
Identification of a Ubiquinone–Ubiquinol Quinhydrone Complex in Bacterial Photosynthetic Membranes and Isolated Reaction Centers by Time-Resolved Infrared Spectroscopy


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
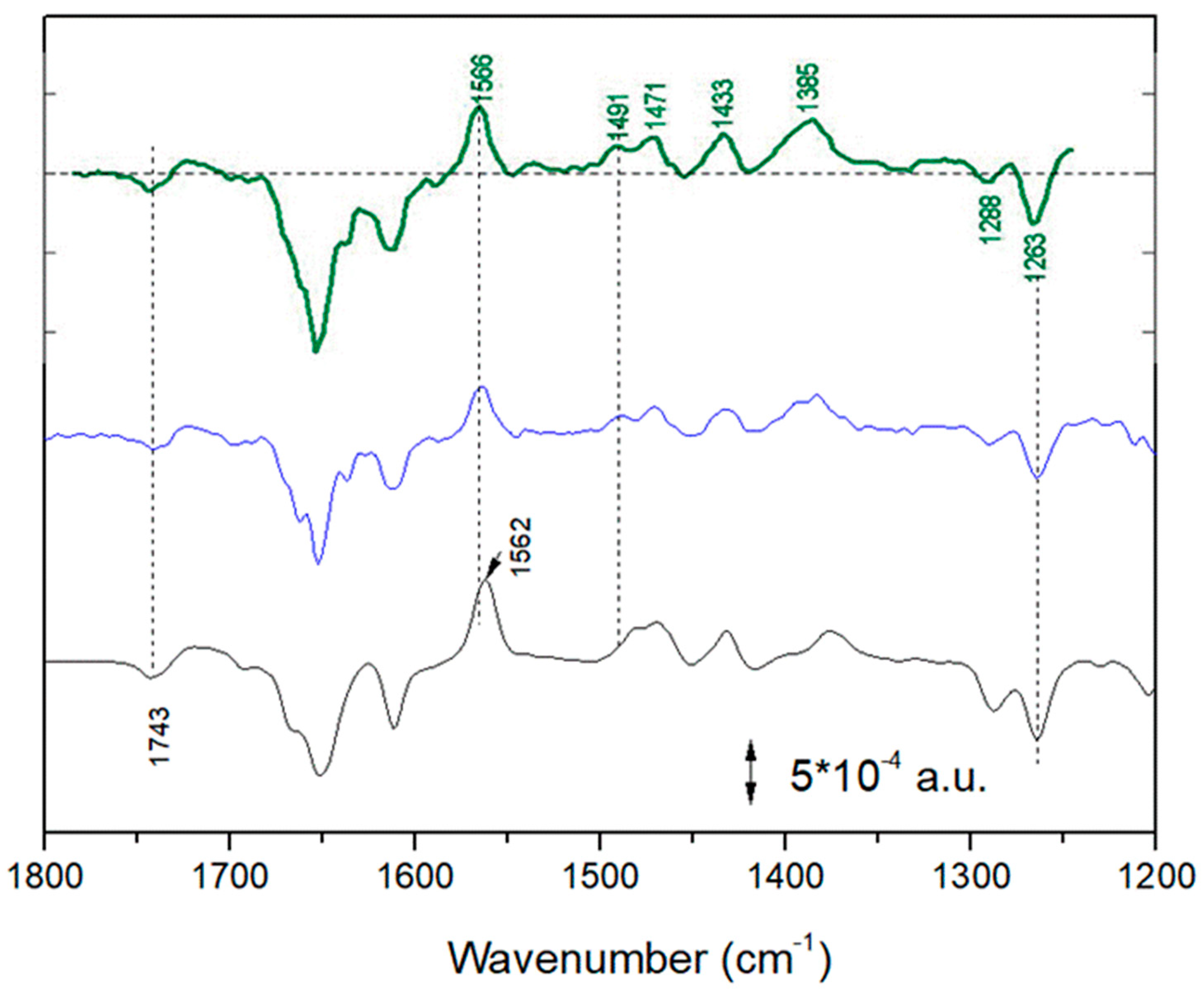
2.1. Light-Induced Fourier Transform Infrared (FTIR) Difference Spectroscopy on Chromatophores
- (1)
- A spectral component, characterized by a strong ~1564 cm−1 positive peak, is present only under a strong illumination condition of chromatophores;
- (2)
- A previous detailed chemometric analysis [7] demonstrated that this spectral component represents a reduction of (a fraction of) the ubiquinone pool;
- (3)
- The most characteristic band of this spectral component, the ~1564 (+) cm−1 peak, shows only a small downshift (~2 cm−1) upon D/H exchange.
