
Specific Signal Transduction of Constitutively Activating (D576G) and Inactivating (R476H) Mutants of Agonist-Stimulated Luteinizing Hormone Receptor in Eel
 , ,
, ,
Abstract
:1. Introduction
2. Results
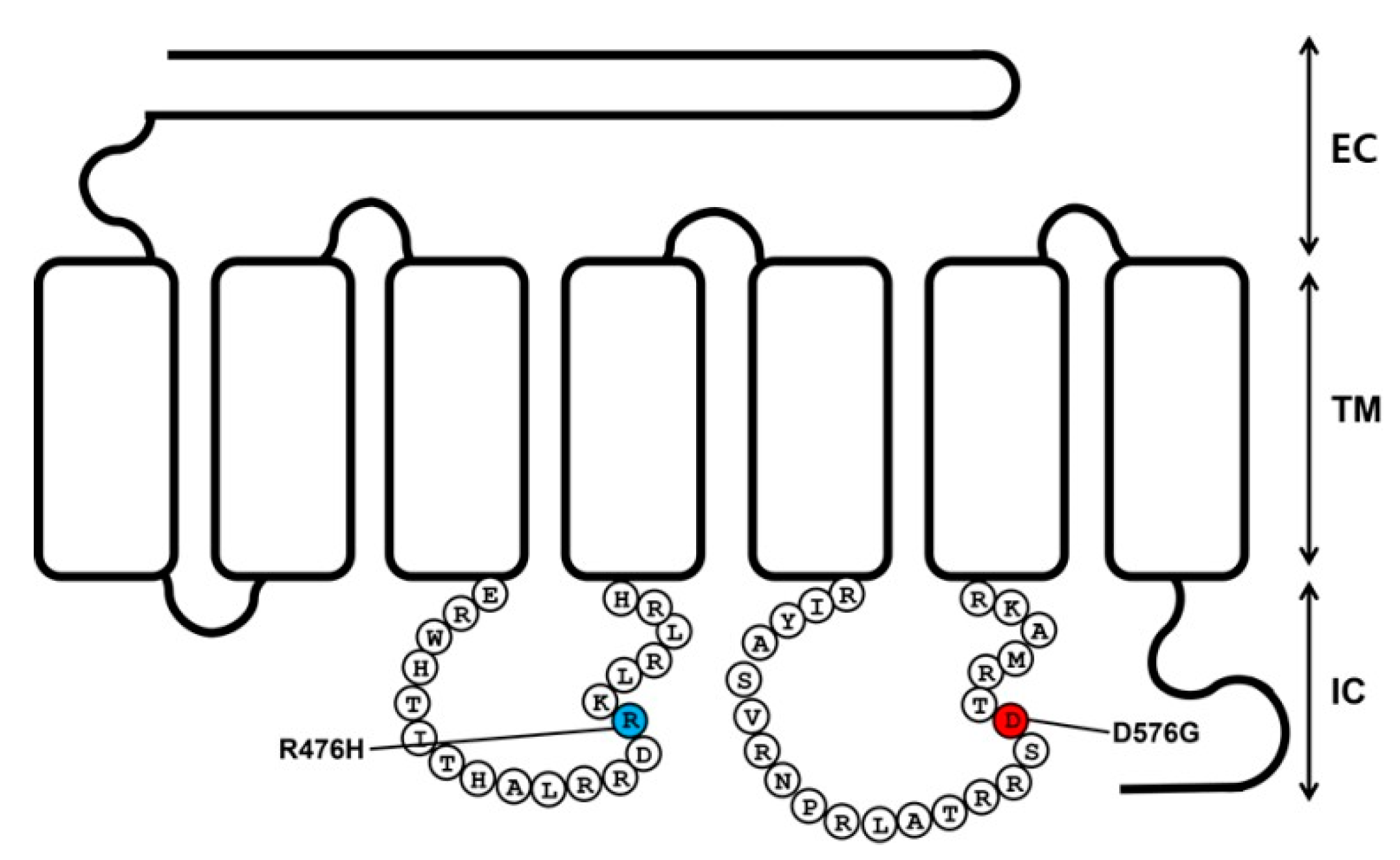
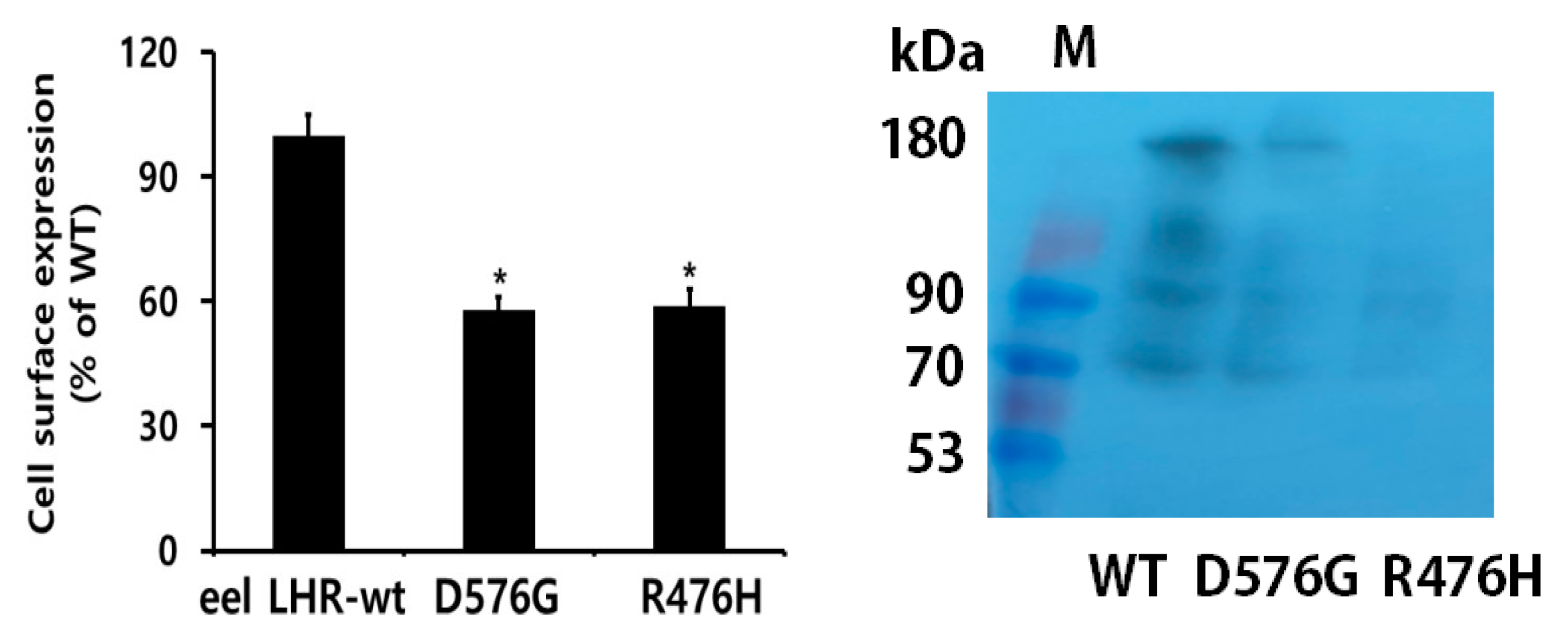
2.1. Preparation of Eel LHR Mutants and Their Cell-Surface Expression
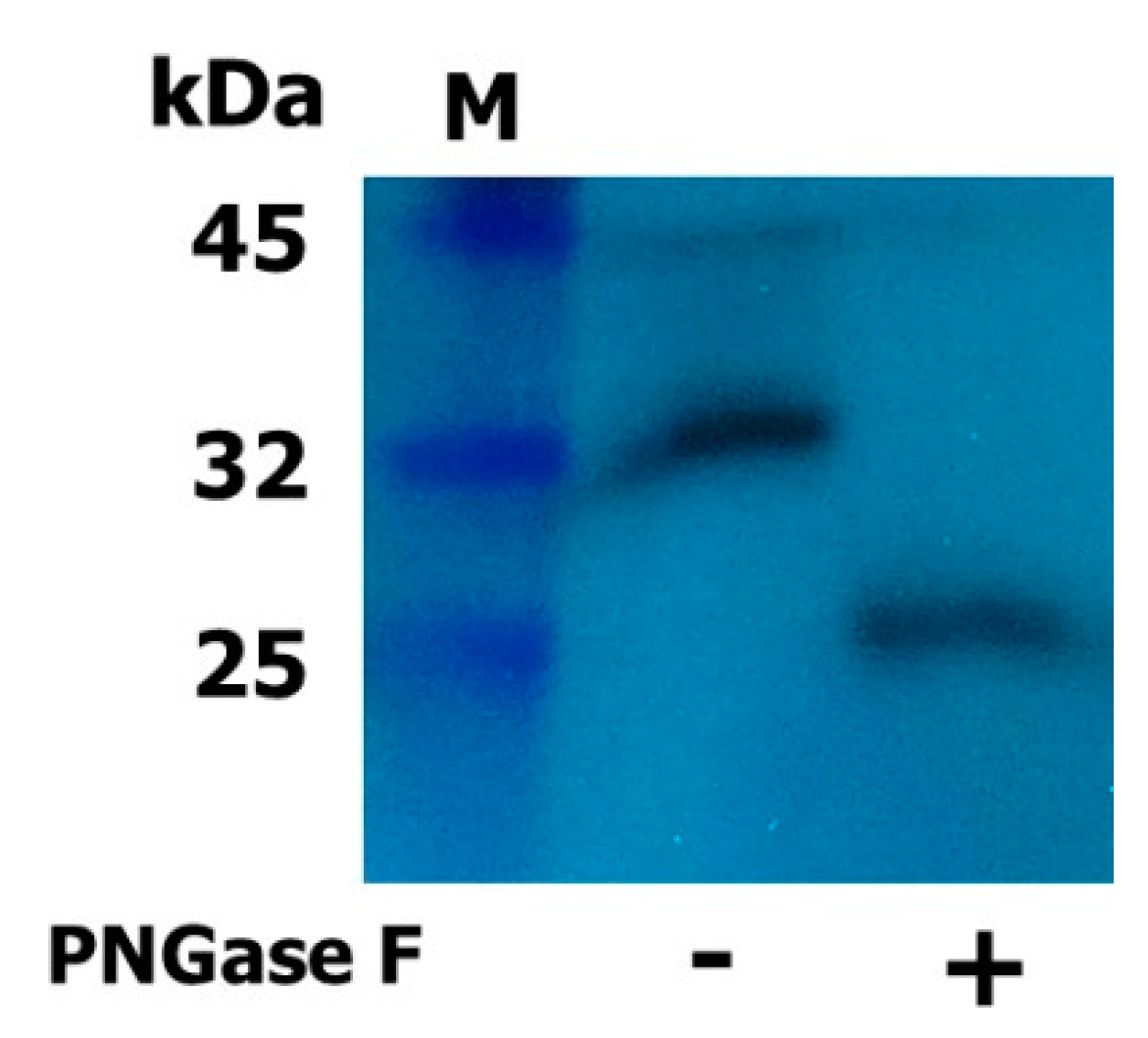
2.2. Expression and Western Blot Analysis of Rec-Eel LH
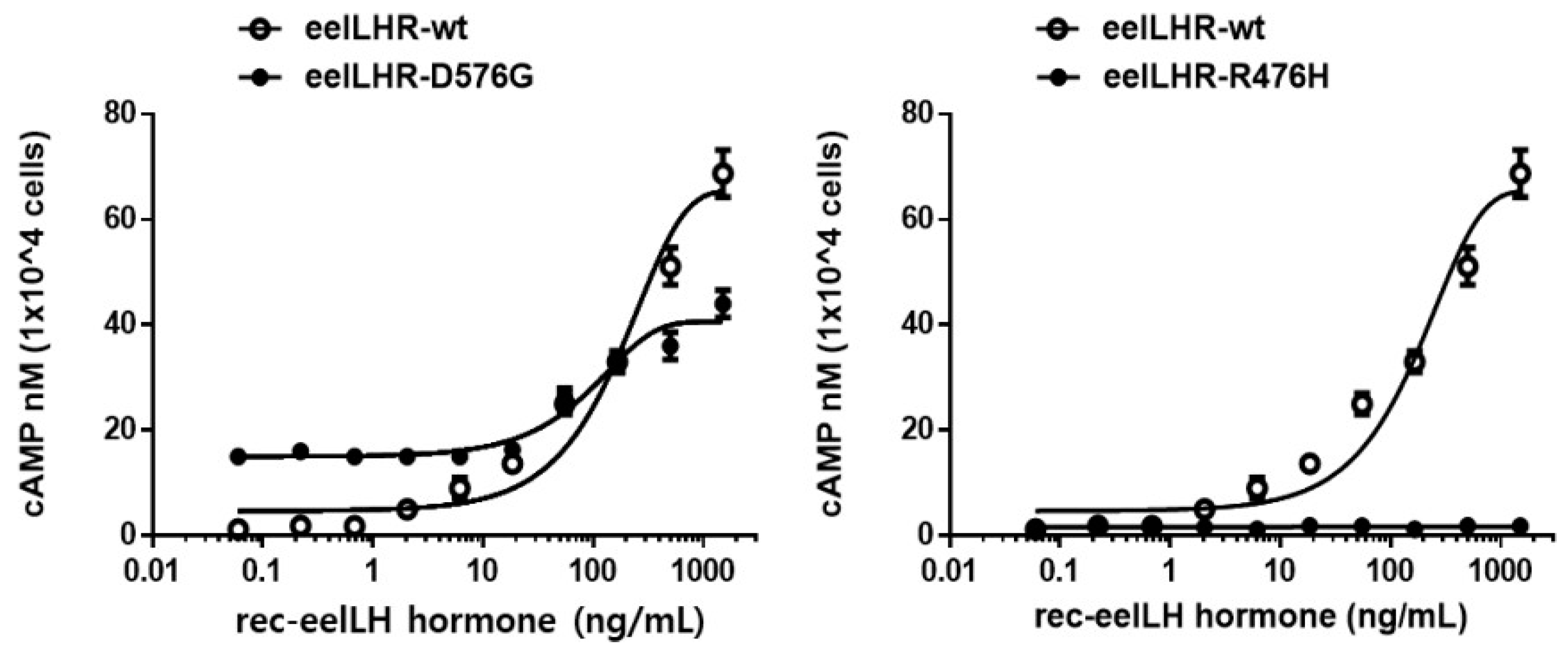
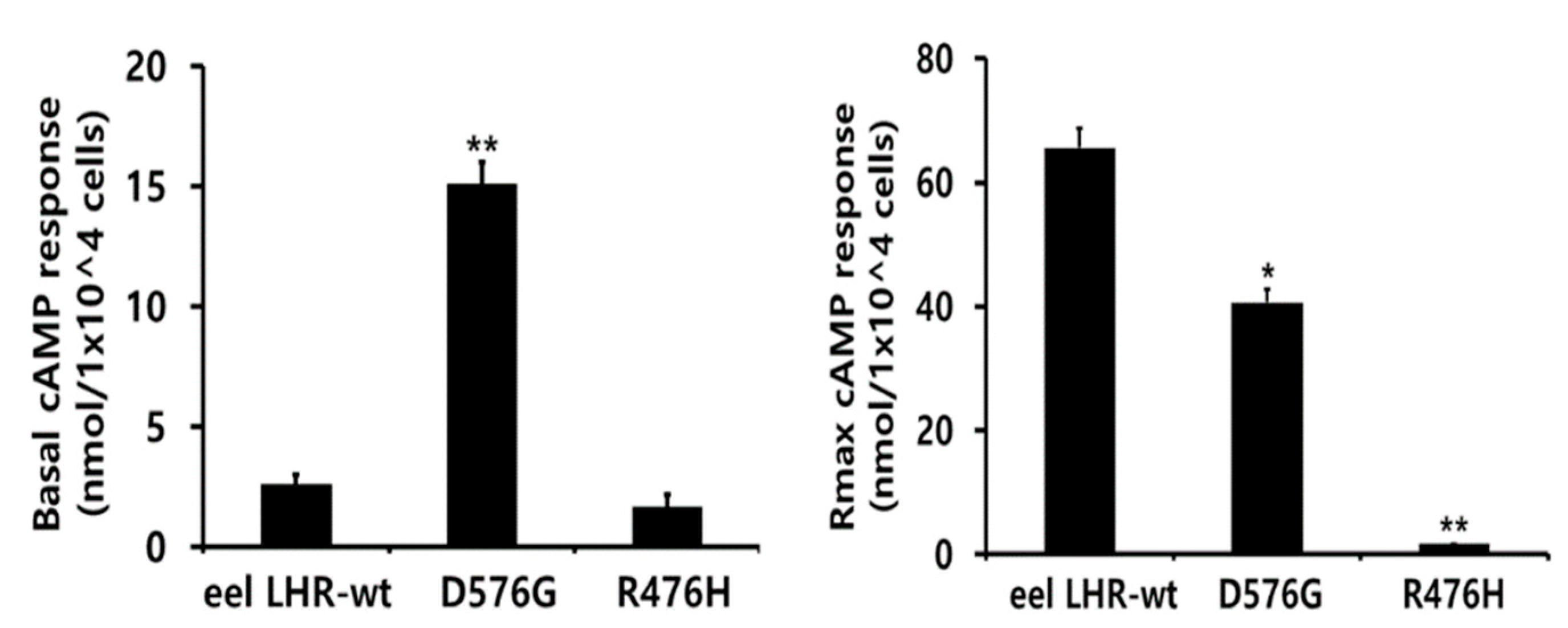
2.3. cAMP Responsiveness Induced by Agonists in Activating and Inactivating Mutants
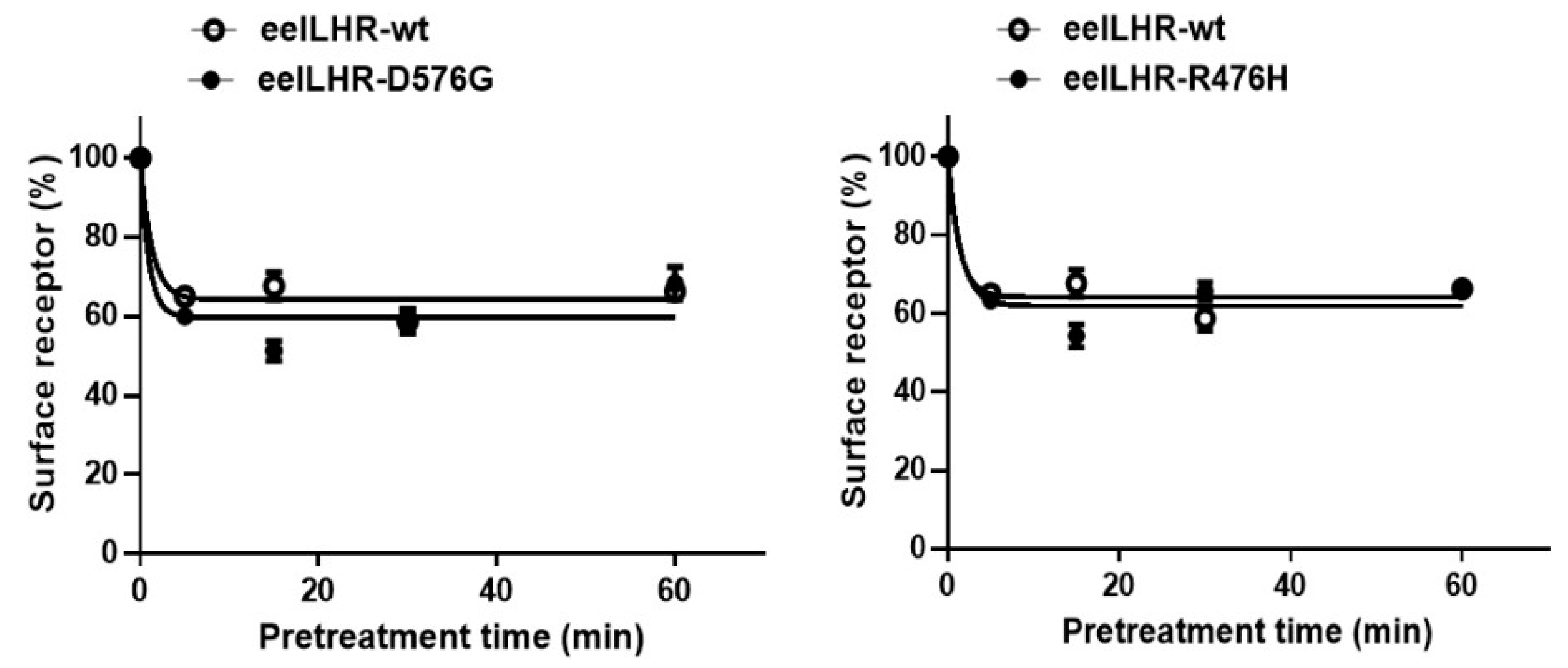
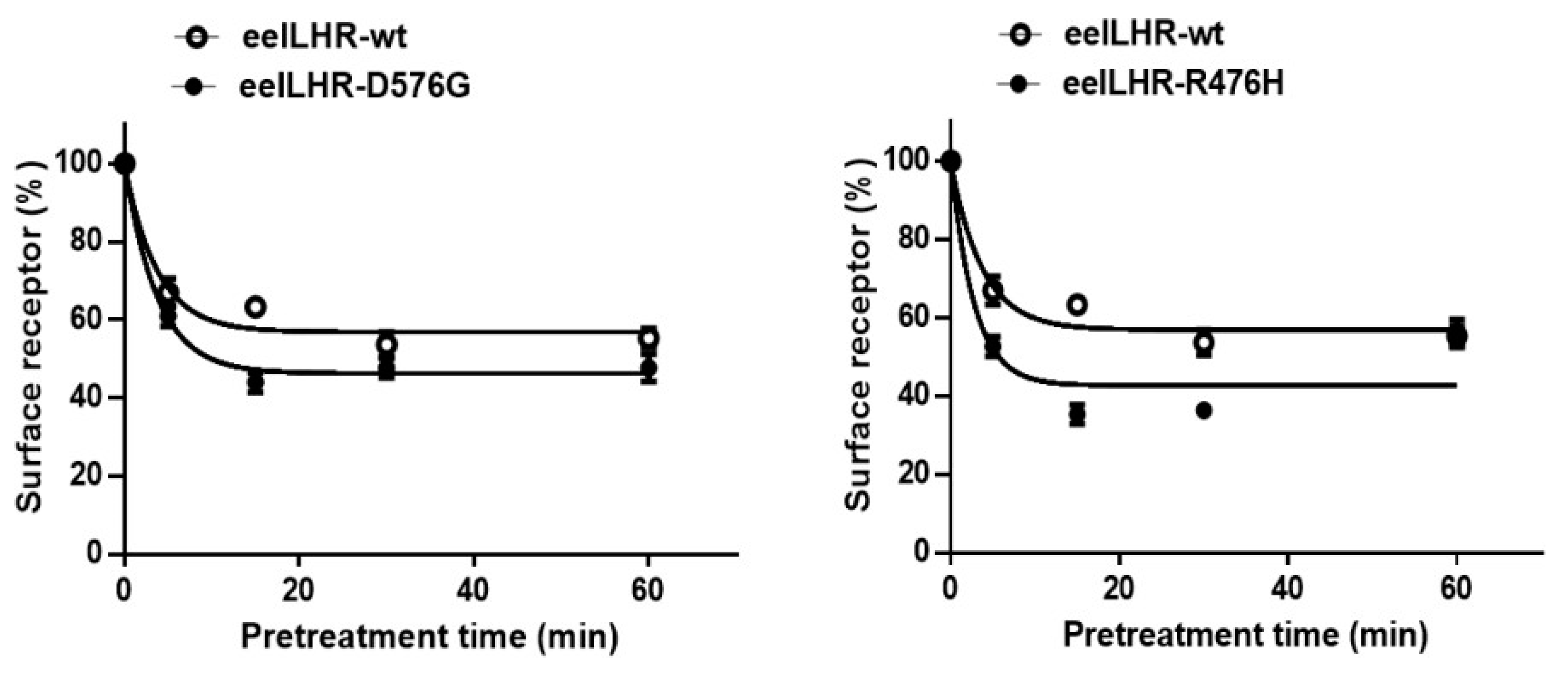
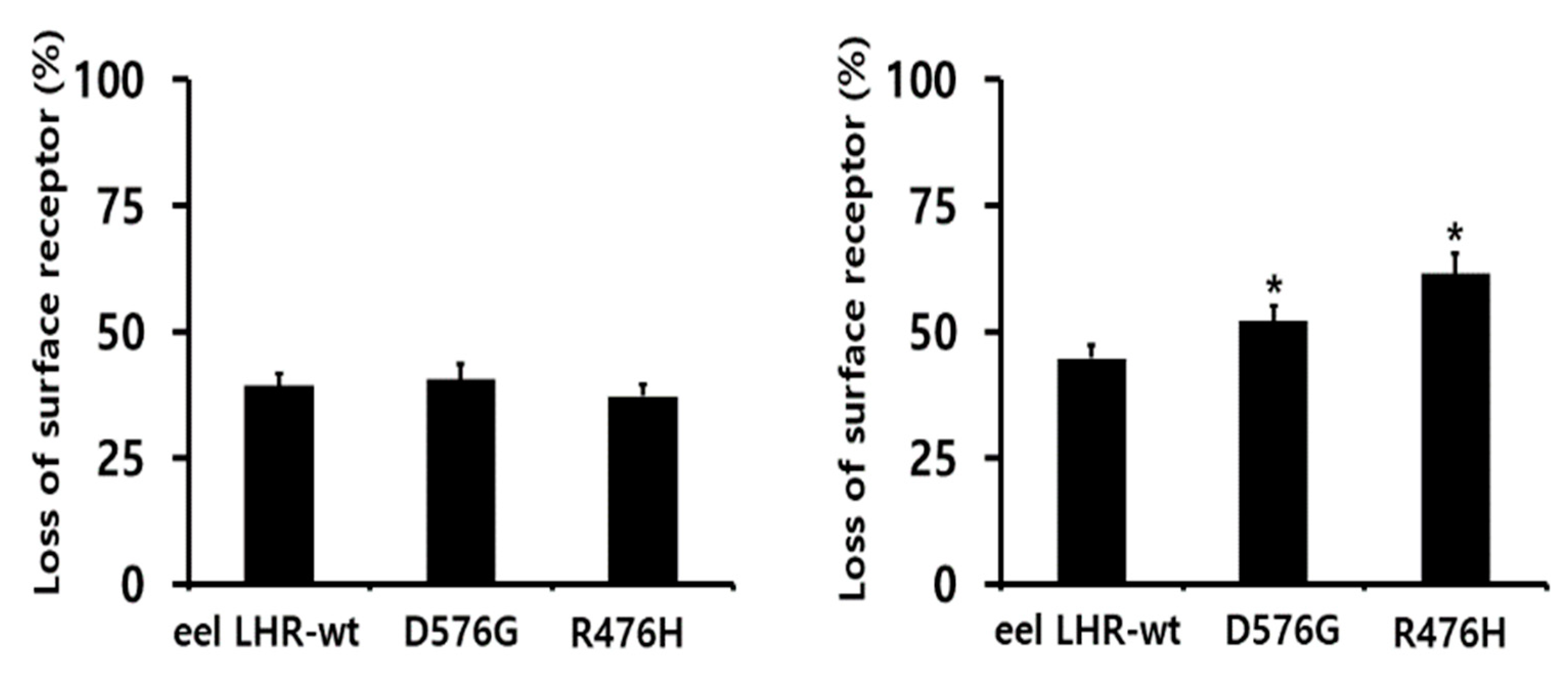
2.4. Loss in Cell-Surface Receptor by Agonist Treatment
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Production and ELISA Analysis of Recombinant Eel LH (Rec-Eel LH)
4.3. Enzymatic Deglycosylation and Western Blotting of Rec-Eel LH
4.4. Site-Directed Mutagenesis and Vector Construction
4.5. Transient Transfection
4.6. SDS-Polyacrylamide Gel Electrophoresis and Western Blotting of Eel LH Receptors
4.7. The cAMP Analysis by Homogeneous Time-Resolved Fluorescence (HTRF)
4.8. Agonist-Induced Loss in Cell-Surface Expression of Receptors
4.9. Data Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Min, K.S.; Liu, X.; Fabritz, J.; Jaquette, J.; Abell, A.N.; Ascoli, M. Mutations that induce constitutive activations and mutations signal transduction modulate the basal and/or agonist-stimulated internalization of the lutropin/choriogonadotropin receptor. J. Biol. Chem. 1998, 273, 34911–34919. [Google Scholar] [CrossRef] [PubMed]
