
Heart-Type Fatty Acid Binding Protein Binds Long-Chain Acylcarnitines and Protects against Lipotoxicity

 , , , , , and
, , , , , and
Abstract
:1. Introduction
2. Results
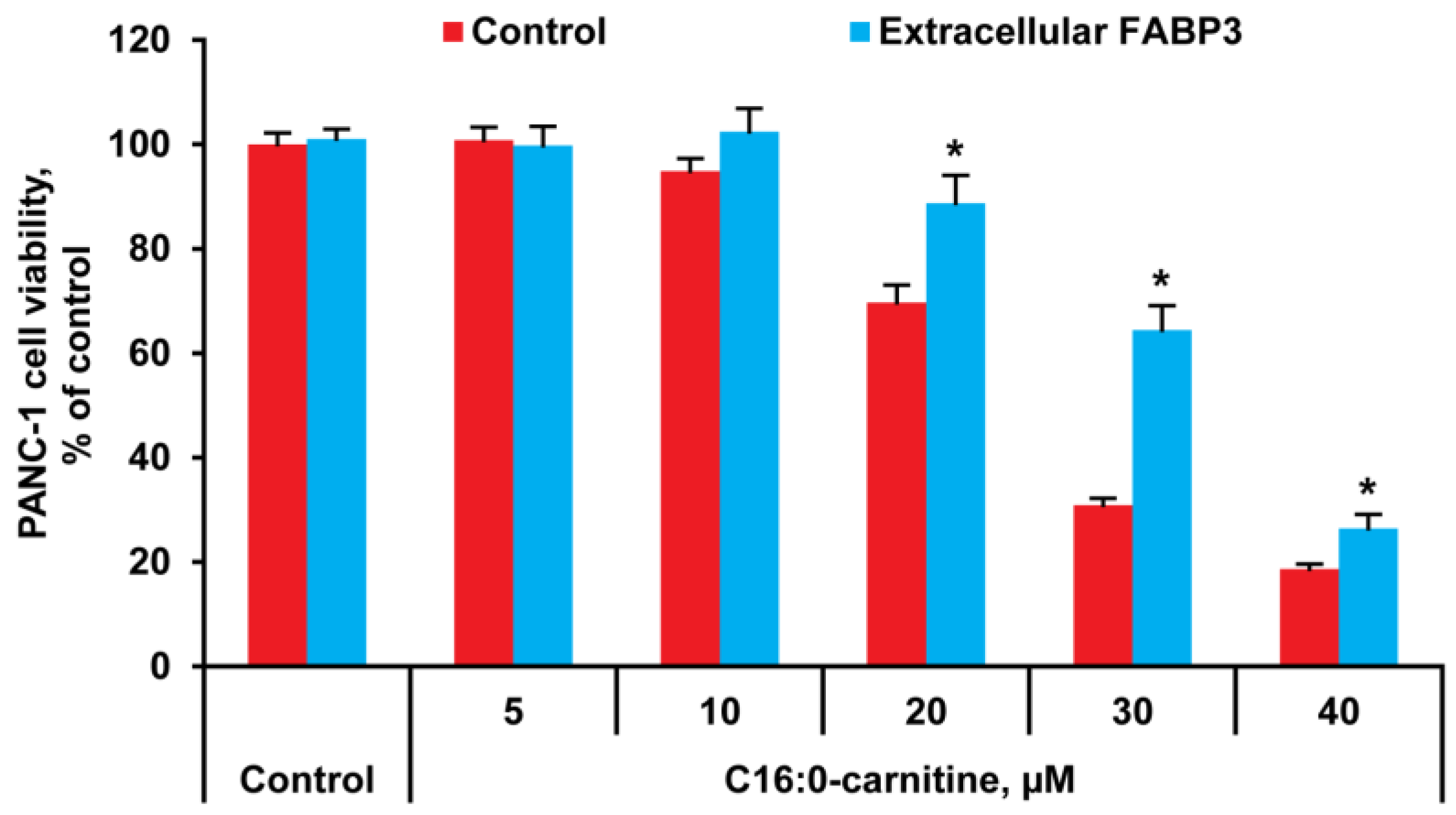
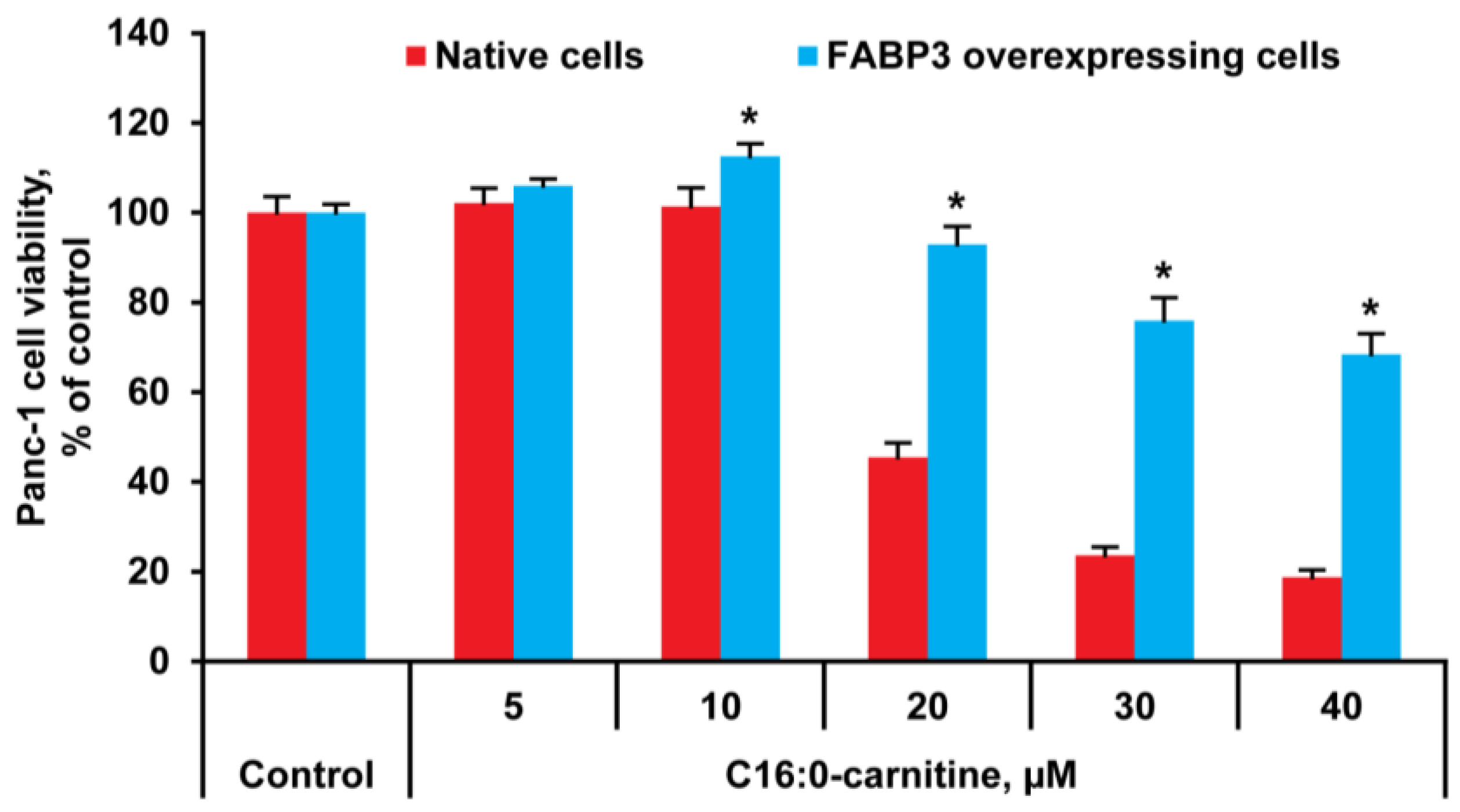
2.1. LCAC Cytotoxicity Assay
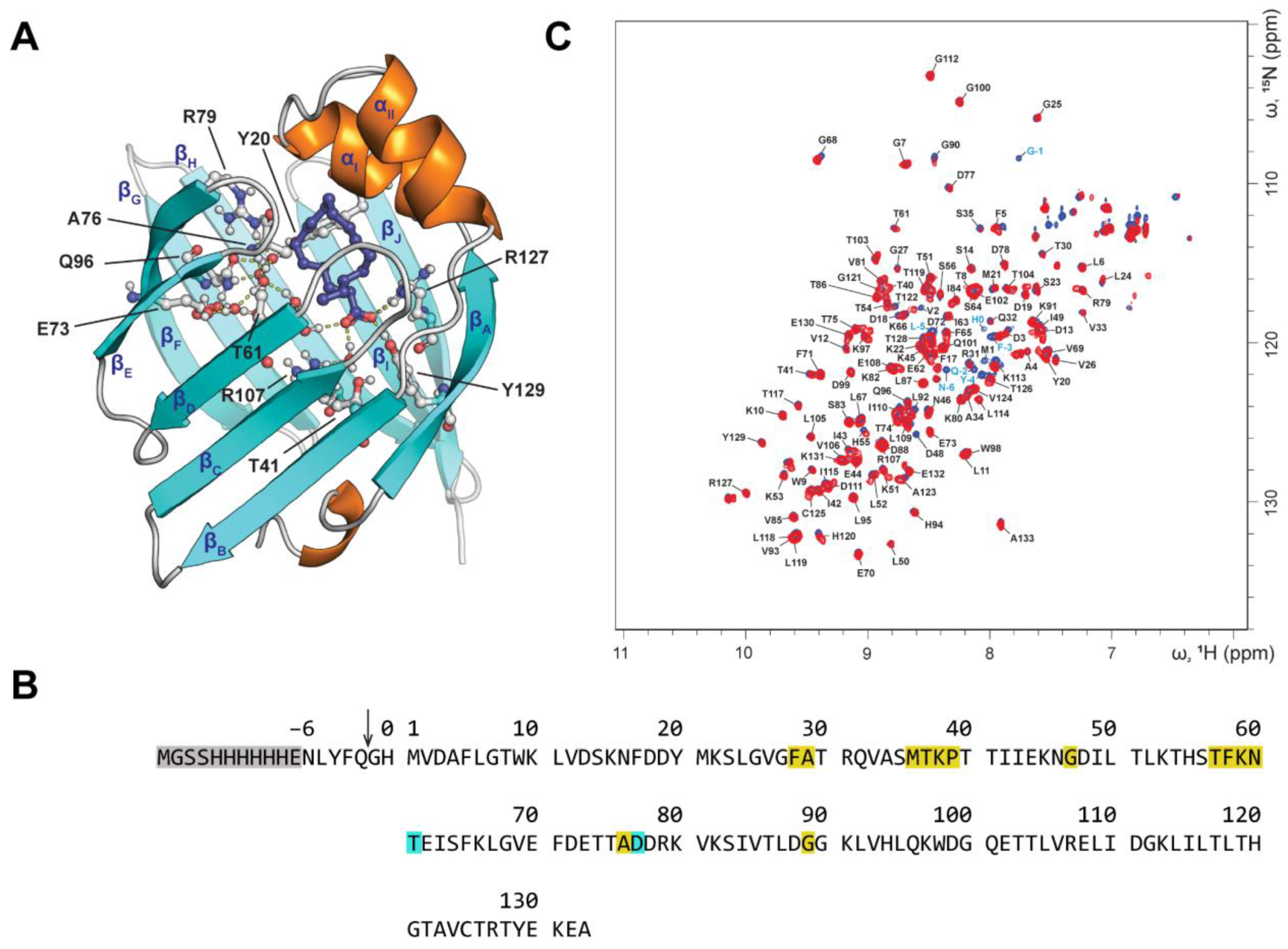
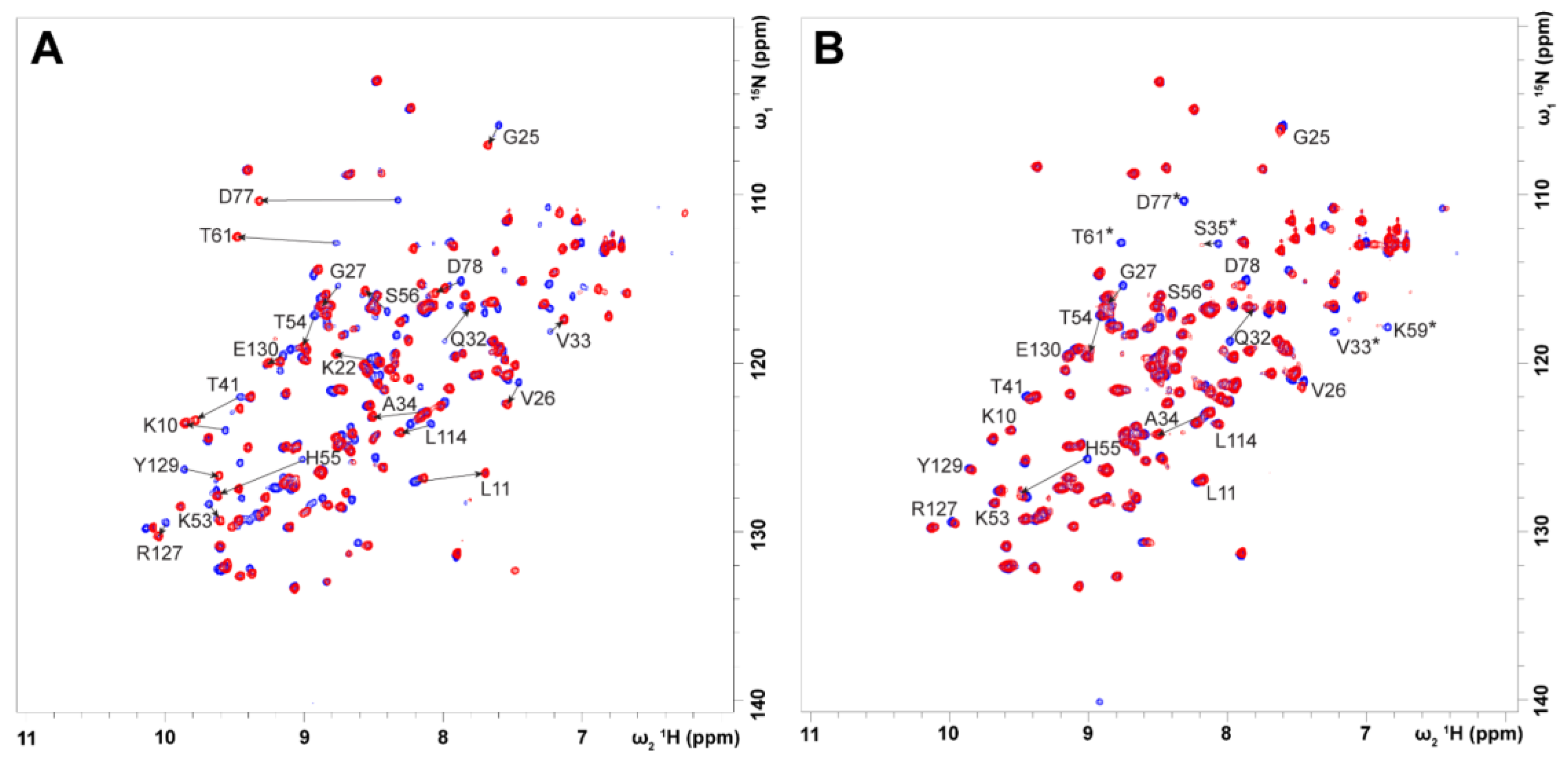
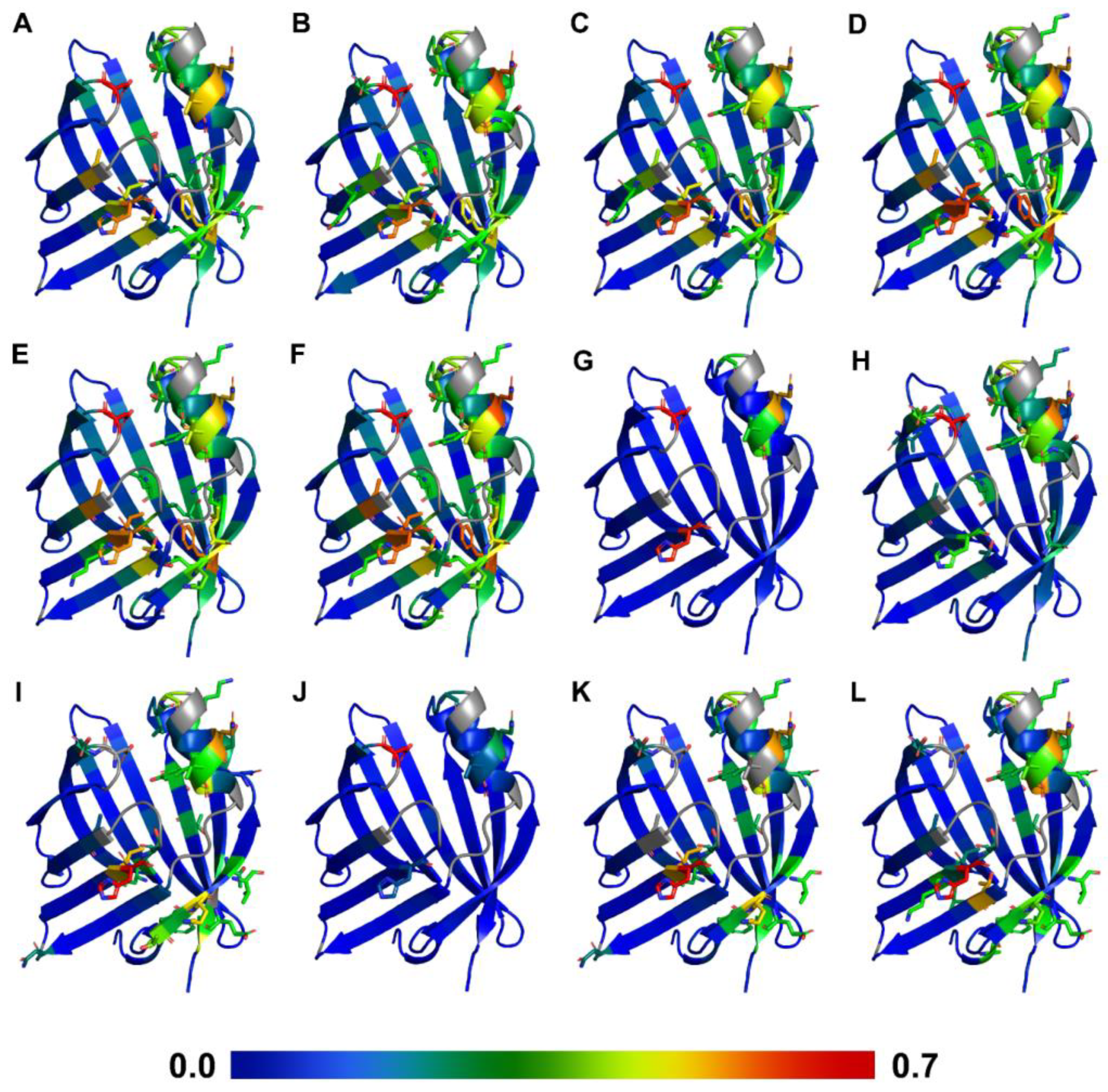
2.2. FA and AC Binding Studies by NMR Spectroscopy
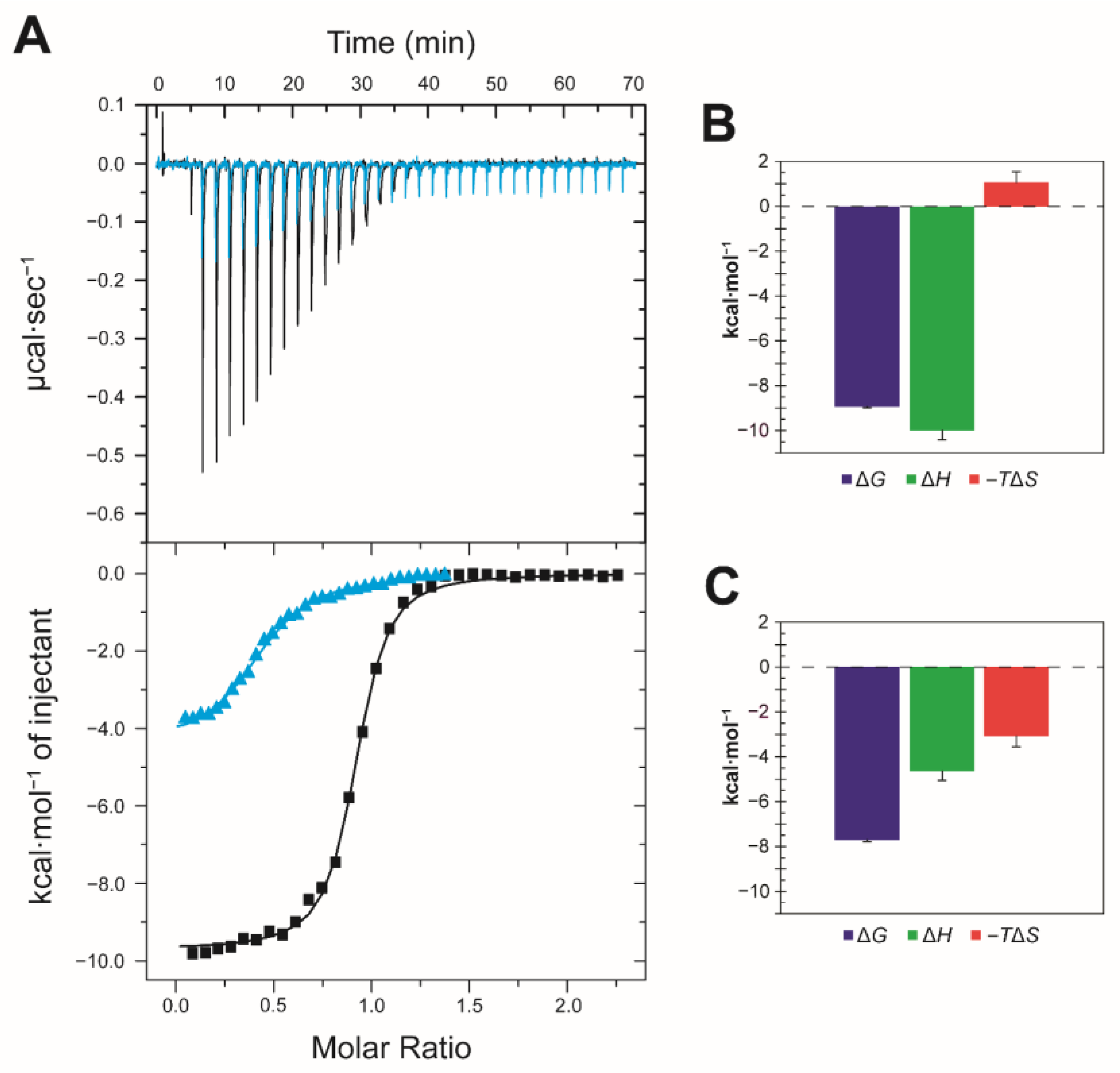
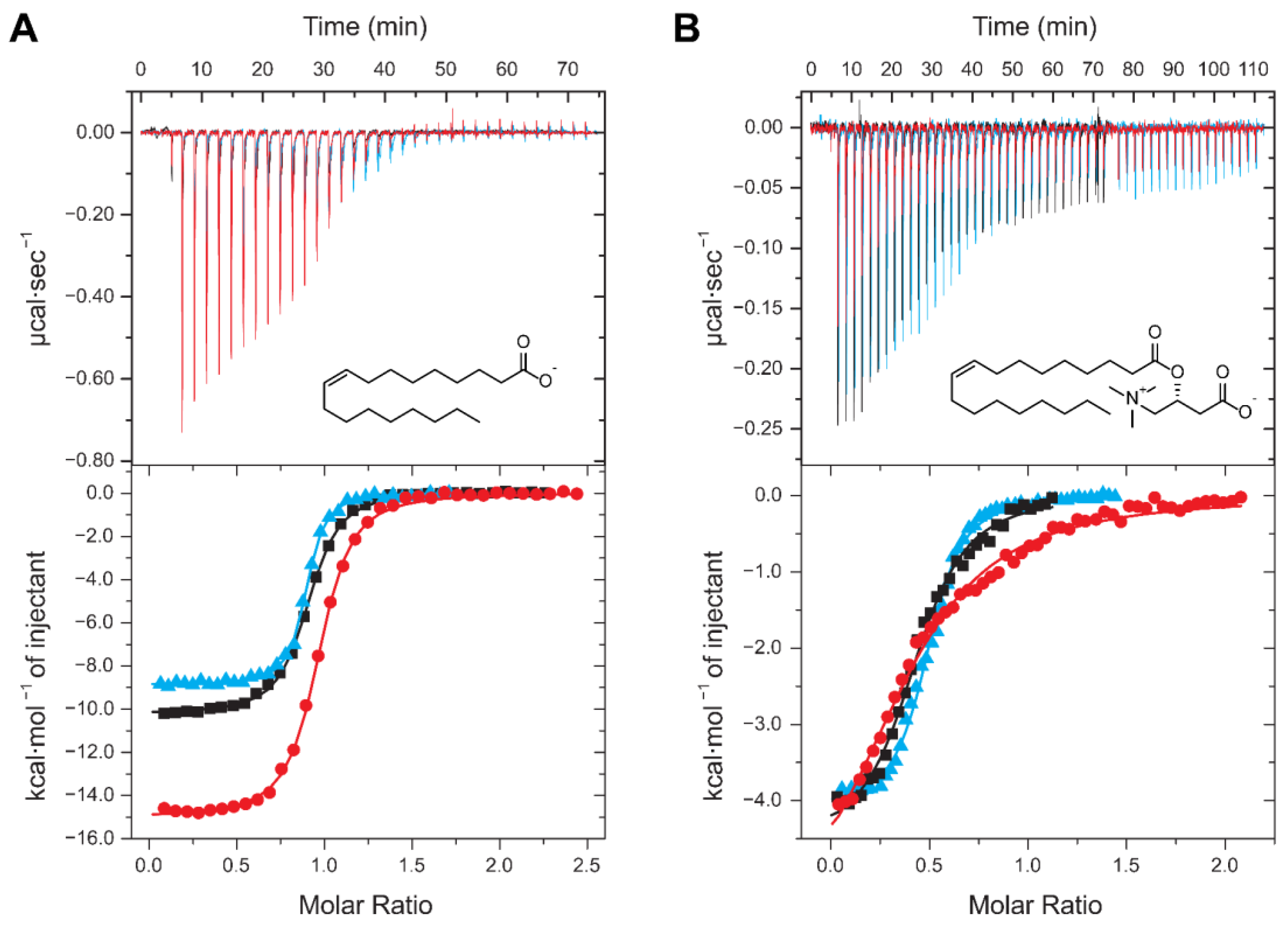
2.3. Thermodynamics of LCFA Binding to FABP3
2.4. Thermodynamics of LCAC Binding to FABP3
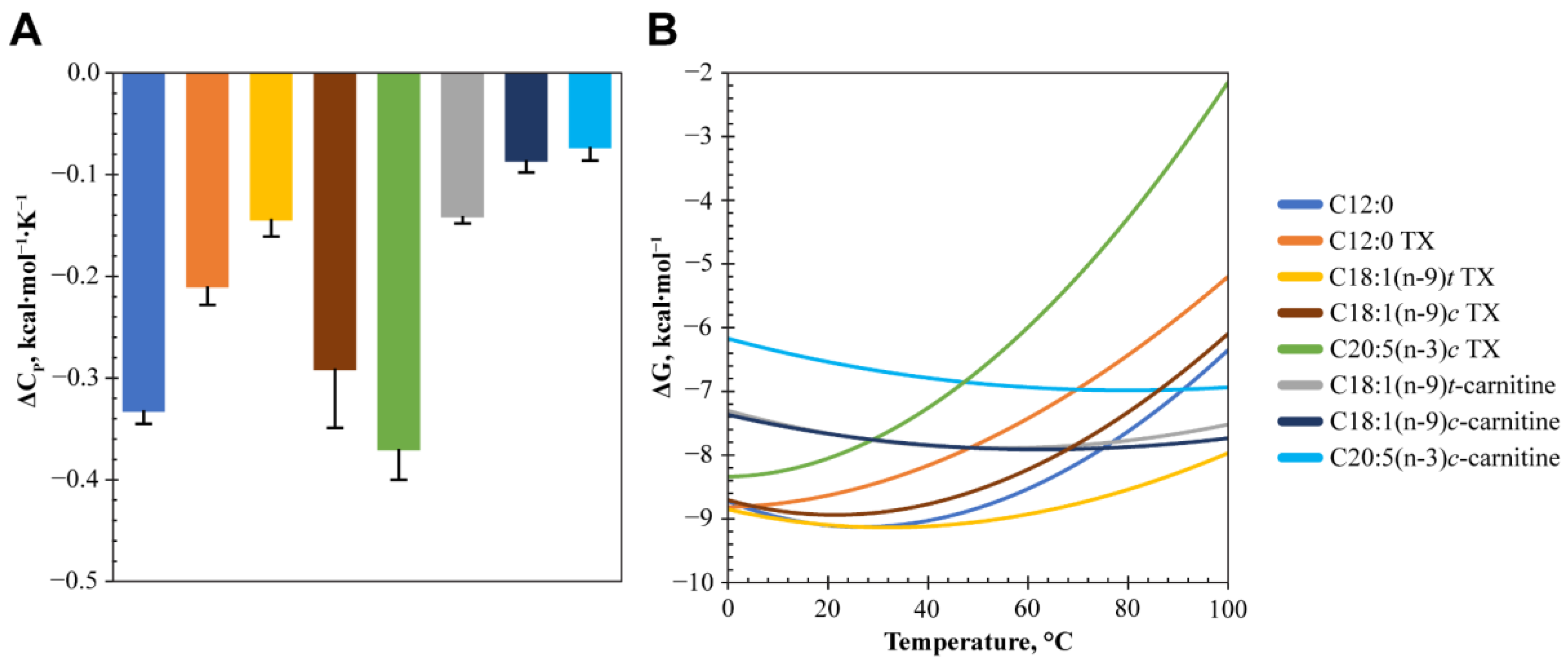
2.5. Determination of Changes in the Heat Capacity for LCFA and LCAC Binding to FABP3
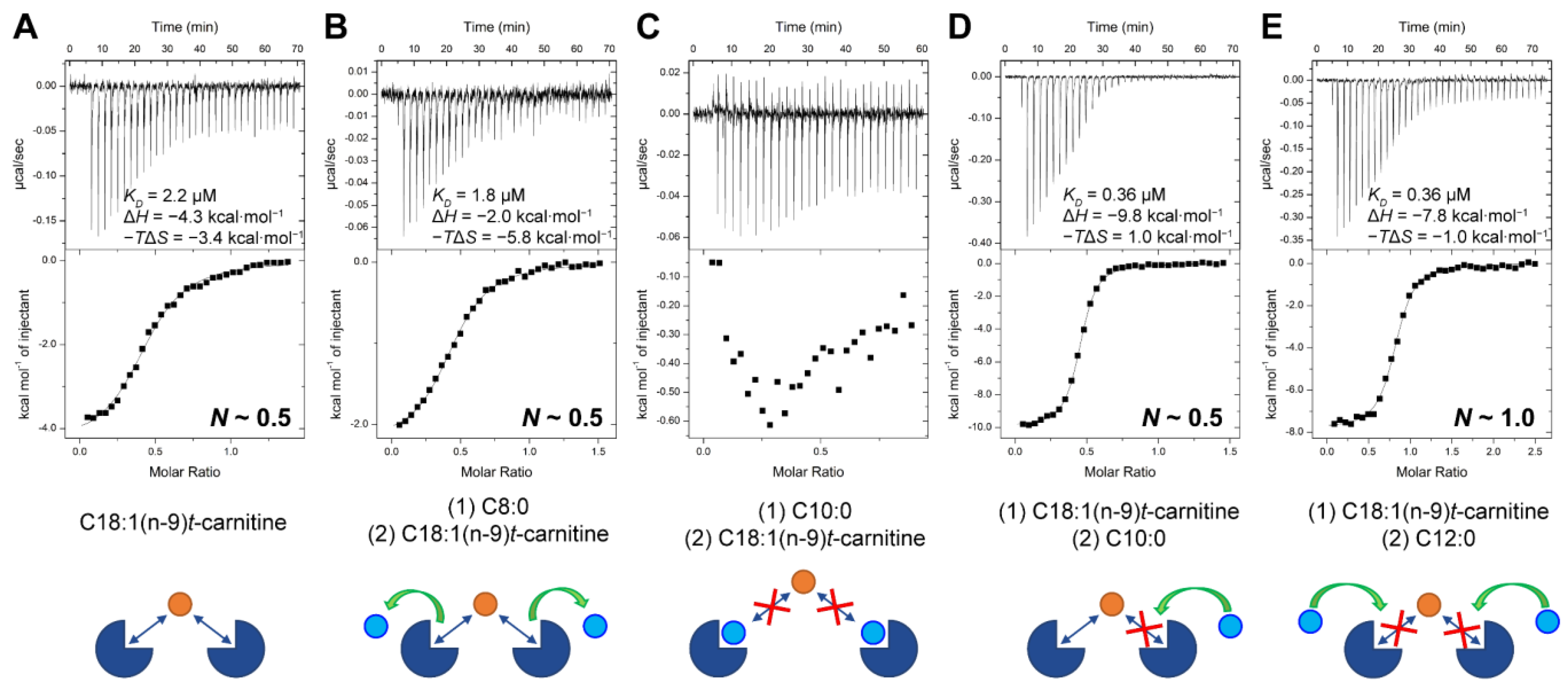
2.6. Competitive Binding Experiments for ACs
3. Discussion
4. Materials and Methods
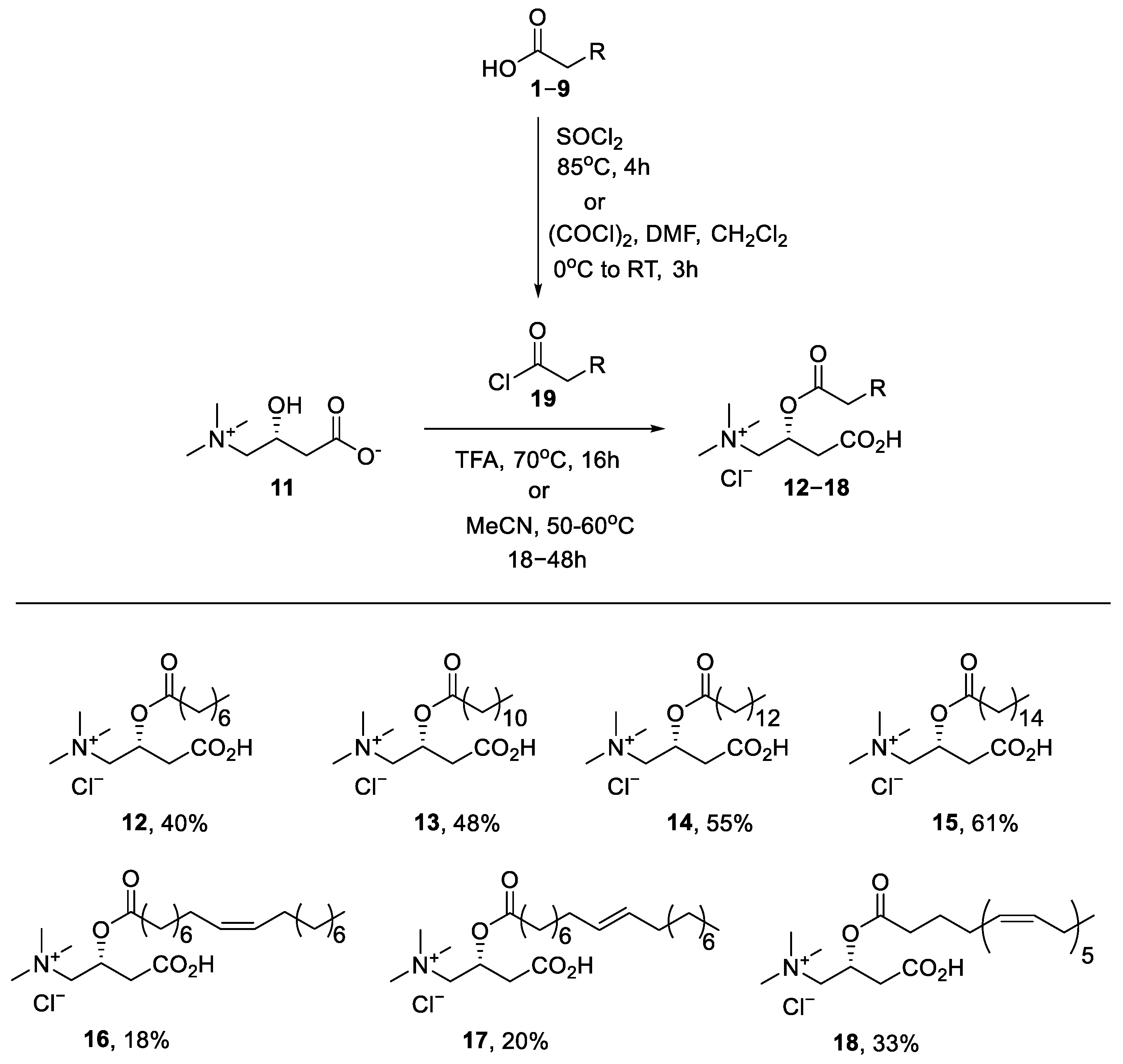
4.1. Chemistry
4.2. Protein Production
4.3. Protein Purification and Delipidation
4.4. LCAC Cytotoxicity Assays
4.5. NMR Spectroscopy
4.6. ITC Assay
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| AC | acylcarnitine |
| ACBP | acyl-CoA-binding protein |
| CMC | critical micelle concentration |
| CSP | chemical shift perturbation |
| FA | fatty acid |
| FABP | fatty acid binding protein |
| FABP3 | heart-type fatty acid binding protein |
| ITC | isothermal titration calorimetry |
| KPi | potassium phosphate buffer |
| LCAC | long-chain acylcarnitine |
| LCFA | long-chain fatty acid |
| NMR | nuclear magnetic resonance |
| SASA | solvent-accessible surface area |
| SEC | size-exclusion chromatography |
| TX | Triton™ X-100 |
References
