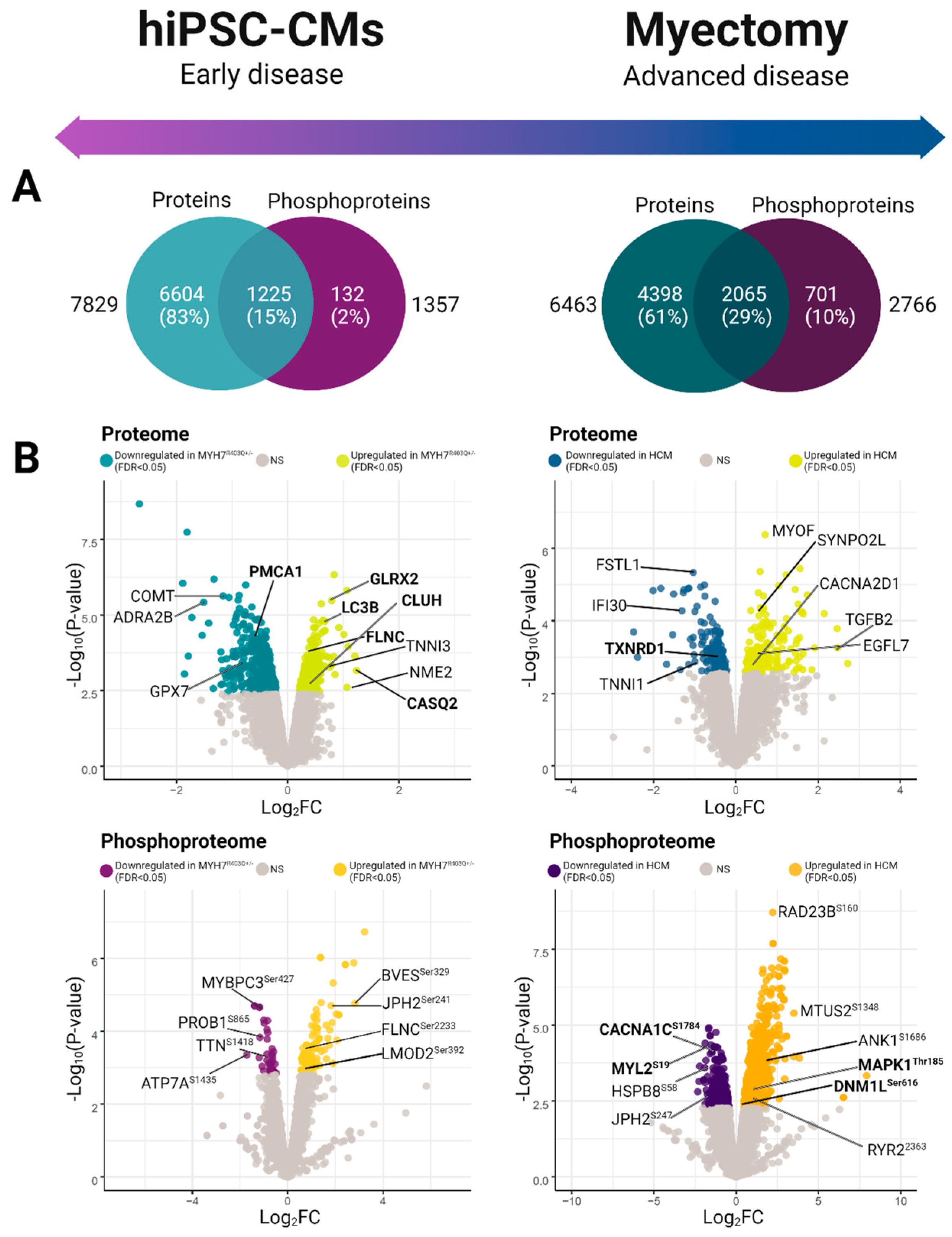
2.1. Differential Phosphoproteomics in Early HCM Specimens
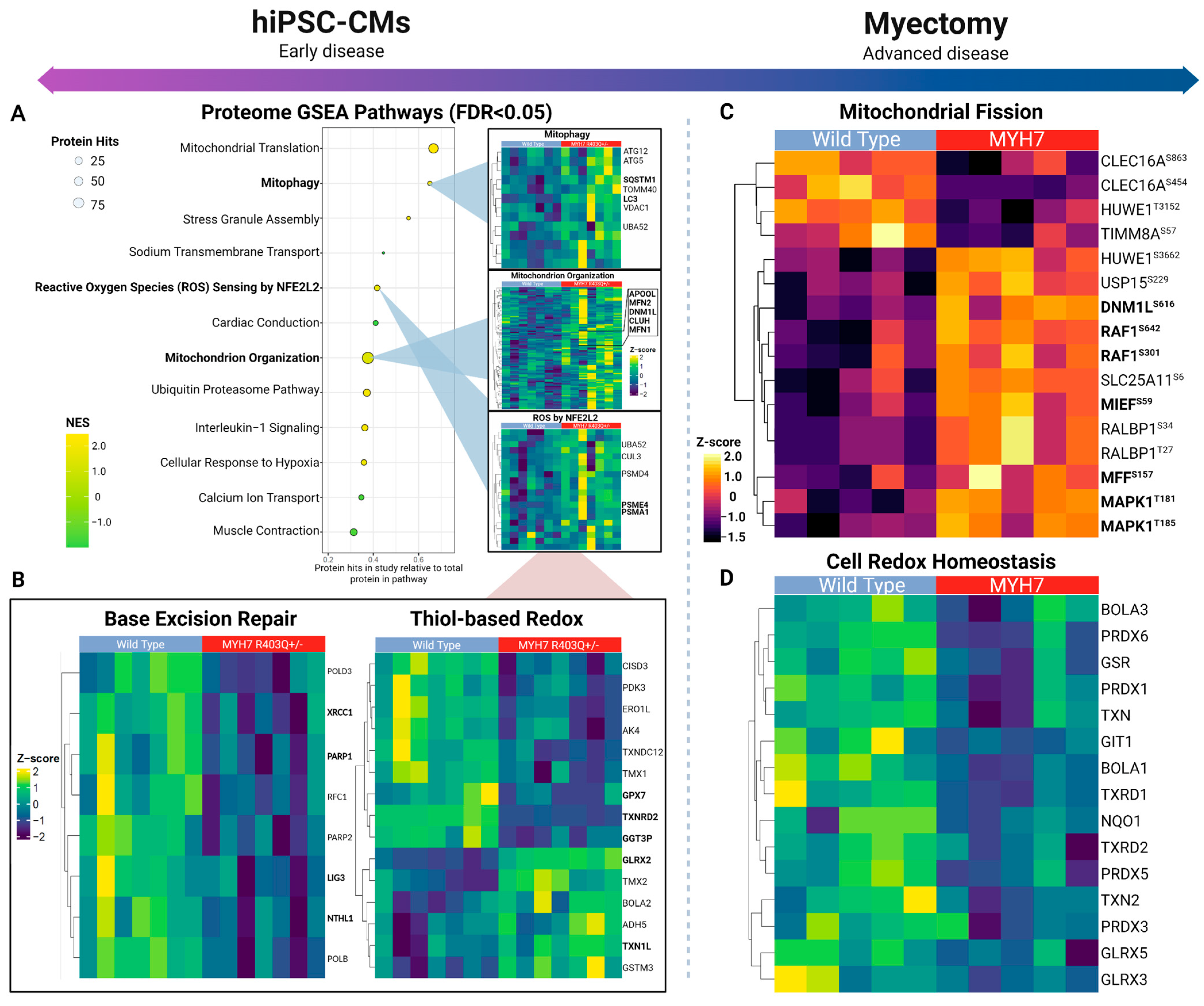
2.2. Pathway-Level Changes in Early Model of HCM Reveal Altered Mitochondrial Dynamics in Early HCM Specimens
2.3. Insufficient Oxidative Stress Response Associated with Mutant hiPSC-CMs
2.4. Overlapping Mechanisms Driving Mitochondrial Dynamics and Downregulation of Oxidative Stress Response in Advanced HCM
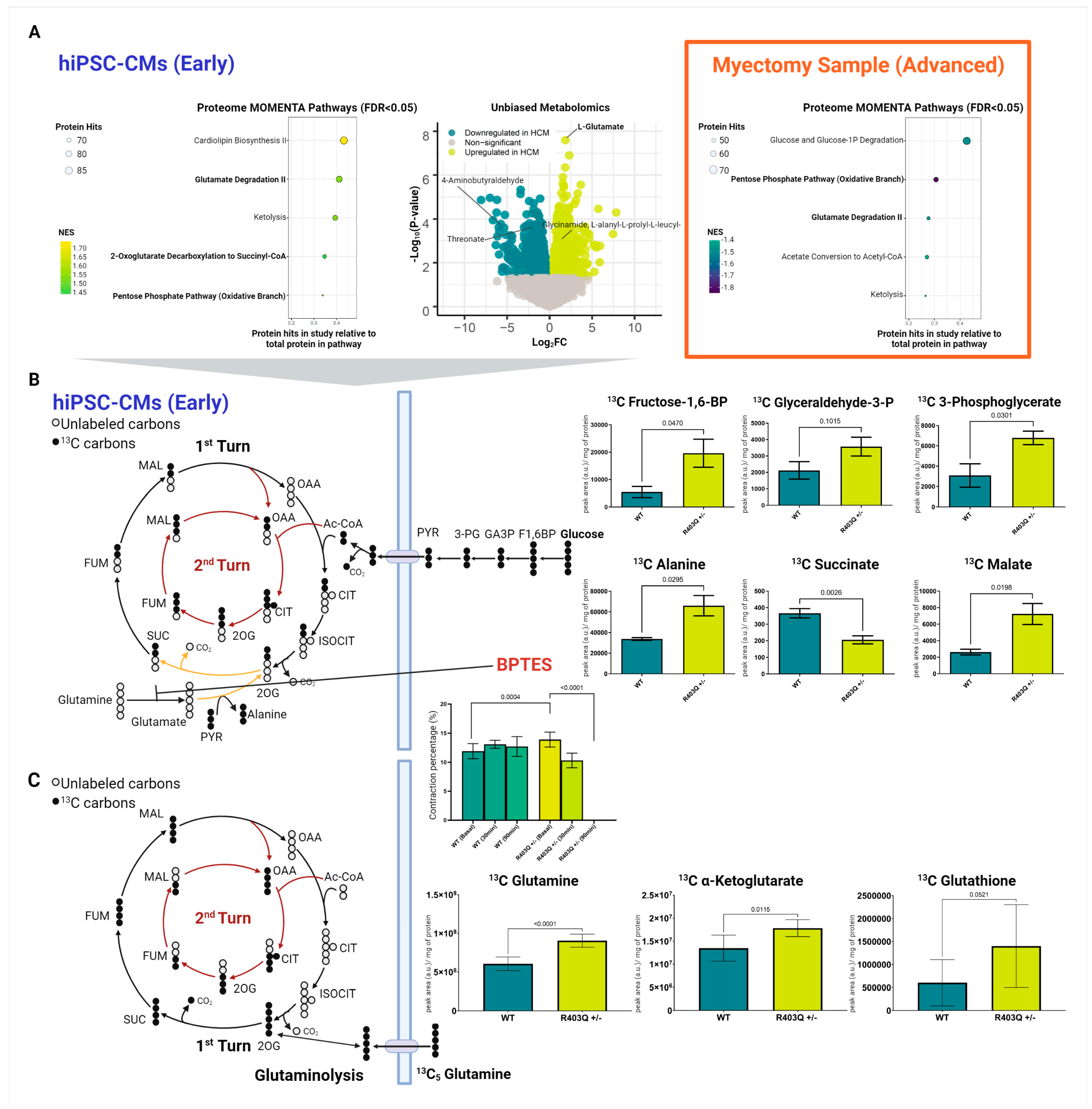
2.5. Integrated Metabolic Network Analysis Reveals Increased Dependence on Glutaminolysis in Early HCM
2.6. Metabolic Network Analysis Reveals Decompensated Metabolism in Advanced Specimens
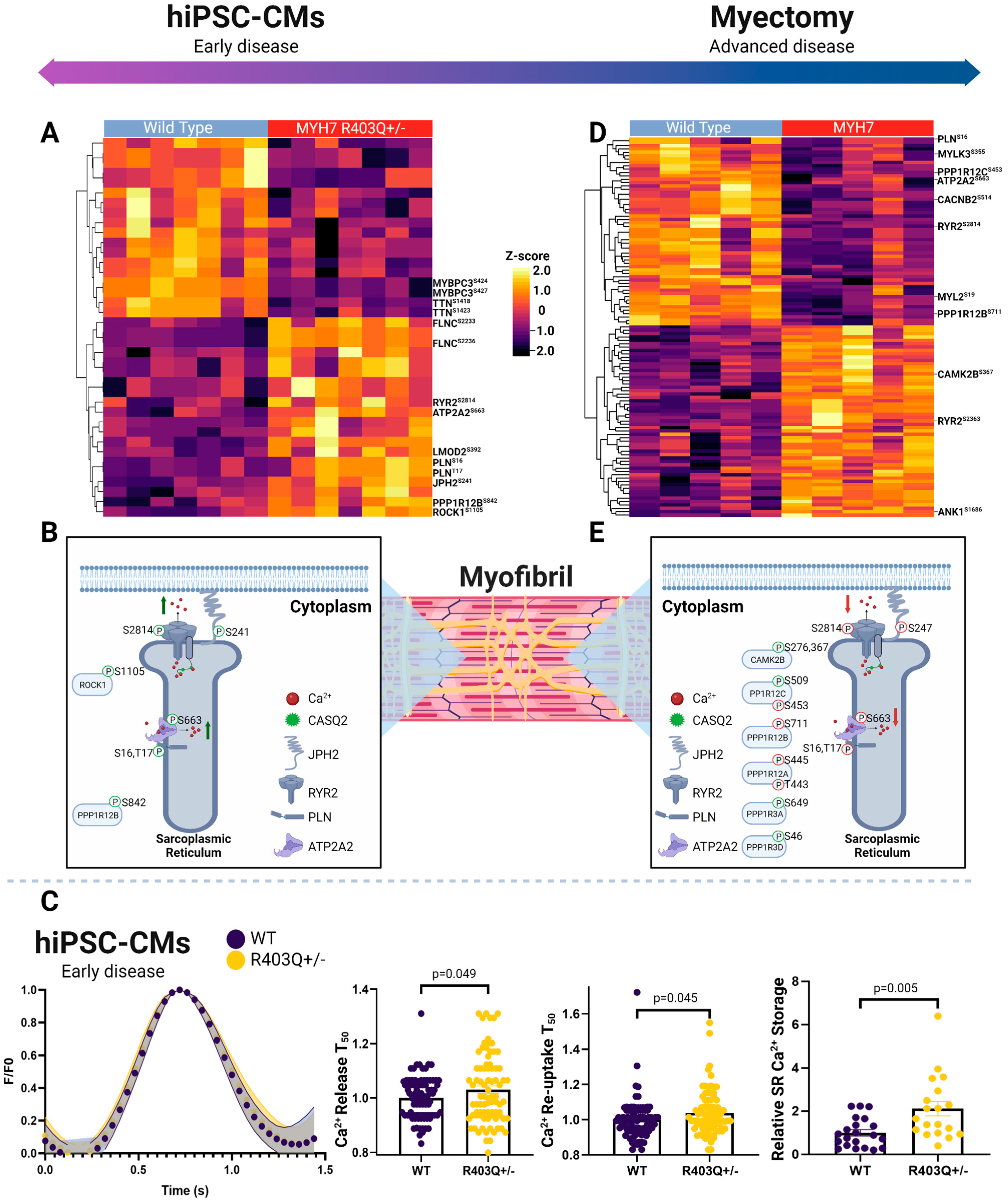
2.7. Phosphorylation-Dependent Regulation of Excitation Contraction Coupling in hiPSC-CMs
2.8. Phosphorylation-Dependent Regulation of Excitation Contraction Coupling in Myectomy Specimens
3. Discussion
4. Materials and Methods
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Geske, J.B.; Ommen, S.R.; Gersh, B.J. Hypertrophic Cardiomyopathy: Clinical Update. JACC Heart Fail 2018, 6, 364–375. [Google Scholar] [CrossRef] [PubMed]
- Writing Committee Members; Ommen, S.R.; Mital, S.; Burke, M.A.; Day, S.M.; Deswal, A.; Elliott, P.; Evanovich, L.L.; Hung, J.; Joglar, J.A.; et al. 2020 AHA/ACC Guideline for the Diagnosis and Treatment of Patients With Hypertrophic Cardiomyopathy. Circulation 2020, 142, e558–e631. [Google Scholar] [CrossRef] [PubMed]
- Marian Ali, J. Braunwald Eugene Hypertrophic Cardiomyopathy. Circ. Res. 2017, 121, 749–770. [Google Scholar] [CrossRef]
- Akhtar, M.; Elliott, P. The Genetics of Hypertrophic Cardiomyopathy. Glob. Cardiol. Sci. Pract. 2018, 2018. [Google Scholar] [CrossRef] [Green Version]
- Viola, H.M.; Hool, L.C. Impaired Calcium Handling and Mitochondrial Metabolic Dysfunction as Early Markers of Hypertrophic Cardiomyopathy. Arch. Biochem. Biophys. 2019, 665, 166–174. [Google Scholar] [CrossRef]
- Geisterfer-Lowrance, A.A.T.; Kass, S.; Tanigawa, G.; Vosberg, H.-P.; McKenna, W.; Seidman, C.E.; Seidman, J.G. A Molecular Basis for Familial Hypertrophic Cardiomyopathy: A β Cardiac Myosin Heavy Chain Gene Missense Mutation. Cell 1990, 62, 999–1006. [Google Scholar] [CrossRef] [PubMed]
- Landstrom, A.P.; Ackerman, M.J. Mutation Type Is Not Clinically Useful in Predicting Prognosis in Hypertrophic Cardiomyopathy. Circulation 2010, 122, 2441–2450. [Google Scholar] [CrossRef] [Green Version]
- Witjas-Paalberends, E.R.; Ferrara, C.; Scellini, B.; Piroddi, N.; Montag, J.; Tesi, C.; Stienen, G.J.M.; Michels, M.; Ho, C.Y.; Kraft, T.; et al. Faster Cross-Bridge Detachment and Increased Tension Cost in Human Hypertrophic Cardiomyopathy with the R403Q MYH7 Mutation. J. Physiol. 2014, 592, 3257–3272. [Google Scholar] [CrossRef]
- Ranjbarvaziri, S.; Kooiker, K.B.; Ellenberger, M.; Fajardo, G.; Zhao, M.; Vander Roest, A.S.; Woldeyes, R.A.; Koyano, T.T.; Fong, R.; Ma, N.; et al. Altered Cardiac Energetics and Mitochondrial Dysfunction in Hypertrophic Cardiomyopathy. Circulation 2021, 144, 1714–1731. [Google Scholar] [CrossRef]
- Anderson, R.L.; Trivedi, D.V.; Sarkar, S.S.; Henze, M.; Ma, W.; Gong, H.; Rogers, C.S.; Gorham, J.M.; Wong, F.L.; Morck, M.M.; et al. Deciphering the Super Relaxed State of Human β-Cardiac Myosin and the Mode of Action of Mavacamten from Myosin Molecules to Muscle Fibers. Proc. Natl. Acad. Sci. USA 2018, 115, E8143–E8152. [Google Scholar] [CrossRef] [Green Version]
- McNamara, J.W.; Li, A.; Lal, S.; Bos, J.M.; Harris, S.P.; van der Velden, J.; Ackerman, M.J.; Cooke, R.; Remedios, C.G. dos MYBPC3 Mutations Are Associated with a Reduced Super-Relaxed State in Patients with Hypertrophic Cardiomyopathy. PLoS ONE 2017, 12, e0180064. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coppini, R.; Ferrantini, C.; Mugelli, A.; Poggesi, C.; Cerbai, E. Altered Ca2+ and Na+ Homeostasis in Human Hypertrophic Cardiomyopathy: Implications for Arrhythmogenesis. Front. Physiol. 2018, 9, 1391. [Google Scholar] [CrossRef] [PubMed]