- Dufau, M.L. The luteinizing hormone receptor. Annu. Rev. Physiol. 1998, 60, 461–496. [Google Scholar] [CrossRef] [PubMed]
- Tao, Y.X.; Abell, A.N.; Liu, X.; Nakamura, K.; Segaloff, D.L. Constitutive activation of G protein-coupled receptors as a result of selective substitution of a conserved leucine residue in transmembrane helix lll. Mol. Endocrinol. 2000, 14, 1272–1282. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Mizrachi, D.; Fanelli, F.; Segaloff, D.L. The formation of a salt bridge between helices 3 and 6 is responsible for the constitutive activity and lack of hormone responsiveness of the naturally occurring L457R mutation of the human lutropin receptor. J. Biol. Chem. 2005, 280, 26169–26176. [Google Scholar] [CrossRef]
- Zhang, M.; Tao, Y.X.; Ryan, G.L.; Feng, X.; Fanelli, F.; Segaloff, D.L. Intrinsic differences in the response of the human lutropin receptor versus the human follitropin receptor to activating mutations. J. Biol. Chem. 2007, 282, 25527–25539. [Google Scholar] [CrossRef]
- Tao, X.Y. Inactivating mutations of G protein-coupled receptors and diseases: Structure-function insights and therapeutic implications. Pharmacol. Ther. 2006, 111, 949–973. [Google Scholar] [CrossRef]
- Meehan, T.P.; Narayan, P. Constitutive active luteinizing hormone receptors: Consequences of in vivo expression. Mol. Cell. Endocrinol. 2007, 260–262, 294–300. [Google Scholar] [CrossRef]
- McGee, S.R.; Narayan, P. Precocious puberty and Leydig cell hyperplasia in male mice with a gain of function mutation in the LH receptor gene. Endocrinology 2013, 154, 3900–3913. [Google Scholar] [CrossRef]
- Daussac, A.; Barat, P.; Servant, N.; Yacoub, M.; Missonier, S.; Lavran, F.; Gaspari, L.; Sultan, C.; Paris, F. Testotoxicosis without testicular mass: Revealed by peripheral precocious puberty and confirmed by somatic LHCGR gene mutation. Endocr. Res. 2020, 45, 32–40. [Google Scholar] [CrossRef]
