
Ursodeoxycholic Acid Binds PERK and Ameliorates Neurite Atrophy in a Cellular Model of GM2 Gangliosidosis
 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
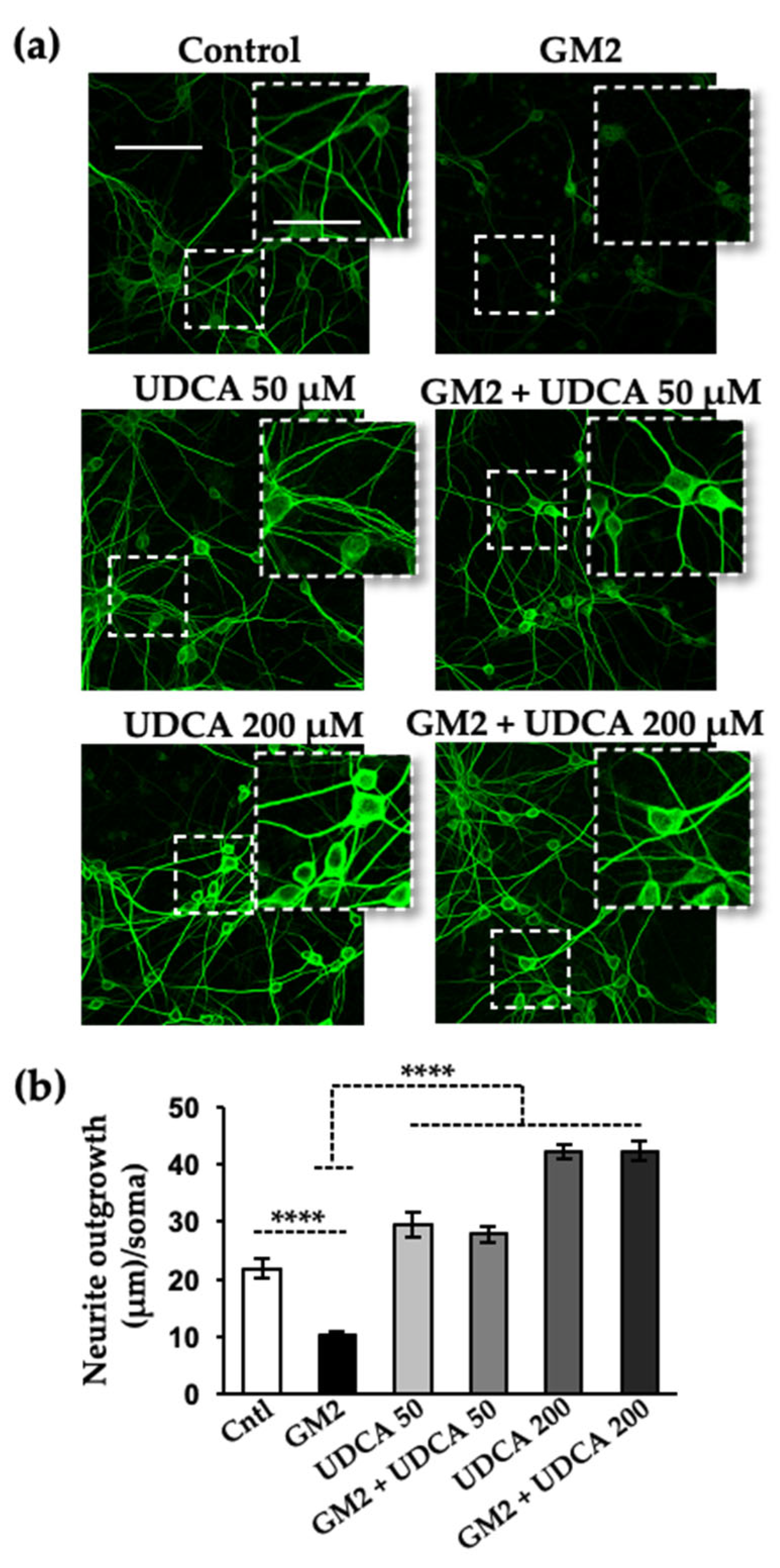
2.1. UDCA Treatment Reverses the Decreased Neurite Outgrowth following GM2 Loading
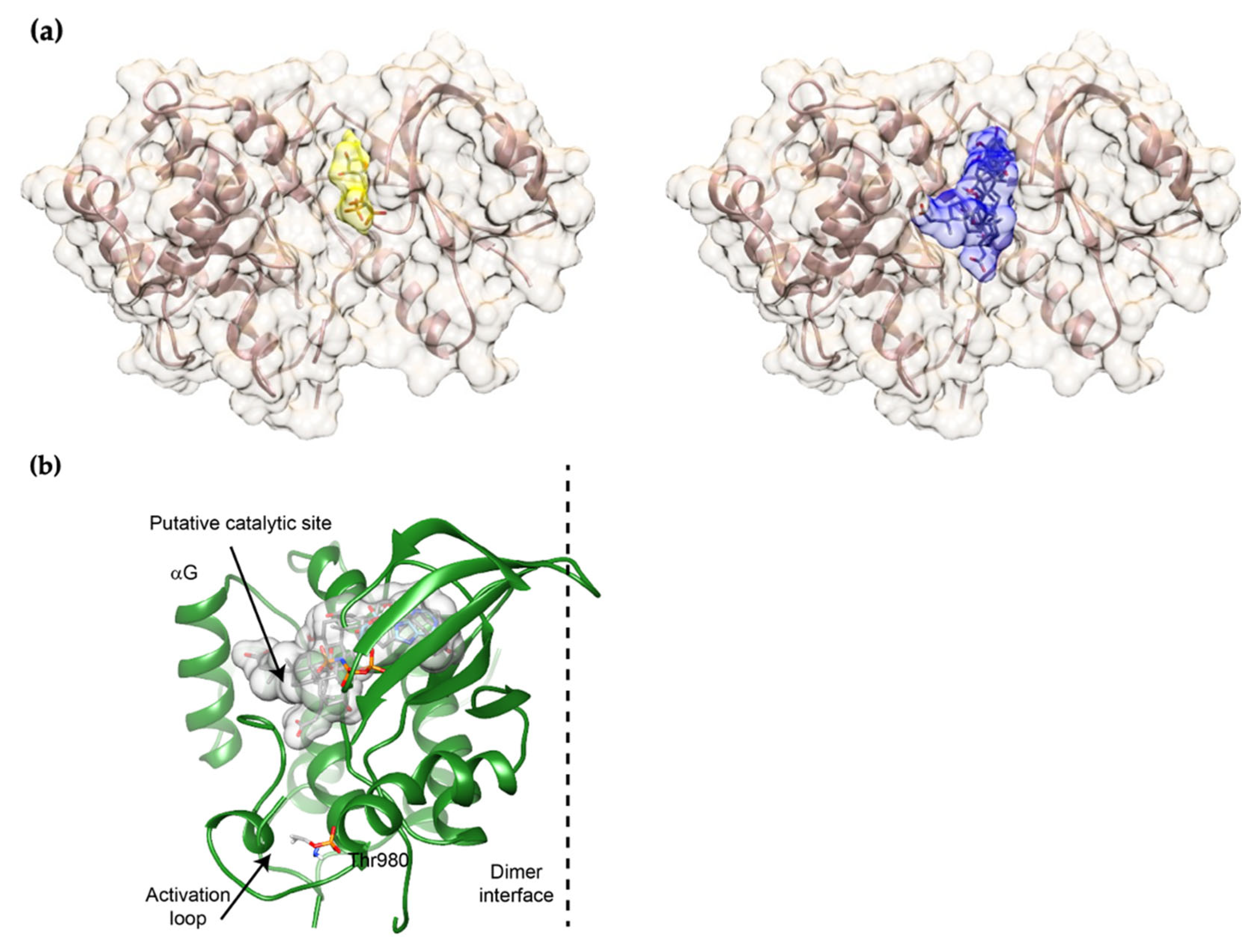
2.2. UDCA Fits into the ATP-Binding Site of mPERK KD
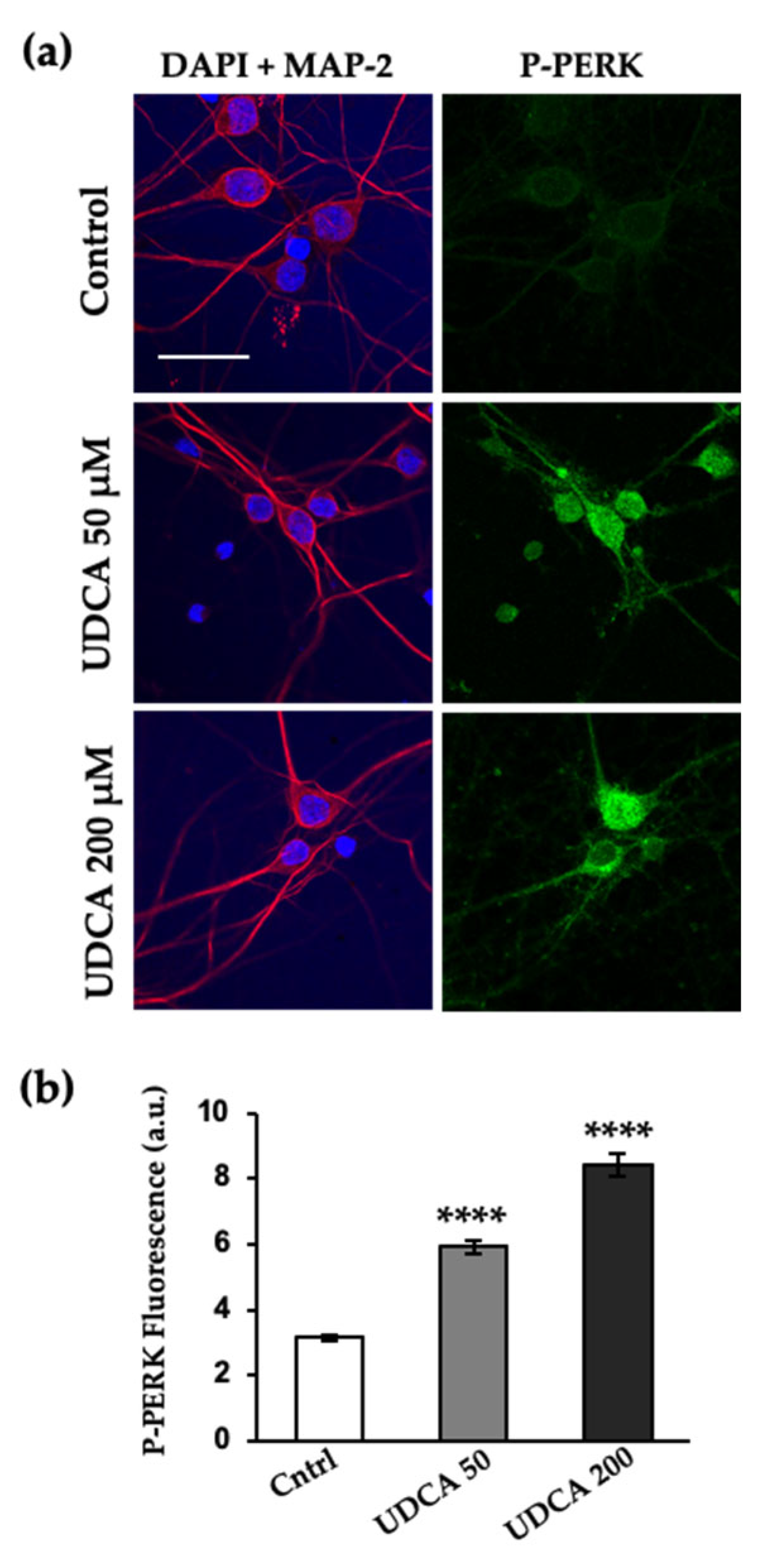
2.3. UDCA Prompts PERK Activation at Cellular Level and In Vitro
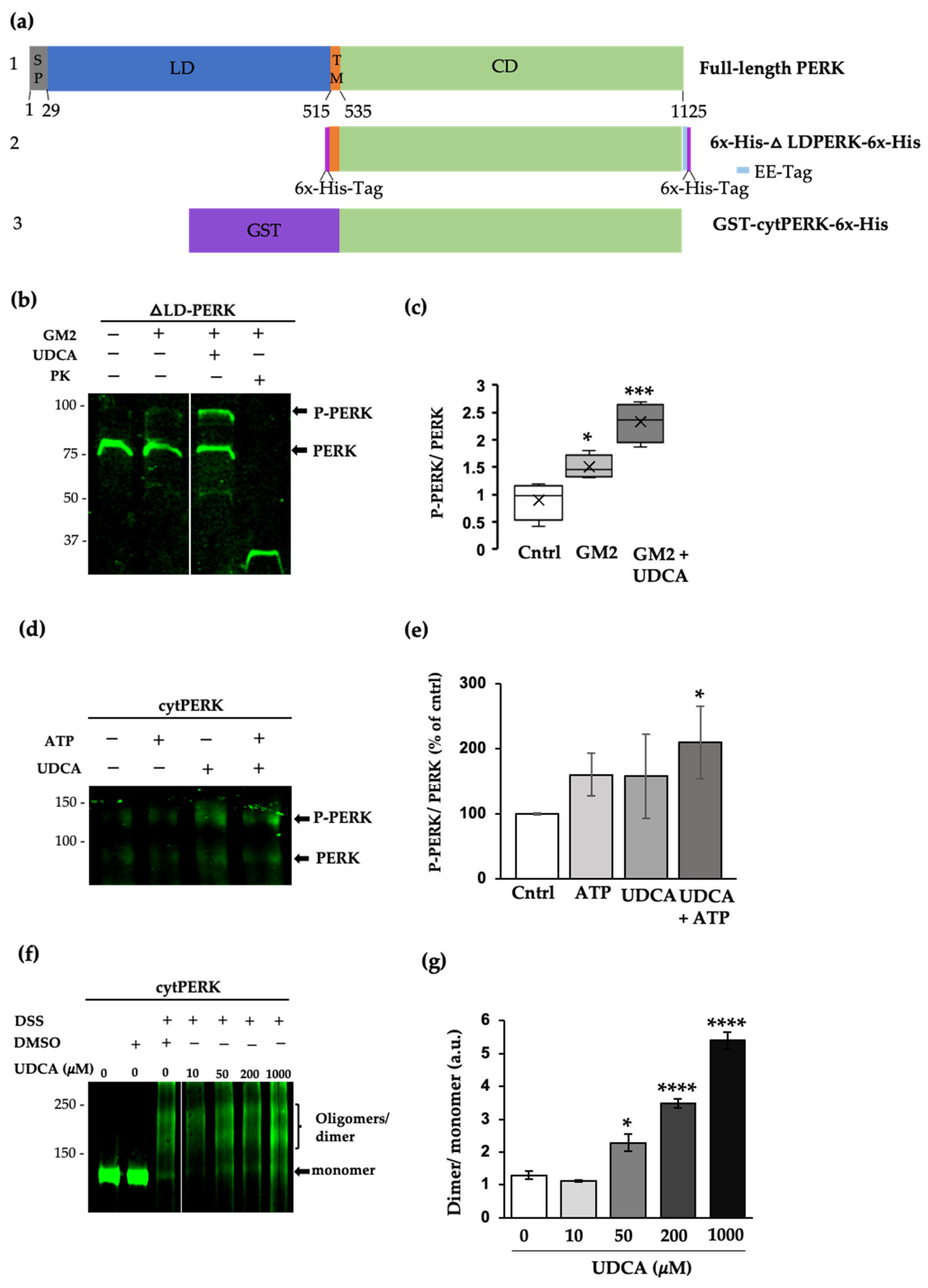
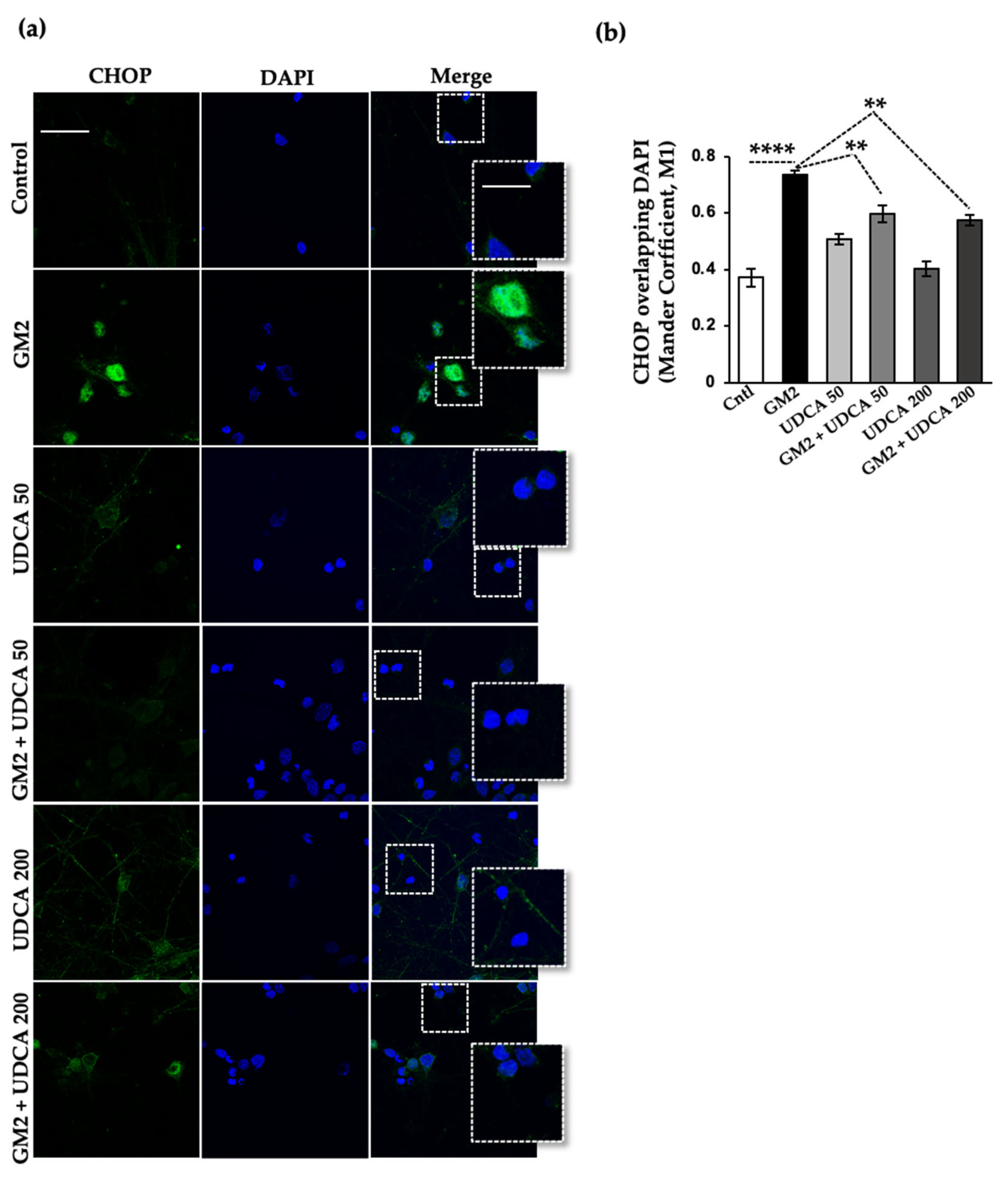
2.4. UDCA Modulates PERK Signaling Activated by GM2 Accumulation
3. Discussion
4. Materials and Methods
4.1. Reagents and Antibodies
4.2. Cell Cultures
4.3. Immunocytochemistry
4.4. Bacterial Protein Expression
4.5. Preparation of Liposomes with Incorporated ΔLD-PERK
4.6. In Vitro Kinase Assays
4.7. GST-cytPERK Crosslinking Assay
4.8. Western Blotting
4.9. Protein-Ligand Docking
4.10. Data Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Van Ziel, A.M.; Scheper, W. The UPR in Neurodegenerative Disease: Not Just an Inside Job. Biomolecules 2020, 10, 1090. [Google Scholar] [CrossRef] [PubMed]
- Moreno, J.A.; Radford, H.; Peretti, D.; Steinert, J.R.; Verity, N.; Martin, M.G.; Halliday, M.; Morgan, J.; Dinsdale, D.; Ortori, C.A.; et al. Sustained translational repression by eIF2alpha-P mediates prion neurodegeneration. Nature 2012, 485, 507–511. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hetz, C.; Saxena, S. ER stress and the unfolded protein response in neurodegeneration. Nat. Rev. Neurol. 2017, 13, 477–491. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.H.; Li, H.; Yasumura, D.; Cohen, H.R.; Zhang, C.; Panning, B.; Shokat, K.M.; Lavail, M.M.; Walter, P. IRE1 signaling affects cell fate during the unfolded protein response. Science 2007, 318, 944–949. [Google Scholar] [CrossRef] [Green Version]