2.2. Light-Induced FTIR Difference Spectroscopy on Isolated Reaction Centers
2.3. Comparison between Experiments in Different Conditions
2.4. Theoretical Calculations
3. Discussion
4. Materials and Methods
4.1. Sample Preparation
4.2. FTIR Spectroscopy
4.2.1. FTIR Spectra in Cyclohexane
4.2.2. Time-Resolved FTIR Experiments under and after Continuous Illumination in Chromatophores
4.2.3. Two-Flash Rapid-Scan FTIR Experiments on Isolated RCs
4.3. Theoretical Calculations
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Trumpower, B.L. Function of Quinones in Energy Conserving Systems; Academic Press: New York, NY, USA, 1982. [Google Scholar]
- Feher, G.; Allen, J.P.; Okamura, M.Y.; Rees, D.C. Structure and function of bacterial photosynthetic reaction centres. Nature 1989, 339, 111–116. [Google Scholar] [CrossRef]
- Crofts, A.R.; Meinhardt, S.W.; Jones, K.R.; Snozzi, M. The role of the quinone pool in the cyclic electron transfer chain of Rhodopseudomonas sphaeroides. A modified Q-cycle mechanism. Biochim. Biophys. Acta—Bioenergy 1983, 723, 202–218. [Google Scholar] [CrossRef] [Green Version]
- Berthomieu, C.; Hienerwadel, R. Fourier Transform Infrared (FTIR) spectroscopy. Photosynth. Res. 2009, 101, 157–170. [Google Scholar] [CrossRef]
- Lorenz-Fonfria, V.A. Infrared difference spectroscopy of proteins: From bands to bonds. Chem. Rev. 2020, 120, 3466–3576. [Google Scholar] [CrossRef]
- Mezzetti, A.; Leibl, W. Investigation of ubiquinol formation in isolated photosynthetic reaction centers by rapid-scan Fourier transform IR spectroscopy. Eur. Biophys. J. 2005, 34, 921–936. [Google Scholar] [CrossRef]
- Blanchet, L.; Ruckebusch, C.; Mezzetti, A.; Huvenne, J.-P.; de Juan, A. Monitoring and Interpretation of Photoinduced Biochemical Processes by Rapid-Scan FTIR Difference Spectroscopy and Hybrid Hard and Soft Modeling. J. Phys. Chem. B 2009, 113, 6031–6040. [Google Scholar] [CrossRef]
- Nedelkovski, V.; Schwaighofer, A.; Wraight, C.A.; Nowak, C.; Naumann, R.L.C. Surface-Enhanced Infrared Absorption Spectroscopy (SEIRAS) of Light-Activated Photosynthetic Reaction Centers from Rhodobacter sphaeroides Reconstituted in a Biomimetic Membrane System. J. Phys. Chem. C 2013, 117, 16357–16363. [Google Scholar] [CrossRef]
- Kishi, R.; Imanishi, M.; Kobayashi, M.; Takenaka, S.; Madigan, M.T.; Wang-Otomo, Z.-U.; Kimura, Y. Quinone transport in the closed light-harvesting 1 reaction center complex from the thermophilic purple bacterium Thermocromatium tepidum. Biochim. Biophys. Acta—Bioenergy 2021, 1862, 148307. [Google Scholar] [CrossRef]
- Mezzetti, A.; Schnee, J.; Lapini, A.; Di Donato, M. Time-resolved infrared absorption spectroscopy applied to photoinduced reactions: How and why. Photochem. Photobiol. Sci. 2022, 21, 557–584. [Google Scholar] [CrossRef]
- Mezzetti, A.; Leibl, W. Time-resolved infrared spectroscopy in the study of photosynthetic systems. Photosynth. Res. 2017, 131, 121–144. [Google Scholar] [CrossRef]
- Di Donato, M.; Groot, M.L. Ultrafast infrared spectroscopy in photosynthesis. Biochim. Biophys. Acta—Bioenergy 2015, 1847, 2–11. [Google Scholar] [CrossRef] [Green Version]
- Mezzetti, A.; Leibl, W.; Breton, J.; Nabedryk, E. Photoreduction of the quinone pool in the bacterial photosynthetic membrane: Identification of infrared marker bands for quinol formation. FEBS Lett. 2003, 537, 161–165. [Google Scholar] [CrossRef] [Green Version]
- Blanchet, L.; Mezzetti, A.; Ruckebusch, C.; Huvenne, J.-P.; de Juan, A. Multivariate curve resolution of rapid-scan FTIR difference spectra of quinone photoreduction in bacterial photosynthetic membranes. Anal. Bioanal. Chem. 2007, 387, 1863–1873. [Google Scholar] [CrossRef]