- Shenker, A.; Laue, L.; Kosugi, S.; Merendino, J.J., Jr.; Minegishi, T.; Cutler, G.B., Jr. A constitutively activating mutation of the luteinizing hormone receptor in familial male precocious puberty. Nature 1993, 365, 652–654. [Google Scholar] [CrossRef]
- Senker, A. Activating mutations of the lutropin choriogonadotropin receptor in precocious puberty. Recept. Channels 2002, 8, 3–18. [Google Scholar] [CrossRef]
- Huhtaniemi, I.H.; Themmen, A.P. Mutations in human gonadotropin and gonadotropin-receptor genes. Endocrine 2005, 26, 207–217. [Google Scholar] [CrossRef] [PubMed]
- Boot, A.M.; Lubbroso, S.; Verhoef-Post, M.; Richter-Unruh, A.; Looijenga, L.H.J.; Funaro, A.; Beishuizen, A.; van Marle, A.; Drop, S.L.S.; Themmen, A.P.N. Mutation analysis of the LH receptor gene in Leydig cell adenoma and hyperplasia and functional and biochemical studies of activating mutations of the LH receptor gene. J. Clin. Endocrinol. Metab. 2011, 96, E1197–E1205. [Google Scholar] [CrossRef] [PubMed]
- Latronico, A.C.; Arnhold, I.J. Inactivating mutations of LH and FSH receptors—From genotype to phenotype. Pediatr. Endocrinol. 2006, 4, 28–31. [Google Scholar]
- Latronico, A.C.; Segaloff, D.L. Insight learned from L457R(3.43)R, an activating mutant of the human lutropin receptor. Mol. Cell. Endocrinol. 2007, 260–262, 287–293. [Google Scholar] [CrossRef] [PubMed]
- Latronico, A.C. Naturally occurring mutations of the luteinizing hormone receptor gene affecting reproduction. Semin. Reprod. Med. 2000, 18, 17–20. [Google Scholar] [CrossRef]