- Wijnker, P.J.M.; van der Velden, J. Mutation-Specific Pathology and Treatment of Hypertrophic Cardiomyopathy in Patients, Mouse Models and Human Engineered Heart Tissue. Biochim. Biophys. Acta (BBA)-Mol. Basis Dis. 2020, 1866, 165774. [Google Scholar] [CrossRef] [PubMed]
- Cohn, R.; Thakar, K.; Lowe, A.; Ladha, F.A.; Pettinato, A.M.; Romano, R.; Meredith, E.; Chen, Y.-S.; Atamanuk, K.; Huey, B.D.; et al. A Contraction Stress Model of Hypertrophic Cardiomyopathy Due to Sarcomere Mutations. Stem Cell Rep. 2019, 12, 71–83. [Google Scholar] [CrossRef] [Green Version]
- Wijnker, P.J.M.; Sequeira, V.; Kuster, D.W.D.; van der Velden, J. Hypertrophic Cardiomyopathy: A Vicious Cycle Triggered by Sarcomere Mutations and Secondary Disease Hits. Antioxid. Redox Signal. 2019, 31, 318–358. [Google Scholar] [CrossRef]
- Sequeira, V.; Bertero, E.; Maack, C. Energetic Drain Driving Hypertrophic Cardiomyopathy. FEBS Lett. 2019, 593, 1616–1626. [Google Scholar] [CrossRef] [Green Version]
- Jung, W.-I.; Sieverding, L.; Breuer, J.; Hoess, T.; Widmaier, S.; Schmidt, O.; Bunse, M.; van Erckelens, F.; Apitz, J.; Lutz, O.; et al. 31P NMR Spectroscopy Detects Metabolic Abnormalities in Asymptomatic Patients With Hypertrophic Cardiomyopathy. Circulation 1998, 97, 2536–2542. [Google Scholar] [CrossRef] [Green Version]
- Senthil, V.; Chen, S.N.; Tsybouleva, N.; Halder, T.; Nagueh, S.F.; Willerson, J.T.; Roberts, R.; Marian, A.J. Prevention of Cardiac Hypertrophy by Atorvastatin in a Transgenic Rabbit Model of Human Hypertrophic Cardiomyopathy. Circ. Res. 2005, 97, 285–292. [Google Scholar] [CrossRef] [Green Version]
- Moore, J.; Emili, A. Mass-Spectrometry-Based Functional Proteomic and Phosphoproteomic Technologies and Their Application for Analyzing Ex Vivo and In Vitro Models of Hypertrophic Cardiomyopathy. Int. J. Mol. Sci. 2021, 22, 13644. [Google Scholar] [CrossRef]
- Kuzmanov, U.; Wang, E.Y.; Vanderlaan, R.; Kim, D.H.; Lee, S.-H.; Hadipour-Lakmehsari, S.; Guo, H.; Zhao, Y.; McFadden, M.; Sharma, P.; et al. Mapping signalling perturbations in myocardial fibrosis via the integrative phosphoproteomic profiling of tissue from diverse sources. Nat. Biomed. Eng. 2020, 4, 889–900. [Google Scholar] [CrossRef]
- Lian, X.; Hsiao, C.; Wilson, G.; Zhu, K.; Hazeltine, L.B.; Azarin, S.M.; Raval, K.K.; Zhang, J.; Kamp, T.J.; Palecek, S.P. Robust Cardiomyocyte Differentiation from Human Pluripotent Stem Cells via Temporal Modulation of Canonical Wnt Signaling. Proc. Natl. Acad. Sci. USA 2012, 109, E1848–E1857. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, K.; Cloonan, P.E.; Sundaram, S.; Liu, F.; Das, S.L.; Ewoldt, J.K.; Bays, J.L.; Tomp, S.; Toepfer, C.N.; Marsiglia, J.D.C.; et al. Plakophilin-2 Truncating Variants Impair Cardiac Contractility by Disrupting Sarcomere Stability and Organization. Sci. Adv. 2021, 7, eabh3995. [Google Scholar] [CrossRef] [PubMed]
- Toepfer, C.N.; Garfinkel, A.C.; Venturini, G.; Wakimoto, H.; Repetti, G.; Alamo, L.; Sharma, A.; Agarwal, R.; Ewoldt, J.F.; Cloonan, P.; et al. Myosin Sequestration Regulates Sarcomere Function, Cardiomyocyte Energetics, and Metabolism, Informing the Pathogenesis of Hypertrophic Cardiomyopathy. Circulation 2020, 141, 828–842. [Google Scholar] [CrossRef] [PubMed]
- Cantin, G.T.; Shock, T.R.; Park, S.K.; Madhani, H.D.; Yates, J.R. Optimizing TiO2-Based Phosphopeptide Enrichment for Automated Multidimensional Liquid Chromatography Coupled to Tandem Mass Spectrometry. Anal. Chem. 2007, 79, 4666–4673. [Google Scholar] [CrossRef] [Green Version]
- Blum, B.C.; Emili, A. Omics Notebook: Robust, Reproducible and Flexible Automated Multiomics Exploratory Analysis and Reporting. Bioinform. Adv. 2021, 1, vbab024. [Google Scholar] [CrossRef]
- Candasamy, A.J.; Haworth, R.S.; Cuello, F.; Ibrahim, M.; Aravamudhan, S.; Krüger, M.; Holt, M.R.; Terracciano, C.M.N.; Mayr, M.; Gautel, M.; et al. Phosphoregulation of the Titin-Cap Protein Telethonin in Cardiac Myocytes. J. Biol. Chem. 2014, 289, 1282–1293. [Google Scholar] [CrossRef] [Green Version]