- Mezzetti, A.; Blanchet, L.; de Juan, A.; Leibl, W.; Ruckebusch, C. Ubiquinol formation in isolated photosynthetic reaction centres monitored by time-resolved differential FTIR in combination with 2D correlation spectroscopy and multivariate curve resolution. Anal. Bioanal. Chem. 2011, 399, 1999–2014. [Google Scholar] [CrossRef]
- Slifkin, M.A.; Walmsley, R.H. Infra-red studies of quinhydrone type complexes. Spectrochim. Acta Part A 1970, 26, 1237–1242. [Google Scholar] [CrossRef]
- Kubinyi, M.; Keresztury, G. Infrared and Raman spectroscopic study of molecular interactions in quinhydrone crystals. Spectrochim. Acta Part A 1989, 45, 421–429. [Google Scholar] [CrossRef]
- Kruk, J. Charge-transfer complexes of plastoquinone and α-tocopherol quinone in vitro. Biophys. Chem. 1988, 30, 143–149. [Google Scholar] [CrossRef]
- Kruk, J.; Strzalka, K.; Leblanc, R.M. Fourier transform infrared studies on charge-transfer interactions of plastoquinones and α-tocopherol quinone with their hydroquinone forms and monogalactosyldiacylglycerol. Biophys. Chem. 1993, 45, 235–244. [Google Scholar] [CrossRef]
- Kanesaka, I.; Nagami, H.; Kobayashi, I.; Ohno, K. Infrared intensity study on molecular interactions in quinhydrone. Bull. Chem. Soc. Jpn. 2006, 79, 406–412. [Google Scholar] [CrossRef]
- Foucrault, M.; Lachapelle, M.; Sandorfy, C. The complexes between quinone and hydroquinone and their methoxy derivatives. Spectrosc. Int. J. 1988, 6, 17–32. [Google Scholar]
- Regeimbal, J.; Gleiter, S.; Trumpower, B.L.; Bardwell, J.C.A. Disulfide bond formation involves a quinhydrone-type charge transfer complex. Proc. Natl. Acad. Sci. USA 2003, 100, 13779–13784. [Google Scholar] [CrossRef] [Green Version]
- Brandt, U. Bifurcated ubihydroquinone oxidation in the cytochrome bc1 complex by proton-gated charge-transfer. FEBS Lett. 1996, 387, 1–6. [Google Scholar] [CrossRef] [Green Version]
- Bartoschek, S.; Johansson, J.; Geierstanger, B.H.; Okun, J.G.; Lancaster, C.R.; Humpfer, E.; Yu, L.; Yu, C.-A.; Griesinger, C.; Brandt, U. Three molecules of ubiquinone. bind specifically to mitochondrial cytochrome bc1 complex. J. Biol. Chem. 2001, 276, 35231–35234. [Google Scholar] [CrossRef] [Green Version]
- Cheng, Y.; Cunnane, V.J.; Kontturi, A.K.; Kontturi, K.; Schiffrin, D.J. Potential dependence of transmembrane electron transfer across pospholipid bilayers mediated by ubiquinone 10. J. Phys. Chem. 1996, 100, 15470–15477. [Google Scholar] [CrossRef]
- Gulaboski, R.; Markovski, V.; Jihe, Z. Redox chemistry of coenzyme Q—A short overview of the voltammetric features. J. Solid State Electrochem. 2016, 20, 3229–3238. [Google Scholar] [CrossRef] [Green Version]
- Mezzetti, A.; Nabedryk, E.; Breton, J.; Okamura, M.Y.; Paddock, M.L.; Giacometti, G.; Leibl, W. Rapid-scan Fourier transform infrared spectroscopy shows coupling of Glu-L212 protonation and electron transfer to QB in Rhodobacter sphaeroides reaction centers. Biochim. Biophys. Acta—Bioenergy 2002, 1553, 320–330. [Google Scholar] [CrossRef] [Green Version]
- Iwaki, M.; Osyczka, A.; Moser, C.C.; Dutton, P.L.; Rick, P.R. ATR-FTIR Spectroscopy Studies of Iron-Sulfur Protein and Cytochrome c1 in the Rhodobacter capsulatus Cytochrome bc1 Complex. Biochemistry 2004, 43, 9477–9486. [Google Scholar] [CrossRef]
- Hellwig, P.; Mogi, T.; Tomson, F.L.; Gennis, R.B.; Iwata, J.; Miyoshi, H.; Mantele, W. Vibrational Modes of Ubiquinone in Cytochrome bo3 from Escherichia coli Identified by Fourier Transform Infrared Difference Spectroscopy and Specific 13C Labeling. Biochemistry 1999, 38, 14683–14689. [Google Scholar] [CrossRef]