- Laue, L.; Chan, W.Y.; Hsueh, A.J.W.; Kudo, M.; Hsu, S.Y.; Wu, S.M.; Blomberg, L.; Cutler, G.B., Jr. Genetic heterogeneity of constitutively activating mutations of the human luteinizing hormone receptor in familial male-limited precocious puberty. Proc. Natl. Acad. Sci. USA 1995, 92, 1906–1910. [Google Scholar] [CrossRef]
- Bradbury, F.A.; Kawate, N.; Foster, C.M.; Menon, K.M. Post-translational processing in the Golgi plays a critical role in the trafficking of the luteinizing hormone/human chorionic gonadotropin receptor to the cell surface. J. Biol. Chem. 1997, 272, 5921–5926. [Google Scholar] [CrossRef]
- Byambaragchaa, M.; Seong, H.K.; Choi, S.H.; Kim, D.J.; Kang, M.H.; Min, K.S. Constitutively activating mutants of equine LH/CGR constitutively induce signal transduction and inactivating mutations impair biological activity and cell-surface receptor loss in vitro. Int. J. Mol. Sci. 2021, 22, 10723. [Google Scholar] [CrossRef]
- Dhanwada, K.R.; Vijapurkar, U.; Ascoli, M. Two mutations of the lutropin/choriogonadotropin receptor that impair signal transduction also interfere with receptor-mediated endocytosis. Mol. Endocrinol. 1996, 10, 544–554. [Google Scholar]
- Byambaragchaa, M.; Park, A.; Gil, S.J.; Lee, H.W.; Ko, Y.J.; Choi, S.H.; Min, K.S. Luteinizing hormone-like and follicle- stimulating hormone-like activities of equine chorionic gonadotropin β-subunits in cells expressing rat luteinizing hormone/chorionic gonadotropin receptor and rat follicle-stimulating hormone receptor. Anim. Cells Syst. 2021, 3, 171–181. [Google Scholar] [CrossRef] [PubMed]