- Furuhashi, M.; Hotamisligil, G.S. Fatty Acid-Binding Proteins: Role in Metabolic Diseases and Potential as Drug Targets. Nat. Rev. Drug Discov. 2008, 7, 489–503. [Google Scholar] [CrossRef] [Green Version]
- Santos, A.L.; Preta, G. Lipids in the Cell: Organisation Regulates Function. Cell. Mol. Life Sci. 2018, 75, 1909–1927. [Google Scholar] [CrossRef] [PubMed]
- Munir, R.; Lisec, J.; Swinnen, J.V.; Zaidi, N. Too Complex to Fail? Targeting Fatty Acid Metabolism for Cancer Therapy. Prog. Lipid Res. 2022, 85, 101143. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Wang, K.; Yang, L.; Liu, R.; Chu, Y.; Qin, X.; Yang, P.; Yu, H. Lipid Metabolism in Inflammation-Related Diseases. Analyst 2018, 143, 4526–4536. [Google Scholar] [CrossRef]
- Hotamisligil, G.S. Inflammation and Metabolic Disorders. Nature 2006, 444, 860–867. [Google Scholar] [CrossRef] [PubMed]
- Saltiel, A.R.; Kahn, C.R. Insulin Signalling and the Regulation of Glucose and Lipid Metabolism. Nature 2001, 414, 799–806. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Glatz, J.F.C.; Vork, M.M.; Cistola, D.P.; van der Vusse, G.J. Cytoplasmic Fatty Acid Binding Protein: Significance for Intracellular Transport of Fatty Acids and Putative Role on Signal Transduction Pathways. Prostaglandins Leukot. Essent. Fat. Acids 1993, 48, 33–41. [Google Scholar] [CrossRef] [PubMed]
- Zimmerman, A.W.; Veerkamp, J.H. New Insights into the Structure and Function of Fatty Acid-Binding Proteins. Cell. Mol. Life Sci. 2002, 59, 1096–1116. [Google Scholar] [CrossRef]
- Furuhashi, M. Fatty Acid-Binding Protein 4 in Cardiovascular and Metabolic Diseases. J. Atheroscler. Thromb. 2019, 26, 216–232. [Google Scholar] [CrossRef] [Green Version]