- Knöll, R.; Linke, W.A.; Zou, P.; Miočiċ, S.; Kostin, S.; Buyandelger, B.; Ku, C.-H.; Neef, S.; Bug, M.; Schäfer, K.; et al. Telethonin Deficiency Is Associated With Maladaptation to Biomechanical Stress in the Mammalian Heart. Circ. Res. 2011, 109, 758–769. [Google Scholar] [CrossRef]
- Korotkevich, G.; Sukhov, V.; Sergushichev, A. Fast Gene Set Enrichment Analysis. Bioinformatics 2016. [Google Scholar] [CrossRef] [Green Version]
- Giacomello, M.; Pyakurel, A.; Glytsou, C.; Scorrano, L. The Cell Biology of Mitochondrial Membrane Dynamics. Nat. Rev. Mol. Cell Biol. 2020, 21, 204–224. [Google Scholar] [CrossRef]
- Cho, E.; Jung, W.; Joo, H.-Y.; Park, E.-R.; Kim, M.-Y.; Kim, S.-B.; Kim, K.S.; Lim, Y.B.; Lee, K.H.; Shin, H.J. Cluh Plays a Pivotal Role during Adipogenesis by Regulating the Activity of Mitochondria. Sci. Rep. 2019, 9, 6820. [Google Scholar] [CrossRef] [Green Version]
- Gao, J.; Schatton, D.; Martinelli, P.; Hansen, H.; Pla-Martin, D.; Barth, E.; Becker, C.; Altmueller, J.; Frommolt, P.; Sardiello, M.; et al. CLUH Regulates Mitochondrial Biogenesis by Binding MRNAs of Nuclear-Encoded Mitochondrial Proteins. J. Cell Biol. 2014, 207, 213–223. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fransson, Å.; Ruusala, A.; Aspenström, P. The Atypical Rho GTPases Miro-1 and Miro-2 Have Essential Roles in Mitochondrial Trafficking. Biochem. Biophys. Res. Commun. 2006, 344, 500–510. [Google Scholar] [CrossRef] [PubMed]
- Wakim, J.; Goudenege, D.; Perrot, R.; Gueguen, N.; Desquiret-Dumas, V.; Chao de la Barca, J.M.; Dalla Rosa, I.; Manero, F.; Le Mao, M.; Chupin, S.; et al. CLUH Couples Mitochondrial Distribution to the Energetic and Metabolic Status. J. Cell Sci. 2017, 130, 1940–1951. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, H.; Sibilla, C.; Liu, R.; Yun, J.; Hay, B.A.; Blackstone, C.; Chan, D.C.; Harvey, R.J.; Guo, M. Clueless/CLUH Regulates Mitochondrial Fission by Promoting Recruitment of Drp1 to Mitochondria. Nat Commun 2022, 13, 1582. [Google Scholar] [CrossRef]
- Pla-Martín, D.; Schatton, D.; Wiederstein, J.L.; Marx, M.-C.; Khiati, S.; Krüger, M.; Rugarli, E.I. CLUH Granules Coordinate Translation of Mitochondrial Proteins with MTORC1 Signaling and Mitophagy. EMBO J. 2020, 39, e102731. [Google Scholar] [CrossRef]
- Ashrafi, G.; Schwarz, T.L. The Pathways of Mitophagy for Quality Control and Clearance of Mitochondria. Cell Death Differ. 2013, 20, 31–42. [Google Scholar] [CrossRef] [Green Version]
- Ni, H.-M.; Williams, J.A.; Ding, W.-X. Mitochondrial Dynamics and Mitochondrial Quality Control. Redox Biol. 2015, 4, 6–13. [Google Scholar] [CrossRef] [Green Version]
- Yamada, T.; Dawson, T.M.; Yanagawa, T.; Iijima, M.; Sesaki, H. SQSTM1/P62 Promotes Mitochondrial Ubiquitination Independently of PINK1 and PRKN/Parkin in Mitophagy. Autophagy 2019, 15, 2012–2018. [Google Scholar] [CrossRef]
- Bhujabal, Z.; Birgisdottir, Å.B.; Sjøttem, E.; Brenne, H.B.; Øvervatn, A.; Habisov, S.; Kirkin, V.; Lamark, T.; Johansen, T. FKBP8 Recruits LC3A to Mediate Parkin-Independent Mitophagy. EMBO Rep. 2017, 18, 947–961. [Google Scholar] [CrossRef]
- Hirota, Y.; Yamashita, S.; Kurihara, Y.; Jin, X.; Aihara, M.; Saigusa, T.; Kang, D.; Kanki, T. Mitophagy Is Primarily Due to Alternative Autophagy and Requires the MAPK1 and MAPK14 Signaling Pathways. Autophagy 2015, 11, 332–343. [Google Scholar] [CrossRef] [Green Version]
- Kraft, L.J.; Dowler, J.; Manral, P.; Kenworthy, A.K. Size, Organization, and Dynamics of Soluble SQSTM1 and LC3-SQSTM1 Complexes in Living Cells. Autophagy 2016, 12, 1660–1674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsutsui, H.; Kinugawa, S.; Matsushima, S. Oxidative Stress and Heart Failure. Am. J. Physiol. Heart Circ. Physiol. 2011, 301, H2181–H2190. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szyguła-Jurkiewicz, B.; Szczurek-Wasilewicz, W.; Osadnik, T.; Frycz-Kurek, A.M.; Macioł-Skurk, K.; Małyszek-Tumidajewicz, J.; Skrzypek, M.; Romuk, E.; Gąsior, M.; Banach, M.; et al. Oxidative Stress Markers in Hypertrophic Cardiomyopathy. Medicina 2021, 58, 31. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, K.; Kusano, K.F.; Matsubara, H.; Nakamura, Y.; Miura, A.; Nishii, N.; Banba, K.; Nagase, S.; Miyaji, K.; Morita, H.; et al. Relationship Between Oxidative Stress and Systolic Dysfunction in Patients With Hypertrophic Cardiomyopathy. J. Card. Fail. 2005, 11, 117–123. [Google Scholar] [CrossRef] [PubMed]