- Byambaragchaa, M.; Kim, D.J.; Kang, M.H.; Min, K.S. Site specificity of eel luteinizing hormone N-linked oligosaccharides in signal transduction. Gen. Comp. Endocrinol. 2018, 268, 50–56. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.J.; Park, C.W.; Kim, D.W.; Park, H.K.; Byambaragchaa, M.; Lee, N.S.; Hong, S.M.; Kang, M.H.; Min, K.S. Production and characterization of monoclonal antibodies against recombinant tethered follicle-stimulating hormone from Japanese eel Anguilla japonica. Gen. Comp. Endocrinol. 2016, 233, 8–15. [Google Scholar] [CrossRef] [PubMed]
- Min, K.S.; Park, J.J.; Lee, S.Y.; Byambaragchaa, M.; Kang, M.H. Comparative gene expression profiling of mouse ovaries upon stimulation with natural equine chorionic gonadotropin (N-eCG) and tethered recombinant-eCG (R-eCG). BMC Biotechnol. 2020, 20, 59. [Google Scholar] [CrossRef] [PubMed]
- Min, K.S.; Park, J.J.; Byambaragchaa, M.; Kang, M.H. Characterization of tethered equine chorionic gonadotropin and its deglycosylated mutants by ovulation stimulation in mice. BMC Biotechnol. 2019, 19, 60. [Google Scholar] [CrossRef]
- Lee, S.Y.; Byambaragchaa, M.; Choi, S.H.; Kang, H.J.; Kang, M.H.; Min, K.S. Roles of N-linked and O-linked glycosylation sites in the activity of equine chorionic gonadotropin in cells expressing rat luteinizing hormone/chorionic gonadotropin receptor and follicle-stimulating hormone receptor. BMC Biotechnol. 2021, 21, 52. [Google Scholar] [CrossRef]
- Byambaragchaa, M.; Kim, J.S.; Park, H.K.; Kim, D.J.; Hong, S.M.; Kang, M.H.; Min, K.S. Constitutive activation and inactivation of mutations inducing cell surface loss of receptor and impairing of signal transduction of agonist-stimulated eel follicle-stimulating hormone receptors. Int. J. Mol. Sci. 2020, 21, 7075. [Google Scholar] [CrossRef]