- Smathers, R.L.; Petersen, D.R. The Human Fatty Acid-Binding Protein Family: Evolutionary Divergences and Functions. Hum. Genom. 2011, 5, 170–191. [Google Scholar] [CrossRef] [Green Version]
- Chmurzyńska, A. The Multigene Family of Fatty Acid-Binding Proteins (FABPs): Function, Structure and Polymorphism. J. Appl. Genet. 2006, 47, 39–48. [Google Scholar] [CrossRef] [PubMed]
- Zanotti, G. Muscle Fatty Acid-Binding Protein. Biochim. Biophys. Acta-Mol. Cell Biol. Lipids 1999, 1441, 94–105. [Google Scholar] [CrossRef] [PubMed]
- Binas, B.; Danneberg, H.; Mcwhir, J.; Mullins, L.; Clark, A.J. Requirement for the Heart-type Fatty Acid Binding Protein in Cardiac Fatty Acid Utilization. FASEB J. 1999, 13, 805–812. [Google Scholar] [CrossRef] [Green Version]
- Schaap, F.G.; Binas, B.; Danneberg, H.; Van Der Vusse, G.J.; Glatz, J.F.C. Impaired Long-Chain Fatty Acid Utilization by Cardiac Myocytes Isolated from Mice Lacking the Heart-Type Fatty Acid Binding Protein Gene. Circ. Res. 1999, 85, 329–337. [Google Scholar] [CrossRef] [PubMed]
- Matsuoka, S.; Sugiyama, S.; Matsuoka, D.; Hirose, M.; Lethu, S.; Ano, H.; Hara, T.; Ichihara, O.; Kimura, S.R.; Murakami, S.; et al. Water-Mediated Recognition of Simple Alkyl Chains by Heart-Type Fatty-Acid-Binding Protein. Angew. Chem. Int. Ed. 2015, 54, 1508–1511. [Google Scholar] [CrossRef] [Green Version]
- Liepinsh, E.; Makrecka, M.; Kuka, J.; Makarova, E.; Vilskersts, R.; Cirule, H.; Sevostjanovs, E.; Grinberga, S.; Pugovics, O.; Dambrova, M. The Heart Is Better Protected against Myocardial Infarction in the Fed State Compared to the Fasted State. Metabolism 2014, 63, 127–136. [Google Scholar] [CrossRef]