- Lin, J.H.; Li, H.; Zhang, Y.; Ron, D.; Walter, P. Divergent effects of PERK and IRE1 signaling on cell viability. PLoS ONE 2009, 4, e4170. [Google Scholar] [CrossRef]
- Virgolini, M.J.; Feliziani, C.; Cambiasso, M.J.; Lopez, P.H.; Bollo, M. Neurite atrophy and apoptosis mediated by PERK signaling after accumulation of GM2-ganglioside. Biochim. Biophys. Acta Mol. Cell Res. 2019, 1866, 225–239. [Google Scholar] [CrossRef]
- Harding, H.P.; Zhang, Y.; Ron, D. Protein translation and folding are coupled by an endoplasmic-reticulum-resident kinase. Nature 1999, 397, 271–274. [Google Scholar] [CrossRef]
- Cullinan, S.B.; Zhang, D.; Hannink, M.; Arvisais, E.; Kaufman, R.J.; Diehl, J.A. Nrf2 is a direct PERK substrate and effector of PERK-dependent cell survival. Mol. Cell. Biol. 2003, 23, 7198–7209. [Google Scholar] [CrossRef] [Green Version]
- Cullinan, S.B.; Diehl, J.A. PERK-dependent activation of Nrf2 contributes to redox homeostasis and cell survival following endoplasmic reticulum stress. J. Biol. Chem. 2004, 279, 20108–20117. [Google Scholar] [CrossRef] [Green Version]
- Bertolotti, A.; Zhang, Y.; Hendershot, L.M.; Harding, H.P.; Ron, D. Dynamic interaction of BiP and ER stress transducers in the unfolded-protein response. Nat. Cell Biol. 2000, 2, 326–332. [Google Scholar] [CrossRef]
- Zhou, J.; Liu, C.Y.; Back, S.H.; Clark, R.L.; Peisach, D.; Xu, Z.; Kaufman, R.J. The crystal structure of human IRE1 luminal domain reveals a conserved dimerization interface required for activation of the unfolded protein response. Proc. Natl. Acad. Sci. USA 2006, 103, 14343–14348. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shamu, C.E.; Walter, P. Oligomerization and phosphorylation of the Ire1p kinase during intracellular signaling from the endoplasmic reticulum to the nucleus. EMBO J. 1996, 15, 3028–3039. [Google Scholar] [CrossRef] [PubMed]