- Scherz-Shouval, R.; Elazar, Z. ROS, Mitochondria and the Regulation of Autophagy. Trends Cell Biol. 2007, 17, 422–427. [Google Scholar] [CrossRef]
- Aiken, C.T.; Kaake, R.M.; Wang, X.; Huang, L. Oxidative Stress-Mediated Regulation of Proteasome Complexes. Mol. Cell. Proteom. 2011, 10, R110.006924. [Google Scholar] [CrossRef] [Green Version]
- Seco-Cervera, M.; González-Cabo, P.; Pallardó, F.V.; Romá-Mateo, C.; García-Giménez, J.L. Thioredoxin and Glutaredoxin Systems as Potential Targets for the Development of New Treatments in Friedreich’s Ataxia. Antioxidants 2020, 9, 1257. [Google Scholar] [CrossRef]
- Ribas, V.; García-Ruiz, C.; Fernández-Checa, J.C. Glutathione and Mitochondria. Front. Pharmacol. 2014, 5, 151. [Google Scholar] [CrossRef] [Green Version]
- Van Houten, B.; Woshner, V.; Santos, J.H. Role of Mitochondrial DNA in Toxic Responses to Oxidative Stress. DNA Repair 2006, 5, 145–152. [Google Scholar] [CrossRef]
- Guo, C.; Sun, L.; Chen, X.; Zhang, D. Oxidative Stress, Mitochondrial Damage and Neurodegenerative Diseases. Neural Regen. Res. 2013, 8, 2003–2014. [Google Scholar] [CrossRef]
- Szczesny, B.; Tann, A.W.; Longley, M.J.; Copeland, W.C.; Mitra, S. Long Patch Base Excision Repair in Mammalian Mitochondrial Genomes. J. Biol. Chem. 2008, 283, 26349–26356. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krokan, H.E.; Bjørås, M. Base Excision Repair. Cold Spring Harb. Perspect. Biol. 2013, 5, a012583. [Google Scholar] [CrossRef] [PubMed]
- Prakash, A.; Doublié, S. Base Excision Repair in the Mitochondria. J. Cell. Biochem. 2015, 116, 1490–1499. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Campalans, A.; Kortulewski, T.; Amouroux, R.; Menoni, H.; Vermeulen, W.; Radicella, J.P. Distinct Spatiotemporal Patterns and PARP Dependence of XRCC1 Recruitment to Single-Strand Break and Base Excision Repair. Nucleic Acids Res. 2013, 41, 3115–3129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ronson, G.E.; Piberger, A.L.; Higgs, M.R.; Olsen, A.L.; Stewart, G.S.; McHugh, P.J.; Petermann, E.; Lakin, N.D. PARP1 and PARP2 Stabilise Replication Forks at Base Excision Repair Intermediates through Fbh1-Dependent Rad51 Regulation. Nat. Commun. 2018, 9, 746. [Google Scholar] [CrossRef] [Green Version]
- Rossi, M.N.; Carbone, M.; Mostocotto, C.; Mancone, C.; Tripodi, M.; Maione, R.; Amati, P. Mitochondrial Localization of PARP-1 Requires Interaction with Mitofilin and Is Involved in the Maintenance of Mitochondrial DNA Integrity. J. Biol. Chem. 2009, 284, 31616–31624. [Google Scholar] [CrossRef] [Green Version]
- Schreiber, V.; Amé, J.-C.; Dollé, P.; Schultz, I.; Rinaldi, B.; Fraulob, V.; Murcia, J.M.; de Murcia, G. Poly(ADP-Ribose) Polymerase-2 (PARP-2) Is Required for Efficient Base Excision DNA Repair in Association with PARP-1 and XRCC1. J. Biol. Chem. 2002, 277, 23028–23036. [Google Scholar] [CrossRef] [Green Version]
- Taguchi, N.; Ishihara, N.; Jofuku, A.; Oka, T.; Mihara, K. Mitotic Phosphorylation of Dynamin-Related GTPase Drp1 Participates in Mitochondrial Fission *. J. Biol. Chem. 2007, 282, 11521–11529. [Google Scholar] [CrossRef] [Green Version]
- Perdiz, D.; Lorin, S.; Leroy-Gori, I.; Poüs, C. Stress-Induced Hyperacetylation of Microtubule Enhances Mitochondrial Fission and Modulates the Phosphorylation of Drp1 at 616Ser. Cell. Signal. 2017, 39, 32–43. [Google Scholar] [CrossRef]
- Kraus, I.; Besong Agbo, D.; Otto, M.; Wiltfang, J.; Klafki, H. Detection and Differentiation of Threonine- and Tyrosine-Monophosphorylated Forms of ERK1/2 by Capillary Isoelectric Focusing-Immunoassay. Sci. Rep. 2015, 5, 12767. [Google Scholar] [CrossRef] [Green Version]