- Byambaragchaa, M.; Ahn, T.Y.; Choi, S.H.; Kang, M.H.; Min, K.S. Functional characterization of naturally-occurring constitutively activating/inactivating mutations in equine follicle-stimulating hormone receptor. Anim. Biosci. 2022, 3, 399–409. [Google Scholar] [CrossRef]
- Kosugi, S.; Mori, T.; Shenker, A. The role of Asp578 in maintaining the inactive conformation of the human lutropin/choriogonadotropin receptor. J. Biol. Chem. 1996, 271, 31813–31817. [Google Scholar] [CrossRef]
- Kosugi, S.; Mori, T.; Shenker, A. An anionic residue at position 564 is important for maintaining the inactive conformation of the human lutropin/choriogonadotropin receptor. Mol. Pharmacol. 1998, 53, 894–901. [Google Scholar]
- Yano, K.; Kohn, L.D.; Saji, M.; Kataoka, N.; Okuno, A.; Cutler, G.B., Jr. A case of male-limited precocious puberty caused by a point mutation in the second transmembrane domain of the luteinizing hormone choriogonadotropin receptor gene. Biochem. Biophys. Res. Commun. 1996, 220, 1036–1042. [Google Scholar] [CrossRef] [PubMed]
- Kraaij, R.; Post, M.; Kremer, H.; Milgrom, E.; Epping, W.; Brunner, H.G.; Grootegoed, J.A.; Themmen, A.P. A missense mutation in the second transmembrane segment of the luteinizing hormone receptor causes familial male-limited precocious puberty. J. Clin. Endocrinol. Metab. 1995, 80, 3168–3172. [Google Scholar] [PubMed]
- Foster, S.R.; Brauner-Osborne, H. Investigating internalization and intracellular trafficking of GPCRs: New techniques and real-time experimental approaches. Handb. Exp. Pharmacol. 2018, 245, 41–61. [Google Scholar] [PubMed]
- Slosky, L.M.; Bai, Y.; Toth, K.; Ray, C.; Rochelle, L.K.; Badea, A.; Chandrasekhar, R.; Pogorelov, V.M.; Abraham, D.M.; Atluri, N.; et al. β-arrestin-biased allosteric modulated of NTSR1 selectively attenuates addictive behaviors. Cell 2020, 181, 1364–1379. [Google Scholar] [CrossRef] [PubMed]