- Volmer, R.; van der Ploeg, K.; Ron, D. Membrane lipid saturation activates endoplasmic reticulum unfolded protein response transducers through their transmembrane domains. Proc. Natl. Acad. Sci. USA 2013, 110, 4628–4633. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, W.; Frank, C.L.; Korth, M.J.; Sopher, B.L.; Novoa, I.; Ron, D.; Katze, M.G. Control of PERK eIF2alpha kinase activity by the endoplasmic reticulum stress-induced molecular chaperone P58IPK. Proc. Natl. Acad. Sci. USA 2002, 99, 15920–15925. [Google Scholar] [CrossRef] [Green Version]
- Bollo, M.; Paredes, R.M.; Holstein, D.; Zheleznova, N.; Camacho, P.; Lechleiter, J.D. Calcineurin interacts with PERK and dephosphorylates calnexin to relieve ER stress in mammals and frogs. PLoS ONE 2010, 5, e11925. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Holstein, D.M.; Aime, S.; Bollo, M.; Lechleiter, J.D. Calcineurin beta protects brain after injury by activating the unfolded protein response. Neurobiol. Dis. 2016, 94, 139–156. [Google Scholar] [CrossRef] [Green Version]
- Gravel, R.; Kaback, M.; Proia, R.; Sandhoff, K.; Suzuki, K.; Suzuki, K. The GM2 gangliosidoses. In The Metabolic and Molecular Bases of Inherited Disease; McGraw-Hill: New York, NY, USA, 2000; pp. 3371–3388. [Google Scholar]
- Sandhoff, K.; Harzer, K. Gangliosides and gangliosidoses: Principles of molecular and metabolic pathogenesis. J. Neurosci. 2013, 33, 10195–10208. [Google Scholar] [CrossRef] [Green Version]
- Lawson, C.A.; Martin, D.R. Animal models of GM2 gangliosidosis: Utility and limitations. Appl. Clin. Genet. 2016, 9, 111–120. [Google Scholar] [CrossRef] [Green Version]
- Welch, W.J.; Brown, C.R. Influence of molecular and chemical chaperones on protein folding. Cell Stress Chaperones 1996, 1, 109–115. [Google Scholar] [CrossRef]
- Berger, E.; Haller, D. Structure-function analysis of the tertiary bile acid TUDCA for the resolution of endoplasmic reticulum stress in intestinal epithelial cells. Biochem. Biophys. Res. Commun. 2011, 409, 610–615. [Google Scholar] [CrossRef]