- Roe, A.J.; Qi, X. Drp1 Phosphorylation by MAPK1 Causes Mitochondrial Dysfunction in Cell Culture Model of Huntington’s Disease. Biochem. Biophys. Res. Commun. 2018, 496, 706–711. [Google Scholar] [CrossRef] [PubMed]
- Kashatus, J.A.; Nascimento, A.; Myers, L.J.; Sher, A.; Byrne, F.L.; Hoehn, K.L.; Counter, C.M.; Kashatus, D.F. Erk2 Phosphorylation of Drp1 Promotes Mitochondrial Fission and MAPK-Driven Tumor Growth. Mol. Cell 2015, 57, 537–551. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, R.; Liu, T.; Jin, S.-B.; Ankarcrona, M.; Lendahl, U.; Nistér, M.; Zhao, J. MIEF1/2 Orchestrate Mitochondrial Dynamics through Direct Engagement with Both the Fission and Fusion Machineries. BMC Biol. 2021, 19, 229. [Google Scholar] [CrossRef] [PubMed]
- Ambruso, D.R. Peroxiredoxin-6 and NADPH Oxidase Activity. Methods Enzymol. 2013, 527, 145–167. [Google Scholar] [CrossRef]
- Kudin, A.P.; Augustynek, B.; Lehmann, A.K.; Kovács, R.; Kunz, W.S. The Contribution of Thioredoxin-2 Reductase and Glutathione Peroxidase to H2O2 Detoxification of Rat Brain Mitochondria. Biochim. Biophys. Acta (BBA)-Bioenerget. 2012, 1817, 1901–1906. [Google Scholar] [CrossRef] [Green Version]
- Winn, L.M.; Kim, P.M.; Nickoloff, J.A. Oxidative Stress-Induced Homologous Recombination As a Novel Mechanism for Phenytoin-Initiated Toxicity. J. Pharmacol. Exp. Ther. 2003, 306, 523–527. [Google Scholar] [CrossRef]
- Chaanine, A.H.; LeJemtel, T.H.; Delafontaine, P. Mitochondrial Pathobiology and Metabolic Remodeling in Progression to Overt Systolic Heart Failure. J. Clin. Med. 2020, 9, 3582. [Google Scholar] [CrossRef]
- Blum, B.C.; Lin, W.; Lawton, M.L.; Liu, Q.; Kwan, J.; Turcinovic, I.; Hekman, R.; Hu, P.; Emili, A. Multiomic Metabolic Enrichment Network Analysis Reveals Metabolite–Protein Physical Interaction Subnetworks Altered in Cancer. Mol. Cell. Proteom. 2022, 21, 100189. [Google Scholar] [CrossRef]
- Gansemer, E.R.; McCommis, K.S.; Martino, M.; King-McAlpin, A.Q.; Potthoff, M.J.; Finck, B.N.; Taylor, E.B.; Rutkowski, D.T. NADPH and Glutathione Redox Link TCA Cycle Activity to Endoplasmic Reticulum Homeostasis. iScience 2020, 23, 101116. [Google Scholar] [CrossRef]
- Ge, T.; Yang, J.; Zhou, S.; Wang, Y.; Li, Y.; Tong, X. The Role of the Pentose Phosphate Pathway in Diabetes and Cancer. Front. Endocrinol. 2020, 11, 365. [Google Scholar] [CrossRef]
- Liu, M.; Zhou, X.; Li, Y.; Ma, S.; Pan, L.; Zhang, X.; Zheng, W.; Wu, Z.; Wang, K.; Ahsan, A.; et al. TIGAR Alleviates Oxidative Stress in Brain with Extended Ischemia via a Pentose Phosphate Pathway-Independent Manner. Redox Biol. 2022, 53, 102323. [Google Scholar] [CrossRef] [PubMed]
- Grynberg, A.; Demaison, L. Fatty Acid Oxidation in the Heart. J. Cardiovasc. Pharmacol. 1996, 28 (Suppl. 1), S11–S17. [Google Scholar] [CrossRef] [PubMed]
- Baier, M.J.; Noack, J.; Seitz, M.T.; Maier, L.S.; Neef, S. Phosphorylation of RyR2 Ser-2814 by CaMKII Mediates Β1-adrenergic Stress Induced Ca2+-leak from the Sarcoplasmic Reticulum. FEBS Open Bio 2021, 11, 2756–2762. [Google Scholar] [CrossRef] [PubMed]
- Mattiazzi, A.; Kranias, E. The Role of CaMKII Regulation of Phospholamban Activity in Heart Disease. Front. Pharmacol. 2014, 5, 5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McNamara, J.W.; Singh, R.R.; Sadayappan, S. Cardiac Myosin Binding Protein-C Phosphorylation Regulates the Super-Relaxed State of Myosin. Proc. Natl. Acad. Sci. USA 2019, 116, 11731–11736. [Google Scholar] [CrossRef] [Green Version]
- Sadayappan, S.; Gulick, J.; Osinska, H.; Barefield, D.; Cuello, F.; Avkiran, M.; Lasko, V.M.; Lorenz, J.N.; Maillet, M.; Martin, J.L.; et al. A Critical Function for Ser-282 in Cardiac Myosin Binding Protein-C Phosphorylation and Cardiac Function. Circ. Res. 2011, 109, 141–150. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.; Ni, F.; Kondrashkina, E.; Ma, J.; Wang, Q. Mechanisms of Leiomodin 2-Mediated Regulation of Actin Filament in Muscle Cells. Proc. Natl. Acad. Sci. USA 2015, 112, 12687–12692. [Google Scholar] [CrossRef] [Green Version]