- Byambaragchaa, M.; Park, H.K.; Kim, D.J.; Lee, J.H.; Kang, M.H.; Min, K.S. The N-linked glycosylation site N191 is necessary for PKA signal transduction in eel follicle-stimulating hormone receptor. Int. J. Mol. Sci. 2022, 23, 12792. [Google Scholar] [CrossRef]
- Bhaskaran, R.S.; Ascoli, M. The post-endocytotic fate of the gonadotropin receptors is an important determinant of the desensitization of gonadotropin responses. J. Mol. Endocrinol. 2005, 34, 447–457. [Google Scholar] [CrossRef]
- Mundell, S.J.; Matharu, A.L.; Nisar, S.; Palmer, T.M.; Benovic, J.L.; Kelly, E. Deletion of the distal COOH-terminus of the A2B adenosine receptor switches internalization to an arrestin- and clathrin-independent pathway and inhibits recycling. Br. J. Pharmacol. 2010, 159, 518–533. [Google Scholar] [CrossRef]
- Norskov-Lauritsen, L.; Jorgensen, S.; Brauner-Osborne, H. N-glycosylation and disulfide bonding affects GPRC6A receptor expression, function, and dimerization. FEBS Lett. 2015, 589, 588–597. [Google Scholar] [CrossRef]
- Jacobsen, S.E.; Ammendrup-Johnsen, I.A.; Jansen, A.M.; Gether, U.; Madsen, K.L.; Brauner-Osborne, H. The GPRC6A receptor displays constitutive internalization and sorting to the slow recycling pathway. J. Biol. Chem. 2017, 292, 6910–6926. [Google Scholar] [CrossRef]
- Jean-Alphonse, F.; Bowersox, S.; Chen, S.; Beard, G.; Puthenveedu, M.A.; Hanyaloglu, A.C. Spatially restricted G protein- coupled receptor activity via divergent endocytic compartments. J. Biol. Chem. 2014, 289, 3960–3977. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Eel LH Receptors | cAMP Responses | ||
|---|---|---|---|
| Basal a (nM/104 Cells) | EC50 (ng/mL) | Rmax b (nM/104 Cells) | |