- Makrecka, M.; Kuka, J.; Volska, K.; Antone, U.; Sevostjanovs, E.; Cirule, H.; Grinberga, S.; Pugovics, O.; Dambrova, M.; Liepinsh, E. Long-Chain Acylcarnitine Content Determines the Pattern of Energy Metabolism in Cardiac Mitochondria. Mol. Cell. Biochem. 2014, 395, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Korge, P.; Honda, H.M.; Weiss, J.N. Effects of Fatty Acids in Isolated Mitochondria: Implications for Ischemic Injury and Cardioprotection. Am. J. Physiol. Circ. Physiol. 2003, 285, H259–H269. [Google Scholar] [CrossRef] [Green Version]
- Tominaga, H.; Katoh, H.; Odagiri, K.; Takeuchi, Y.; Kawashima, H.; Saotome, M.; Urushida, T.; Satoh, H.; Hayashi, H. Different Effects of Palmitoyl-L-Carnitine and Palmitoyl-CoA on Mitochondrial Function in Rat Ventricular Myocytes. Am. J. Physiol. Circ. Physiol. 2008, 295, H105–H112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McCoin, C.S.; Knotts, T.A.; Adams, S.H. Acylcarnitines—Old Actors Auditioning for New Roles in Metabolic Physiology. Nat. Rev. Endocrinol. 2015, 11, 617–625. [Google Scholar] [CrossRef] [Green Version]
- Liepinsh, E.; Makrecka-Kuka, M.; Volska, K.; Kuka, J.; Makarova, E.; Antone, U.; Sevostjanovs, E.; Vilskersts, R.; Strods, A.; Tars, K.; et al. Long-Chain Acylcarnitines Determine Ischaemia/Reperfusion-Induced Damage in Heart Mitochondria. Biochem. J. 2016, 473, 1191–1202. [Google Scholar] [CrossRef] [PubMed]
- Aitken-Buck, H.M.; Krause, J.; Zeller, T.; Jones, P.P.; Lamberts, R.R. Long-Chain Acylcarnitines and Cardiac Excitation-Contraction Coupling: Links to Arrhythmias. Front. Physiol. 2020, 11, 577856. [Google Scholar] [CrossRef]
- Dambrova, M.; Makrecka-Kuka, M.; Kuka, J.; Vilskersts, R.; Nordberg, D.; Attwood, M.M.; Smesny, S.; Sen, Z.D.; Guo, A.C.; Oler, E.; et al. Acylcarnitines: Nomenclature, Biomarkers, Therapeutic Potential, Drug Targets, and Clinical Trials. Pharmacol. Rev. 2022, 74, 506–551. [Google Scholar] [CrossRef] [PubMed]