- Arimura, T.; Suematsu, N.; Zhou, Y.-B.; Nishimura, J.; Satoh, S.; Takeshita, A.; Kanaide, H.; Kimura, A. Identification, Characterization, and Functional Analysis of Heart-Specific Myosin Light Chain Phosphatase Small Subunit. J. Biol. Chem. 2001, 276, 6073–6082. [Google Scholar] [CrossRef] [Green Version]
- Grassie, M.E.; Moffat, L.D.; Walsh, M.P.; MacDonald, J.A. The Myosin Phosphatase Targeting Protein (MYPT) Family: A Regulated Mechanism for Achieving Substrate Specificity of the Catalytic Subunit of Protein Phosphatase Type 1δ. Arch. Biochem. Biophys. 2011, 510, 147–159. [Google Scholar] [CrossRef]
- Buraei, Z.; Yang, J. Structure and Function of the β Subunit of Voltage-Gated Ca2+ Channels. Biochim. Biophys. Acta 2013, 1828, 1530–1540. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, F.; Pu, W.T. The Architecture and Function of Cardiac Dyads. Biophys. Rev. 2020, 12, 1007–1017. [Google Scholar] [CrossRef]
- Schmidt, T.A.; Kjeldsen, K. Human Myocardial Na,K-ATPase—Quantification, Regulation and Relation to Ca. Cardiovasc. Res. 1998, 37, 335–345. [Google Scholar] [CrossRef] [PubMed]
- Knollmann, B.C. New Roles of Calsequestrin and Triadin in Cardiac Muscle. J. Physiol. 2009, 587, 3081–3087. [Google Scholar] [CrossRef] [PubMed]
- Terentyev, D.; Viatchenko-Karpinski, S.; Györke, I.; Volpe, P.; Williams, S.C.; Györke, S. Calsequestrin Determines the Functional Size and Stability of Cardiac Intracellular Calcium Stores: Mechanism for Hereditary Arrhythmia. Proc. Natl. Acad. Sci. USA 2003, 100, 11759–11764. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spindler, M.; Saupe, K.W.; Christe, M.E.; Sweeney, H.L.; Seidman, C.E.; Seidman, J.G.; Ingwall, J.S. Diastolic Dysfunction and Altered Energetics in the AlphaMHC403/+ Mouse Model of Familial Hypertrophic Cardiomyopathy. J. Clin. Investig. 1998, 101, 1775–1783. [Google Scholar] [CrossRef] [Green Version]
- Chang, A.N.; Kamm, K.E.; Stull, J.T. Role of Myosin Light Chain Phosphatase in Cardiac Physiology and Pathophysiology. J. Mol. Cell. Cardiol. 2016, 101, 35–43. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sheikh, F.; Lyon, R.C.; Chen, J. Functions of Myosin Light Chain-2 (MYL2) in Cardiac Muscle and Disease. Gene 2015, 569, 14–20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Warren, S.A.; Briggs, L.E.; Zeng, H.; Chuang, J.; Chang, E.I.; Terada, R.; Li, M.; Swanson, M.S.; Lecker, S.H.; Willis, M.S.; et al. Myosin Light Chain Phosphorylation Is Critical for Adaptation to Cardiac Stress. Circulation 2012, 126, 2575–2588. [Google Scholar] [CrossRef] [Green Version]
- Sang, L.; Dick, I.E.; Yue, D.T. Protein Kinase A Modulation of CaV1.4 Calcium Channels. Nat. Commun. 2016, 7, 12239. [Google Scholar] [CrossRef] [Green Version]
- Wang, W.; Wang, J.; Yao, K.; Wang, S.; Nie, M.; Zhao, Y.; Wang, B.; Pang, H.; Xu, J.; Wu, G.; et al. Metabolic Characterization of Hypertrophic Cardiomyopathy in Human Heart. Nat. Cardiovasc. Res. 2022, 1, 445–461. [Google Scholar] [CrossRef]
- Shimada, Y.J.; Raita, Y.; Liang, L.W.; Maurer, M.S.; Hasegawa, K.; Fifer, M.A.; Reilly, M.P. Comprehensive Proteomics Profiling Reveals Circulating Biomarkers of Hypertrophic Cardiomyopathy. Circ. Heart Fail. 2021, 14, e007849. [Google Scholar] [CrossRef] [PubMed]
- Valko, M.; Rhodes, C.J.; Moncol, J.; Izakovic, M.; Mazur, M. Free Radicals, Metals and Antioxidants in Oxidative Stress-Induced Cancer. Chem.-Biol. Interact. 2006, 160, 1–40. [Google Scholar] [CrossRef] [PubMed]
- Garrido Ruiz, D.; Sandoval-Perez, A.; Rangarajan, A.V.; Gunderson, E.L.; Jacobson, M.P. Cysteine Oxidation in Proteins: Structure, Biophysics, and Simulation. Biochemistry 2022, 61, 2165–2176. [Google Scholar] [CrossRef] [PubMed]
- Kwon, S.H.; Pimentel, D.R.; Remondino, A.; Sawyer, D.B.; Colucci, W.S. H2O2 Regulates Cardiac Myocyte Phenotype via Concentration-Dependent Activation of Distinct Kinase Pathways. J. Mol. Cell. Cardiol. 2003, 35, 615–621. [Google Scholar] [CrossRef]
- Lu, S.C. Glutathione Synthesis. Biochim. Biophys. Acta 2013, 1830, 3143–3153. [Google Scholar] [CrossRef] [Green Version]