- Song, S.; Liang, J.J.; Mulhern, M.L.; Madson, C.J.; Shinohara, T. Cholesterol-derived bile acids enhance the chaperone activity of alpha-crystallins. Cell Stress Chaperones 2011, 16, 475–480. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, Q.; Khaoustov, V.I.; Chung, C.C.; Sohn, J.; Krishnan, B.; Lewis, D.E.; Yoffe, B. Effect of tauroursodeoxycholic acid on endoplasmic reticulum stress-induced caspase-12 activation. Hepatology 2002, 36, 592–601. [Google Scholar] [CrossRef] [PubMed]
- Cao, A.L.; Wang, L.; Chen, X.; Wang, Y.M.; Guo, H.J.; Chu, S.; Liu, C.; Zhang, X.M.; Peng, W. Ursodeoxycholic acid and 4-phenylbutyrate prevent endoplasmic reticulum stress-induced podocyte apoptosis in diabetic nephropathy. Lab. Investig. 2016, 96, 610–622. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, Y.J.; Kim, S.J.; Heo, T.H. Protective effect of catechin in type I Gaucher disease cells by reducing endoplasmic reticulum stress. Biochem. Biophys. Res. Commun. 2011, 413, 254–258. [Google Scholar] [CrossRef] [PubMed]
- Monticelli, M.; Liguori, L.; Allocca, M.; Bosso, A.; Andreotti, G.; Lukas, J.; Monti, M.C.; Morretta, E.; Cubellis, M.V.; Hay Mele, B. Drug Repositioning for Fabry Disease: Acetylsalicylic Acid Potentiates the Stabilization of Lysosomal Alpha-Galactosidase by Pharmacological Chaperones. Int. J. Mol. Sci. 2022, 23, 5105. [Google Scholar] [CrossRef]
- Wei, H.; Kim, S.J.; Zhang, Z.; Tsai, P.C.; Wisniewski, K.E.; Mukherjee, A.B. ER and oxidative stresses are common mediators of apoptosis in both neurodegenerative and non-neurodegenerative lysosomal storage disorders and are alleviated by chemical chaperones. Hum. Mol. Genet. 2008, 17, 469–477. [Google Scholar] [CrossRef]
- Ferreira, A.; Busciglio, J.; Landa, C.; Caceres, A. Ganglioside-enhanced neurite growth: Evidence for a selective induction of high-molecular-weight MAP-2. J. Neurosci. 1990, 10, 293–302. [Google Scholar] [CrossRef] [Green Version]