- Becke, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648. [Google Scholar] [CrossRef] [Green Version]
- Hatefi, Y.; Lester, R.L.; Crane, F.L.; Widmer, C. Studies on the electron transport system: XVI. Enzymic oxidoreduction reactions of coenzyme Q. Biochim. Biophys. Acta 1959, 31, 490–501. [Google Scholar] [CrossRef]
- Woronowicz, K.; Sha, D.; Frese, R.N.; Sturgis, J.N.; Nanda, V.; Niederman, R.A. The effects of protein crowding in bacterial photosynthetic membranes on the flow of quinone redox species between the photochemical reaction center and the ubiquinol-cytochrome c2 oxidoreductase. Metallomics 2011, 3, 765–774. [Google Scholar] [CrossRef]
- Shinkarev, V.P.; Wraight, C.A. The interaction of quinone and detergent with reaction centers of purple bacteria. I. Slow quinone exchange between reaction center micelles and pure detergent micelles. Biophys. J. 1997, 72, 2304–2319. [Google Scholar] [CrossRef] [Green Version]
- Slifkin, M.A. Charge Transfer Interactions of Biomolecules; Academic Press: London, UK; New York, NY, USA, 1971. [Google Scholar]
- James, A.M.; Smith, R.A.J.; Murphy, M.P. Antioxidant and prooxidant properties of mithocondrial Coenzyme Q. Arch. Biochem. Biophys. 2004, 423, 47–56. [Google Scholar] [CrossRef] [PubMed]
- Clayton, R.K. The induced synthesis of catalase in Rhodopseudomonas spheroides. Biochim. Biophys. Acta 1960, 37, 503–512. [Google Scholar] [CrossRef] [PubMed]
- Isaacson, R.A.; Lendzian, F.; Abresch, E.C.; Lubitz, W.; Feher, G. Electronic structure of QA− in reaction centers from Rhodobacter sphaeroides. I. Electron paramagnetic resonance in single crystals. Biophys. J. 1995, 69, 311–322. [Google Scholar] [CrossRef]
- Clayton, R.K.; Wang, R.T. Photochemical reaction centers from Rhodopseudomonas spheroides. Methods Enzymol. 1971, 23, 696–704. [Google Scholar] [CrossRef]
- Mezzetti, A.; Leibl, W. Proton and electron transfer in wild-type and mutant reaction centers from Rhodobacter sphaeroides followed by rapid-scan FTIR spectroscopy. Vib. Spectr. 2008, 48, 126–134. [Google Scholar] [CrossRef]
- Mochnik, P.A.; Frei, B.; Ames, B.N. Measurement of antioxidants in human blood plasma. Methods Enzymol. 1994, 234, 269–279. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16, Revision C.01; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef] [Green Version]
- Krishnan, R.; Binkley, J.S.; Seeger, R.; Pople, J.A. Self-consistent molecular orbital methods. XX. A basis set for correlated wave functions. J. Chem. Phys. 1980, 72, 650. [Google Scholar] [CrossRef]









Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mezzetti, A.; Paul, J.-F.; Leibl, W. Identification of a Ubiquinone–Ubiquinol Quinhydrone Complex in Bacterial Photosynthetic Membranes and Isolated Reaction Centers by Time-Resolved Infrared Spectroscopy. Int. J. Mol. Sci. 2023, 24, 5233. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms24065233
Mezzetti A, Paul J-F, Leibl W. Identification of a Ubiquinone–Ubiquinol Quinhydrone Complex in Bacterial Photosynthetic Membranes and Isolated Reaction Centers by Time-Resolved Infrared Spectroscopy. International Journal of Molecular Sciences. 2023; 24(6):5233. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms24065233
Chicago/Turabian StyleMezzetti, Alberto, Jean-François Paul, and Winfried Leibl. 2023. "Identification of a Ubiquinone–Ubiquinol Quinhydrone Complex in Bacterial Photosynthetic Membranes and Isolated Reaction Centers by Time-Resolved Infrared Spectroscopy" International Journal of Molecular Sciences 24, no. 6: 5233. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms24065233