- Watanabe, K.; Nagao, M.; Toh, R.; Irino, Y.; Shinohara, M.; Iino, T.; Yoshikawa, S.; Tanaka, H.; Satomi-Kobayashi, S.; Ishida, T.; et al. Critical Role of Glutamine Metabolism in Cardiomyocytes under Oxidative Stress. Biochem. Biophys. Res. Commun. 2021, 534, 687–693. [Google Scholar] [CrossRef]
- Lynch, T.L.; Sivaguru, M.; Velayutham, M.; Cardounel, A.J.; Michels, M.; Barefield, D.; Govindan, S.; dos Remedios, C.; van der Velden, J.; Sadayappan, S. Oxidative Stress in Dilated Cardiomyopathy Caused by MYBPC3 Mutation. Oxid. Med. Cell. Longev. 2015, 2015, 424751. [Google Scholar] [CrossRef] [Green Version]
- Bhatti, J.S.; Bhatti, G.K.; Reddy, P.H. Mitochondrial Dysfunction and Oxidative Stress in Metabolic Disorders—A Step towards Mitochondria Based Therapeutic Strategies. Biochim. Biophys. Acta (BBA)-Mol. Basis Dis. 2017, 1863, 1066–1077. [Google Scholar] [CrossRef]
- Singh, G.; Pachouri, U.C.; Khaidem, D.C.; Kundu, A.; Chopra, C.; Singh, P. Mitochondrial DNA Damage and Diseases. F1000Research 2015, 4, 176. [Google Scholar] [CrossRef]
- Taylor, R.W.; Turnbull, D.M. Mitochondrial DNA mutations in human disease. Nat. Rev. Genet. 2005, 6, 389–402. [Google Scholar] [CrossRef] [Green Version]
- Zhou, B.; Tian, R. Mitochondrial Dysfunction in Pathophysiology of Heart Failure. J. Clin. Investig 2018, 128, 3716–3726. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adebayo, M.; Singh, S.; Singh, A.P.; Dasgupta, S. Mitochondrial Fusion and Fission: The Fine-tune Balance for Cellular Homeostasis. FASEB J. 2021, 35, e21620. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Vermulst, M.; Wang, Y.E.; Chomyn, A.; Prolla, T.A.; McCaffery, J.M.; Chan, D.C. Mitochondrial Fusion Is Required for MtDNA Stability in Skeletal Muscle and Tolerance of MtDNA Mutations. Cell 2010, 141, 280–289. [Google Scholar] [CrossRef] [Green Version]
- Pangou, E.; Bielska, O.; Guerber, L.; Schmucker, S.; Agote-Arán, A.; Ye, T.; Liao, Y.; Puig-Gamez, M.; Grandgirard, E.; Kleiss, C.; et al. A PKD-MFF Signaling Axis Couples Mitochondrial Fission to Mitotic Progression. Cell Rep. 2021, 35, 109129. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Nie, P.; Zhou, C.; Hu, Y.; Duan, S.; Gu, M.; Jiang, D.; Wang, Y.; Deng, Z.; Chen, J.; et al. Oxidative Stress-Induced Mitophagy Is Suppressed by the MiR-106b-93-25 Cluster in a Protective Manner. Cell Death Dis. 2021, 12, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Frohman, M.A. Role of Mitochondrial Lipids in Guiding Fission and Fusion. J. Mol. Med. 2015, 93, 263–269. [Google Scholar] [CrossRef] [Green Version]
- Paradies, G.; Paradies, V.; Ruggiero, F.M.; Petrosillo, G. Role of Cardiolipin in Mitochondrial Function and Dynamics in Health and Disease: Molecular and Pharmacological Aspects. Cells 2019, 8, 728. [Google Scholar] [CrossRef] [Green Version]
- Van der Velden, J.; Tocchetti, C.G.; Varricchi, G.; Bianco, A.; Sequeira, V.; Hilfiker-Kleiner, D.; Hamdani, N.; Leite-Moreira, A.F.; Mayr, M.; Falcão-Pires, I.; et al. Metabolic Changes in Hypertrophic Cardiomyopathies: Scientific Update from the Working Group of Myocardial Function of the European Society of Cardiology. Cardiovasc. Res. 2018, 114, 1273–1280. [Google Scholar] [CrossRef]
- Michas, C.; Karakan, M.Ç.; Nautiyal, P.; Seidman, J.G.; Seidman, C.E.; Agarwal, A.; Ekinci, K.; Eyckmans, J.; White, A.E.; Chen, C.S. Engineering a Living Cardiac Pump on a Chip Using High-Precision Fabrication. Sci. Adv. 2022, 8, eabm3791. [Google Scholar] [CrossRef]
- Sharma, A.; Toepfer, C.N.; Ward, T.; Wasson, L.; Agarwal, R.; Conner, D.A.; Hu, J.H.; Seidman, C.E. CRISPR/Cas9-Mediated Fluorescent Tagging of Endogenous Proteins in Human Pluripotent Stem Cells. Curr. Protoc. Hum. Genet. 2018, 96, 21.11.1–21.11.20. [Google Scholar] [CrossRef]
- Larson, A.; Chin, M.T. A Method for Cryopreservation and Single Nucleus RNA-Sequencing of Normal Adult Human Interventricular Septum Heart Tissue Reveals Cellular Diversity and Function. BMC Med. Genom. 2021, 14, 161. [Google Scholar] [CrossRef] [PubMed]