| LHR-WT | 2.6 ± 0.4 (1-fold) | 176.6 (138.7 to 242.7) c | 65.6 ± 3.2 (1-fold) |
| LHR-D576G | 15.1 ± 0.9 (5.8-fold) | 94.8 (72.9 to 135.5) | 40.6 ± 2.1 (0.62-fold) |
| LHR-R476H | 1.7 ± 0.5 | - d | - d |
| Ligand Treatment | Eel LHR Cell Lines | t1/2 (min) | Plateau (% of Control) |
|---|---|---|---|
| rec-eel LH | eel LHR-WT | 0.9 ± 0.1 | 64.2 ± 1.5 |
| eel LHR-D576G | 0.6 ± 0.1 | 59.8 ± 2.5 | |
| eel LHR-R476H | 0.6 ± 0.1 | 61.9 ± 2.1 | |
| rec-eCG | eel LHR-WT | 2.5 ± 0.2 | 56.8 ± 1.4 |
| eel LHR-D576G | 2.1 ± 0.1 | 46.3 ± 1.0 | |
| eel LHR-R476H | 1.9 ± 0.1 | 42.6 ± 1.2 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Choi, S.-H.; Byambaragchaa, M.; Kim, D.-J.; Lee, J.-H.; Kang, M.-H.; Min, K.-S. Specific Signal Transduction of Constitutively Activating (D576G) and Inactivating (R476H) Mutants of Agonist-Stimulated Luteinizing Hormone Receptor in Eel. Int. J. Mol. Sci. 2023, 24, 9133. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms24119133
Choi S-H, Byambaragchaa M, Kim D-J, Lee J-H, Kang M-H, Min K-S. Specific Signal Transduction of Constitutively Activating (D576G) and Inactivating (R476H) Mutants of Agonist-Stimulated Luteinizing Hormone Receptor in Eel. International Journal of Molecular Sciences. 2023; 24(11):9133. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms24119133
Chicago/Turabian StyleChoi, Seung-Hee, Munkhzaya Byambaragchaa, Dae-Jung Kim, Jong-Hyuk Lee, Myung-Hwa Kang, and Kwan-Sik Min. 2023. "Specific Signal Transduction of Constitutively Activating (D576G) and Inactivating (R476H) Mutants of Agonist-Stimulated Luteinizing Hormone Receptor in Eel" International Journal of Molecular Sciences 24, no. 11: 9133. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms24119133