- Tahara, E.B.; Navarete, F.D.T.; Kowaltowski, A.J. Tissue-, Substrate-, and Site-Specific Characteristics of Mitochondrial Reactive Oxygen Species Generation. Free Radic. Biol. Med. 2009, 46, 1283–1297. [Google Scholar] [CrossRef] [PubMed]
- Dambrova, M.; Zuurbier, C.J.; Borutaite, V.; Liepinsh, E.; Makrecka-Kuka, M. Energy Substrate Metabolism and Mitochondrial Oxidative Stress in Cardiac Ischemia/Reperfusion Injury. Free Radic. Biol. Med. 2021, 165, 24–37. [Google Scholar] [CrossRef] [PubMed]
- Liepinsh, E.; Kuka, J.; Vilks, K.; Svalbe, B.; Stelfa, G.; Vilskersts, R.; Sevostjanovs, E.; Goldins, N.R.; Groma, V.; Grinberga, S.; et al. Low Cardiac Content of Long-Chain Acylcarnitines in TMLHE Knockout Mice Prevents Ischaemia-Reperfusion-Induced Mitochondrial and Cardiac Damage. Free Radic. Biol. Med. 2021, 177, 370–380. [Google Scholar] [CrossRef]
- Lassen, D.; Lucke, C.; Kveder, M.; Mesgarzadeh, A.; Schmidt, J.M.; Specht, B.; Lezius, A.; Spener, F.; Ruterjans, H. Three-Dimensional Structure of Bovine Heart Fatty-Acid-Binding Protein with Bound Palmitic Acid, Determined by Multidimensional NMR Spectroscopy. Eur. J. Biochem. 1995, 230, 266–280. [Google Scholar] [CrossRef]
- Lücke, C.; Rademacher, M.; Zimmerman, A.W.; Van Moerkerk, H.T.B.; Veerkamp, J.H.; Rüterjans, H. Spin-System Heterogeneities Indicate a Selected-Fit Mechanism in Fatty Acid Binding to Heart-Type Fatty Acid-Binding Protein (H-FABP). Biochem. J. 2001, 354, 259–266. [Google Scholar] [CrossRef] [PubMed]