- Codden, C.J.; Chin, M.T. Common and Distinctive Intercellular Communication Patterns in Human Obstructive and Nonobstructive Hypertrophic Cardiomyopathy. Int. J. Mol. Sci. 2022, 23, 946. [Google Scholar] [CrossRef] [PubMed]
- Codden, C.J.; Larson, A.; Awata, J.; Perera, G.; Chin, M.T. Single Nucleus RNA-Sequencing Reveals Altered Intercellular Communication and Dendritic Cell Activation in Nonobstructive Hypertrophic Cardiomyopathy. Cardiol. Cardiovasc. Med. 2022, 6, 398–415. [Google Scholar] [CrossRef] [PubMed]
- Larson, A.; Codden, C.J.; Huggins, G.S.; Rastegar, H.; Chen, F.Y.; Maron, B.J.; Rowin, E.J.; Maron, M.S.; Chin, M.T. Altered Intercellular Communication and Extracellular Matrix Signaling as a Potential Disease Mechanism in Human Hypertrophic Cardiomyopathy. Sci. Rep. 2022, 12, 5211. [Google Scholar] [CrossRef] [PubMed]
- Ritchie, M.E.; Phipson, B.; Wu, D.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. Limma Powers Differential Expression Analyses for RNA-Sequencing and Microarray Studies. Nucleic Acids Res. 2015, 43, e47. [Google Scholar] [CrossRef] [PubMed]
- Blighe, K.; Rana, S.; Turkes, E.; Ostendorf, B.; Grioni, A.; Lewis, M. EnhancedVolcano: Publication-Ready Volcano Plots with Enhanced Colouring and Labeling. 2023. Available online: https://bioconductor.org/packages/devel/bioc/vignettes/EnhancedVolcano/inst/doc/EnhancedVolcano.html (accessed on 12 May 2021).
- Yuan, M.; Kremer, D.M.; Huang, H.; Breitkopf, S.B.; Ben-Sahra, I.; Manning, B.D.; Lyssiotis, C.A.; Asara, J.M. Ex Vivo and in Vivo Stable Isotope Labelling of Central Carbon Metabolism and Related Pathways with Analysis by LC–MS/MS. Nat. Protoc. 2019, 14, 313–330. [Google Scholar] [CrossRef]
- Toepfer, C.N.; Sharma, A.; Cicconet, M.; Garfinkel, A.C.; Mücke, M.; Neyazi, M.; Willcox, J.A.L.; Agarwal, R.; Schmid, M.; Rao, J.; et al. SarcTrack. Circ. Res. 2019, 124, 1172–1183. [Google Scholar] [CrossRef]
- Fan, T.; Kucia, M.; Jankowski, K.; Higashi, R.; Ratajczak, J.; Ratajczak, M.; Lane, A. Rhabdomyosarcoma Cells Show an Energy Producing Anabolic Metabolic Phenotype Compared with Primary Myocytes. Mol. Cancer 2008, 7, 79. [Google Scholar] [CrossRef] [Green Version]
- Mcdonald, T.; Carrasco-Pozo, C.; Hodson, M.; Borges, K. Alterations in Cytosolic and Mitochondrial [U- 13 C]-Glucose Metabolism in a Chronic Epilepsy Mouse Model. eNeuro 2017, 4, ENEURO.0341-16.2017. [Google Scholar] [CrossRef] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample ID | Age | Sex | MYH7 Mutation |
|---|---|---|---|
| Control 1 | 23 | F | - |
| Control 2 | 45 | F | - |
| Control 3 | 57 | M | - |
| Control 4 | 43 | M | - |
| Control 5 | 67 | F | - |
| HCM 1 | 37 | F | Met849Thr |
| HCM 2 | 51 | F | Asp906Gly |
| HCM 3 | 46 | M | Val606Met |
| HCM 4 | 29 | M | Arg143Gln |
| HCM 5 | 65 | F | Gly584Arg |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Moore, J.; Ewoldt, J.; Venturini, G.; Pereira, A.C.; Padilha, K.; Lawton, M.; Lin, W.; Goel, R.; Luptak, I.; Perissi, V.; et al. Multi-Omics Profiling of Hypertrophic Cardiomyopathy Reveals Altered Mechanisms in Mitochondrial Dynamics and Excitation–Contraction Coupling. Int. J. Mol. Sci. 2023, 24, 4724. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms24054724
Moore J, Ewoldt J, Venturini G, Pereira AC, Padilha K, Lawton M, Lin W, Goel R, Luptak I, Perissi V, et al. Multi-Omics Profiling of Hypertrophic Cardiomyopathy Reveals Altered Mechanisms in Mitochondrial Dynamics and Excitation–Contraction Coupling. International Journal of Molecular Sciences. 2023; 24(5):4724. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms24054724
Chicago/Turabian StyleMoore, Jarrod, Jourdan Ewoldt, Gabriela Venturini, Alexandre C. Pereira, Kallyandra Padilha, Matthew Lawton, Weiwei Lin, Raghuveera Goel, Ivan Luptak, Valentina Perissi, and et al. 2023. "Multi-Omics Profiling of Hypertrophic Cardiomyopathy Reveals Altered Mechanisms in Mitochondrial Dynamics and Excitation–Contraction Coupling" International Journal of Molecular Sciences 24, no. 5: 4724. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms24054724