- Viana, R.J.; Nunes, A.F.; Castro, R.E.; Ramalho, R.M.; Meyerson, J.; Fossati, S.; Ghiso, J.; Rostagno, A.; Rodrigues, C.M. Tauroursodeoxycholic acid prevents E22Q Alzheimer’s Abeta toxicity in human cerebral endothelial cells. Cell. Mol. Life Sci. 2009, 66, 1094–1104. [Google Scholar] [CrossRef] [Green Version]
- Gani, A.R.; Uppala, J.K.; Ramaiah, K.V. Tauroursodeoxycholic acid prevents stress induced aggregation of proteins in vitro and promotes PERK activation in HepG2 cells. Arch. Biochem. Biophys. 2015, 568, 8–15. [Google Scholar] [CrossRef]
- Seyhun, E.; Malo, A.; Schafer, C.; Moskaluk, C.A.; Hoffmann, R.T.; Goke, B.; Kubisch, C.H. Tauroursodeoxycholic acid reduces endoplasmic reticulum stress, acinar cell damage, and systemic inflammation in acute pancreatitis. Am. J. Physiol. Gastrointest. Liver Physiol. 2011, 301, G773–G782. [Google Scholar] [CrossRef] [Green Version]
- Sicheri, F.; Moarefi, I.; Kuriyan, J. Crystal structure of the Src family tyrosine kinase Hck. Nature 1997, 385, 602–609. [Google Scholar] [CrossRef]
- Dar, A.C.; Dever, T.E.; Sicheri, F. Higher-order substrate recognition of eIF2alpha by the RNA-dependent protein kinase PKR. Cell 2005, 122, 887–900. [Google Scholar] [CrossRef] [Green Version]
- Cui, W.; Li, J.; Ron, D.; Sha, B. The structure of the PERK kinase domain suggests the mechanism for its activation. Acta Crystallogr. D Biol. Crystallogr. 2011, 67, 423–428. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marciniak, S.J.; Garcia-Bonilla, L.; Hu, J.; Harding, H.P.; Ron, D. Activation-dependent substrate recruitment by the eukaryotic translation initiation factor 2 kinase PERK. J. Cell Biol. 2006, 172, 201–209. [Google Scholar] [CrossRef] [PubMed]