- Young, A.C.; Scapin, G.; Kromminga, A.; Patel, S.B.; Veerkamp, J.H.; Sacchettini, J.C. Structural Studies on Human Muscle Fatty Acid Binding Protein at 1.4 Å Resolution: Binding Interactions with Three C18 Fatty Acids. Structure 1994, 2, 523–534. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Naso, J.N.; Bellesi, F.A.; Pizones Ruiz-Henestrosa, V.M.; Pilosof, A.M.R. A New Methodology to Assess the Solubility of Fatty Acids: Impact of Food Emulsifiers. Food Res. Int. 2020, 139, 109829. [Google Scholar] [CrossRef]
- Sharp, K. Entropy--Enthalpy Compensation: Fact or Artifact? Protein Sci. 2001, 10, 661–667. [Google Scholar] [CrossRef] [PubMed]
- Ford, D.M. Enthalpy-Entropy Compensation Is Not a General Feature of Weak Association. J. Am. Chem. Soc. 2005, 127, 16167–16170. [Google Scholar] [CrossRef]
- Sturtevant, J.M. Heat Capacity and Entropy Changes in Processes Involving Proteins. Proc. Natl. Acad. Sci. USA 1977, 74, 2236–2240. [Google Scholar] [CrossRef] [Green Version]
- Hinz, H.J. Thermodynamics of Protein-Ligand Interactions: Calorimetric Approaches. Annu. Rev. Biophys. Bioeng. 1983, 12, 285–317. [Google Scholar] [CrossRef]
- Firestone, R.S.; Cameron, S.A.; Karp, J.M.; Arcus, V.L.; Schramm, V.L. Heat Capacity Changes for Transition-State Analogue Binding and Catalysis with Human 5′-Methylthioadenosine Phosphorylase. ACS Chem. Biol. 2017, 12, 464–473. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jung, H.-I.; Bowden, S.J.; Cooper, A.; Perham, R.N. Thermodynamic Analysis of the Binding of Component Enzymes in the Assembly of the Pyruvate Dehydrogenase Multienzyme Complex of Bacillus Stearothermophilus. Protein Sci. 2002, 11, 1091–1100. [Google Scholar] [CrossRef] [PubMed]
- Schrödinger Release 2022-1: Maestro, Schrödinger, LLC: New York, USA. 2021. Available online: https://www.schrodinger.com/products/maestro (accessed on 10 March 2023).