- Sood, R.; Porter, A.C.; Ma, K.; Quilliam, L.A.; Wek, R.C. Pancreatic eukaryotic initiation factor-2alpha kinase (PEK) homologues in humans, Drosophila melanogaster and Caenorhabditis elegans that mediate translational control in response to endoplasmic reticulum stress. Biochem. J. 2000, 346 Pt 2, 281–293. [Google Scholar] [CrossRef] [PubMed]
- Korennykh, A.; Walter, P. Structural basis of the unfolded protein response. Annu. Rev. Cell Dev. Biol. 2012, 28, 251–277. [Google Scholar] [CrossRef] [PubMed]
- Korennykh, A.V.; Egea, P.F.; Korostelev, A.A.; Finer-Moore, J.; Zhang, C.; Shokat, K.M.; Stroud, R.M.; Walter, P. The unfolded protein response signals through high-order assembly of Ire1. Nature 2009, 457, 687–693. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, X.; Gureasko, J.; Shen, K.; Cole, P.A.; Kuriyan, J. An allosteric mechanism for activation of the kinase domain of epidermal growth factor receptor. Cell 2006, 125, 1137–1149. [Google Scholar] [CrossRef] [Green Version]
- Ozcan, U.; Yilmaz, E.; Ozcan, L.; Furuhashi, M.; Vaillancourt, E.; Smith, R.O.; Gorgun, C.Z.; Hotamisligil, G.S. Chemical chaperones reduce ER stress and restore glucose homeostasis in a mouse model of type 2 diabetes. Science 2006, 313, 1137–1140. [Google Scholar] [CrossRef] [Green Version]
- de Almeida, S.F.; Picarote, G.; Fleming, J.V.; Carmo-Fonseca, M.; Azevedo, J.E.; de Sousa, M. Chemical chaperones reduce endoplasmic reticulum stress and prevent mutant HFE aggregate formation. J. Biol. Chem. 2007, 282, 27905–27912. [Google Scholar] [CrossRef] [Green Version]
- Xie, W.; Pariollaud, M.; Wixted, W.E.; Chitnis, N.; Fornwald, J.; Truong, M.; Pao, C.; Liu, Y.; Ames, R.S.; Callahan, J.; et al. Identification and characterization of PERK activators by phenotypic screening and their effects on NRF2 activation. PLoS ONE 2015, 10, e0119738. [Google Scholar] [CrossRef] [PubMed]
- van Meer, G.; Voelker, D.R.; Feigenson, G.W. Membrane lipids: Where they are and how they behave. Nat. Rev. Mol. Cell Biol. 2008, 9, 112–124. [Google Scholar] [CrossRef]
- Pelled, D.; Lloyd-Evans, E.; Riebeling, C.; Jeyakumar, M.; Platt, F.M.; Futerman, A.H. Inhibition of calcium uptake via the sarco/endoplasmic reticulum Ca2+-ATPase in a mouse model of Sandhoff disease and prevention by treatment with N-butyldeoxynojirimycin. J. Biol. Chem. 2003, 278, 29496–29501. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jacquemyn, J.; Cascalho, A.; Goodchild, R.E. The ins and outs of endoplasmic reticulum-controlled lipid biosynthesis. EMBO Rep. 2017, 18, 1905–1921. [Google Scholar] [CrossRef] [PubMed]
- Bigay, J.; Antonny, B. Curvature, lipid packing, and electrostatics of membrane organelles: Defining cellular territories in determining specificity. Dev. Cell 2012, 23, 886–895. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marsh, D. Liquid-ordered phases induced by cholesterol: A compendium of binary phase diagrams. Biochim. Biophys. Acta 2010, 1798, 688–699. [Google Scholar] [CrossRef] [Green Version]
- Westerlund, B.; Slotte, J.P. How the molecular features of glycosphingolipids affect domain formation in fluid membranes. Biochim. Biophys. Acta 2009, 1788, 194–201. [Google Scholar] [CrossRef] [Green Version]
- Grauby-Heywang, C.; Turlet, J.M. Behavior of GM3 ganglioside in lipid monolayers mimicking rafts or fluid phase in membranes. Chem. Phys. Lipids 2006, 139, 68–76. [Google Scholar] [CrossRef]
- McIntosh, T.J.; Simon, S.A. Long- and short-range interactions between phospholipid/ganglioside GM1 bilayers. Biochemistry 1994, 33, 10477–10486. [Google Scholar] [CrossRef] [PubMed]
- Marsh, D. Protein modulation of lipids, and vice-versa, in membranes. Biochim. Biophys. Acta 2008, 1778, 1545–1575. [Google Scholar] [CrossRef] [Green Version]