- Goldberg, R.N.; Kishore, N.; Lennen, R.M. Thermodynamic Quantities for the Ionization Reaction of Buffers. J. Phys. Chem. Ref. Data 2002, 31, 231–370. [Google Scholar] [CrossRef] [Green Version]
- North, K.N.; Hoppel, C.L.; De Girolami, U.; Kozakewich, H.P.W.; Korson, M.S. Lethal Neonatal Deficiency of Carnitine Palmitoyltransferase II Associated with Dysgenesis of the Brain and Kidneys. J. Pediatr. 1995, 127, 414–420. [Google Scholar] [CrossRef]
- Merritt, J.L., II; Norris, M.; Kanungo, S. Fatty Acid Oxidation Disorders. Ann. Transl. Med. 2018, 6, 473. [Google Scholar] [CrossRef]
- Wanders, R.J.A.; Visser, G.; Ferdinandusse, S.; Vaz, F.M.; Houtkooper, R.H. Mitochondrial Fatty Acid Oxidation Disorders: Laboratory Diagnosis, Pathogenesis, and the Complicated Route to Treatment. J. Lipid Atheroscler. 2020, 9, 313. [Google Scholar] [CrossRef]
- Vilks, K.; Videja, M.; Makrecka-Kuka, M.; Katkevics, M.; Sevostjanovs, E.; Grandane, A.; Dambrova, M.; Liepinsh, E. Long-Chain Acylcarnitines Decrease the Phosphorylation of the Insulin Receptor at Tyr1151 through a Ptp1b-Dependent Mechanism. Int. J. Mol. Sci. 2021, 22, 6470. [Google Scholar] [CrossRef]
- Zimmerman, A.W.; Van Moerkerk, H.T.B.; Veerkamp, J.H. Ligand Specificity and Conformational Stability of Human Fatty Acid-Binding Proteins. Int. J. Biochem. Cell Biol. 2001, 33, 865–876. [Google Scholar] [CrossRef] [PubMed]
- Yalkowsky, S.H.; Zografi, G. Some Micellar Properties of Long-Chain Acylcarnitines. J. Colloid Interface Sci. 1970, 34, 525–533. [Google Scholar] [CrossRef] [Green Version]
- Stanley, F.E.; Warner, A.M.; Schneiderman, E.; Stalcup, A.M. Rapid Determination of Surfactant Critical Micelle Concentrations Using Pressure-Driven Flow with Capillary Electrophoresis Instrumentation. J. Chromatogr. A 2009, 1216, 8431–8434. [Google Scholar] [CrossRef] [Green Version]
- Ho, J.K.; Duclos, R.I.; Hamilton, J.A. Interactions of Acyl Carnitines with Model Membranes. J. Lipid Res. 2002, 43, 1429–1439. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malik, W.U.; Jain, A.K. Electrometric Determination of Critical Micelle Concentration of Soap Solutions. J. Electroanal. Chem. Interfacial Electrochem. 1967, 14, 37–41. [Google Scholar] [CrossRef]
- Fournier, N.C.; Richard, M.A. Fatty Acid-Binding Protein, a Potential Regulator of Energy Production in the Heart. Investigation of Mechanisms by Electron Spin Resonance. J. Biol. Chem. 1988, 263, 14471–14479. [Google Scholar] [CrossRef]
- Fournier, N.C.; Richard, M.A. Role of Fatty Acid-Binding Protein in Cardiac Fatty Acid Oxidation. Mol. Cell. Biochem. 1990, 98, 149–159. [Google Scholar] [CrossRef]
- Paulussen, R.J.A.; van der Logt, C.P.E.; Veerkamp, J.H. Characterization and Binding Properties of Fatty Acid-Binding Proteins from Human, Pig, and Rat Heart. Arch. Biochem. Biophys. 1988, 264, 533–545. [Google Scholar] [CrossRef]
- Jones, P.D.; Carne, A.; Bass, N.M.; Grigor, M.R. Isolation and Characterization of Fatty Acid Binding Proteins from Mammary Tissue of Lactating Rats. Biochem. J. 1988, 251, 919–925. [Google Scholar] [CrossRef] [Green Version]
- Nielsen, S.U.; Vorum, H.; Spener, F.; Brodersen, R. Two-Dimensional Electrophoresis of the Fatty Acid Binding Protein from Human Heart: Evidence for a Thiol Group Which Can Form an Intermolecular Disulfide Bond. Electrophoresis 1990, 11, 870–877. [Google Scholar] [CrossRef] [PubMed]
- Burton, P.B.J.; Hogben, C.E.; Joannou, C.L.; Clark, A.G.B.; Hsuan, J.J.; Totty, N.F.; Sorensen, C.; Evans, R.W.; Tynan, M.J. Heart Fatty Acid Binding Protein Is a Novel Regulator of Cardiac Myocyte Hypertrophy. Biochem. Biophys. Res. Commun. 1994, 205, 1822–1828. [Google Scholar] [CrossRef] [PubMed]
- Haunerland, N.H.; Jacobson, B.L.; Wesenberg, G.; Rayment, I.; Holden, H.M. Three-Dimensional Structure of the Muscle Fatty-Acid-Binding Protein Isolated from the Desert Locust Schistocerca Gregaria. Biochemistry 1994, 33, 12378–12385. [Google Scholar] [CrossRef]
- Gillilan, R.E.; Ayers, S.D.; Noy, N. Structural Basis for Activation of Fatty Acid-Binding Protein 4. J. Mol. Biol. 2007, 372, 1246–1260. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ory, J.J.; Banaszak, L.J. Studies of the Ligand Binding Reaction of Adipocyte Lipid Binding Protein Using the Fluorescent Probe 1,8-Anilinonaphthalene-8-Sulfonate. Biophys. J. 1999, 77, 1107–1116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hellberg, K.; Grimsrud, P.A.; Kruse, A.C.; Banaszak, L.J.; Ohlendorf, D.H.; Bernlohr, D.A. X-ray Crystallographic Analysis of Adipocyte Fatty Acid Binding Protein (AP2) Modified with 4-Hydroxy-2-Nonenal. Protein Sci. 2010, 19, 1480–1489. [Google Scholar] [CrossRef] [Green Version]
- Sanson, B.; Wang, T.; Sun, J.; Wang, L.; Kaczocha, M.; Ojima, I.; Deutsch, D.; Li, H. Crystallographic Study of FABP5 as an Intracellular Endocannabinoid Transporter. Acta Crystallogr. Sect. D Biol. Crystallogr. 2014, 70, 290–298. [Google Scholar] [CrossRef]
- Liu, X.; Liu, P.; Li, H.; Xu, Z.; Jia, L.; Xia, Y.; Yu, M.; Tang, W.; Zhu, X.; Chen, C.; et al. Excited-State Intermediates in a Designer Protein Encoding a Phototrigger Caught by an X-ray Free-Electron Laser. Nat. Chem. 2022, 14, 1054–1060. [Google Scholar] [CrossRef]