- Liu, C.Y.; Schroder, M.; Kaufman, R.J. Ligand-independent dimerization activates the stress response kinases IRE1 and PERK in the lumen of the endoplasmic reticulum. J. Biol. Chem. 2000, 275, 24881–24885. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morales, C.; Bisbal, M.; Bollo, M. Molecular Modulation by Lentivirus-Delivered Specific shRNAs in Endoplasmic Reticulum Stressed Neurons. J. Vis. Exp. 2021, 170, e61974. [Google Scholar] [CrossRef] [PubMed]
- Honorato, R.V.; Koukos, P.I.; Jimenez-Garcia, B.; Tsaregorodtsev, A.; Verlato, M.; Giachetti, A.; Rosato, A.; Bonvin, A. Structural Biology in the Clouds: The WeNMR-EOSC Ecosystem. Front. Mol. Biosci. 2021, 8, 729513. [Google Scholar] [CrossRef]
- Jin, Y.; Stayrook, S.E.; Albert, R.H.; Palackal, N.T.; Penning, T.M.; Lewis, M. Crystal structure of human type III 3alpha-hydroxysteroid dehydrogenase/bile acid binding protein complexed with NADP(+) and ursodeoxycholate. Biochemistry 2001, 40, 10161–10168. [Google Scholar] [CrossRef] [PubMed]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera--a visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [Green Version]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Morales, C.; Fernandez, M.; Ferrer, R.; Raimunda, D.; Carrer, D.C.; Bollo, M. Ursodeoxycholic Acid Binds PERK and Ameliorates Neurite Atrophy in a Cellular Model of GM2 Gangliosidosis. Int. J. Mol. Sci. 2023, 24, 7209. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms24087209
Morales C, Fernandez M, Ferrer R, Raimunda D, Carrer DC, Bollo M. Ursodeoxycholic Acid Binds PERK and Ameliorates Neurite Atrophy in a Cellular Model of GM2 Gangliosidosis. International Journal of Molecular Sciences. 2023; 24(8):7209. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms24087209
Chicago/Turabian StyleMorales, Carolina, Macarena Fernandez, Rodrigo Ferrer, Daniel Raimunda, Dolores C. Carrer, and Mariana Bollo. 2023. "Ursodeoxycholic Acid Binds PERK and Ameliorates Neurite Atrophy in a Cellular Model of GM2 Gangliosidosis" International Journal of Molecular Sciences 24, no. 8: 7209. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms24087209