- Xu, Z.; Bernlohr, D.A.; Banaszak, L.J. The Adipocyte Lipid-Binding Protein at 1.6-Å Resolution. Crystal Structures of the Apoprotein and with Bound Saturated and Unsaturated Fatty Acids. J. Biol. Chem. 1993, 268, 7874–7884. [Google Scholar] [CrossRef]
- LaLonde, J.M.; Bernlohr, D.A.; Banaszak, L.J. X-ray Crystallographic Structures of Adipocyte Lipid-Binding Protein Complexed with Palmitate and Hexadecanesulfonic Acid. Properties of Cavity Binding Sites. Biochemistry 1994, 33, 4885–4895. [Google Scholar] [CrossRef]
- Nivet, J.; Le Blanc, M.; Riess, J. Synthesis and Preliminary Evaluation of Perfluoroalkylacyl Carnitines as Surfactants for Biomedical Use. Eur. J. Med. Chem. 1991, 26, 953–960. [Google Scholar] [CrossRef]
- Gong, X.; Li, J.; Wu, J.; Xun, L.; Zhai, H.; Feng, B.; Xu, W.; Tang, W. Synthesis of Carnitine Benzyl Esters as Prodrugs. J. Chem. Res. 2008, 2008, 327–330. [Google Scholar] [CrossRef]
- Tropea, J.E.; Cherry, S.; Waugh, D.S. Expression and Purification of Soluble His6-Tagged TEV Protease. Methods Mol. Biol. 2009, 498, 297–307. [Google Scholar] [PubMed]
- Mosmann, T. Rapid Colorimetric Assay for Cellular Growth and Survival: Application to Proliferation and Cytotoxicity Assays. J. Immunol. Methods 1983, 65, 55–63. [Google Scholar] [CrossRef] [PubMed]
- Keller, R. The Computer Aided Resonance Assignment Tutorial; CANTINA Verlag: Goldau, Switzerland, 2004; ISBN 3856001123. [Google Scholar]
- Williamson, M.P. Using Chemical Shift Perturbation to Characterise Ligand Binding. Prog. Nucl. Magn. Reson. Spectrosc. 2013, 73, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Hobbs, B.; Drant, J.; Williamson, M.P. The Measurement of Binding Affinities by NMR Chemical Shift Perturbation. J. Biomol. NMR 2022, 76, 153–163. [Google Scholar] [CrossRef] [PubMed]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| # | Compound 1 | Name | 0.1% TX | KD, μM | ΔG, kcal·mol−1 | ΔH, kcal·mol−1 | −TΔS, kcal·mol−1 | N 2 |
|---|---|---|---|---|---|---|---|---|
| 1 | C8:0 | caprylate | - | 1.86 ± 0.05 | −7.82 ± 0.02 | −8.01 ± 0.09 | 0.19 ± 0.07 | 1.0 |
| + | 1.109 ± 0.001 | −8.13 ± 0.01 | −12.6 ± 0.3 | 4.5 ± 0.3 | 1.0 | |||
| 2 | C10:0 | caprate | - | 0.252 ± 0.004 | −9.01 ± 0.01 | −11.09 ± 0.08 | 2.08 ± 0.08 | 1.0 |
| + | 0.56 ± 0.02 | −8.535 ± 0.007 | −13.2 ± 0.3 | 4.7 ± 0.3 | 1.0 | |||
| 3 | C12:0 | laurate | - | 0.20 ± 0.02 | −9.12 ± 0.06 | −8.60 ± 0.08 | −0.53 ± 0.02 | 1.0 |
| + | 0.57 ± 0.06 | −8.54 ± 0.06 | −14.5 ± 0.4 | 6.0 ± 0.3 | 1.0 | |||
| 4 | C14:0 | myristate | + | 0.3 ± 0.1 | −8.9 ± 0.2 | −10.9 ± 0.4 | 1.9 ± 0.2 | 1.0 |
| 5 | C16:0 | palmitate | + | 0.14 ± 0.02 | −9.34 ± 0.07 | −8.7 ± 0.9 | −0.6 ± 0.8 | 1.0 |
| 6 | C18:0 | stearate | + | 0.7 ± 0.1 | −8.34 ± 0.04 | −6.4 ± 0.4 | −2.0 ± 0.4 | 1.0 |
| 7 | C18:1(n-9)c | oleate | + | 0.29 ± 0.03 | −8.93 ± 0.06 | −10.0 ± 0.4 | 1.1 ± 0.5 | 1.0 |
| 8 | C18:1(n-9)t | elaidate | + | 0.21 ± 0.02 | −9.12 ± 0.06 | −8.0 ± 0.9 | −1.1 ± 0.9 | 1.0 |
| 9 | C20:5(n-3)c | EPA | + | 1.6 ± 0.2 | −7.90 ± 0.07 | −17.4 ± 0.7 | 9.5 ± 0.8 | 1.0 |
| 10 | C16:0-CoA | palmitoyl-CoA | - | 8.9 ± 1.2 | −6.90 ± 0.08 | −5.04 ± 0.09 | −1.86 ± 0.01 | 1.0 |
| 14 | C14:0-carnitine | miristoylcarnitine | - | 11.3 | −6.7 | −1.1 | −5.7 | 0.5 |
| 16 | C18:1(n-9)c-carnitine | oleoylcarnitine | - | 2.21 ± 0.07 | −7.72 ± 0.01 | −4.6 ± 0.2 | −3.1 ± 0.2 | 0.5 |
| 17 | C18:1(n-9)t-carnitine | elaidoylcarnitine | - | 2.19 ± 0.04 | −7.72 ± 0.02 | −4.29 ± 0.06 | −3.43 ± 0.08 | 0.5 |
| 18 | C20:5(n-3)c-carnitine | EPA-carnitine | - | 14.83 ± 0.08 | −6.61 ± 0.03 | −2.43 ± 0.09 | −4.19 ± 0.06 | 1.0 |
| # | Compound | 0.1% TX | ΔCp, kcal∙mol−1∙K−1 | R2 | Polar Surface Area, Å2 * |
|---|---|---|---|---|---|
| 3 | C12:0 | − | −0.33 ± 0.01 | 0.996 | 40.1 |
| + | −0.21 ± 0.02 | 0.981 | |||
| 7 | C18:1(n-9)c | + | −0.29 ± 0.06 | 0.964 | 40.1 |
| 8 | C18:1(n-9)t | + | −0.15 ± 0.02 | 0.988 | 40.1 |
| 9 | C20:5(n-3)c | + | −0.37 ± 0.03 | 0.994 | 40.1 |
| 16 | C18:1(n-9)c-carnitine | − | −0.09 ± 0.01 | 0.998 | 66.4 |
| 17 | C18:1(n-9)t-carnitine | − | −0.14 ± 0.01 | 0.985 | 66.4 |
| 18 | C20:5(n-3)c-carnitine | − | −0.07 ± 0.01 | 0.975 | 66.4 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zelencova-Gopejenko, D.; Videja, M.; Grandane, A.; Pudnika-Okinčica, L.; Sipola, A.; Vilks, K.; Dambrova, M.; Jaudzems, K.; Liepinsh, E. Heart-Type Fatty Acid Binding Protein Binds Long-Chain Acylcarnitines and Protects against Lipotoxicity. Int. J. Mol. Sci. 2023, 24, 5528. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms24065528
Zelencova-Gopejenko D, Videja M, Grandane A, Pudnika-Okinčica L, Sipola A, Vilks K, Dambrova M, Jaudzems K, Liepinsh E. Heart-Type Fatty Acid Binding Protein Binds Long-Chain Acylcarnitines and Protects against Lipotoxicity. International Journal of Molecular Sciences. 2023; 24(6):5528. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms24065528
Chicago/Turabian StyleZelencova-Gopejenko, Diana, Melita Videja, Aiga Grandane, Linda Pudnika-Okinčica, Anda Sipola, Karlis Vilks, Maija Dambrova, Kristaps Jaudzems, and Edgars Liepinsh. 2023. "Heart-Type Fatty Acid Binding Protein Binds Long-Chain Acylcarnitines and Protects against Lipotoxicity" International Journal of Molecular Sciences 24, no. 6: 5528. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms24065528