
Altered Hippocampal and Striatal Expression of Endothelial Markers and VIP/PACAP Neuropeptides in a Mouse Model of Systemic Lupus Erythematosus
 , , , , ,
, , , , ,  and
and 
Abstract
:1. Introduction
2. Results
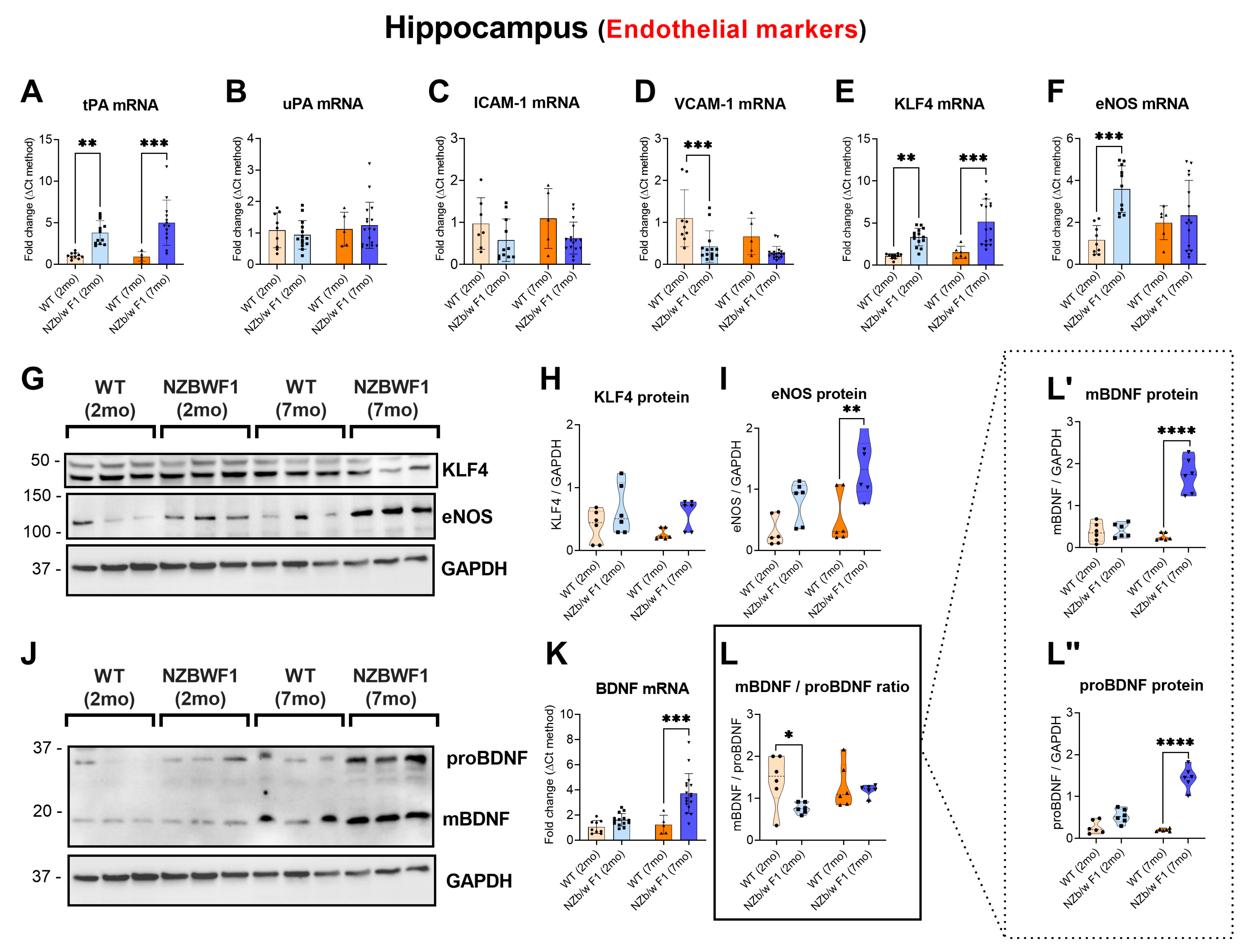
2.1. Evidence of Altered Endothelial Markers in the Hippocampus of NZBWF1 Mice
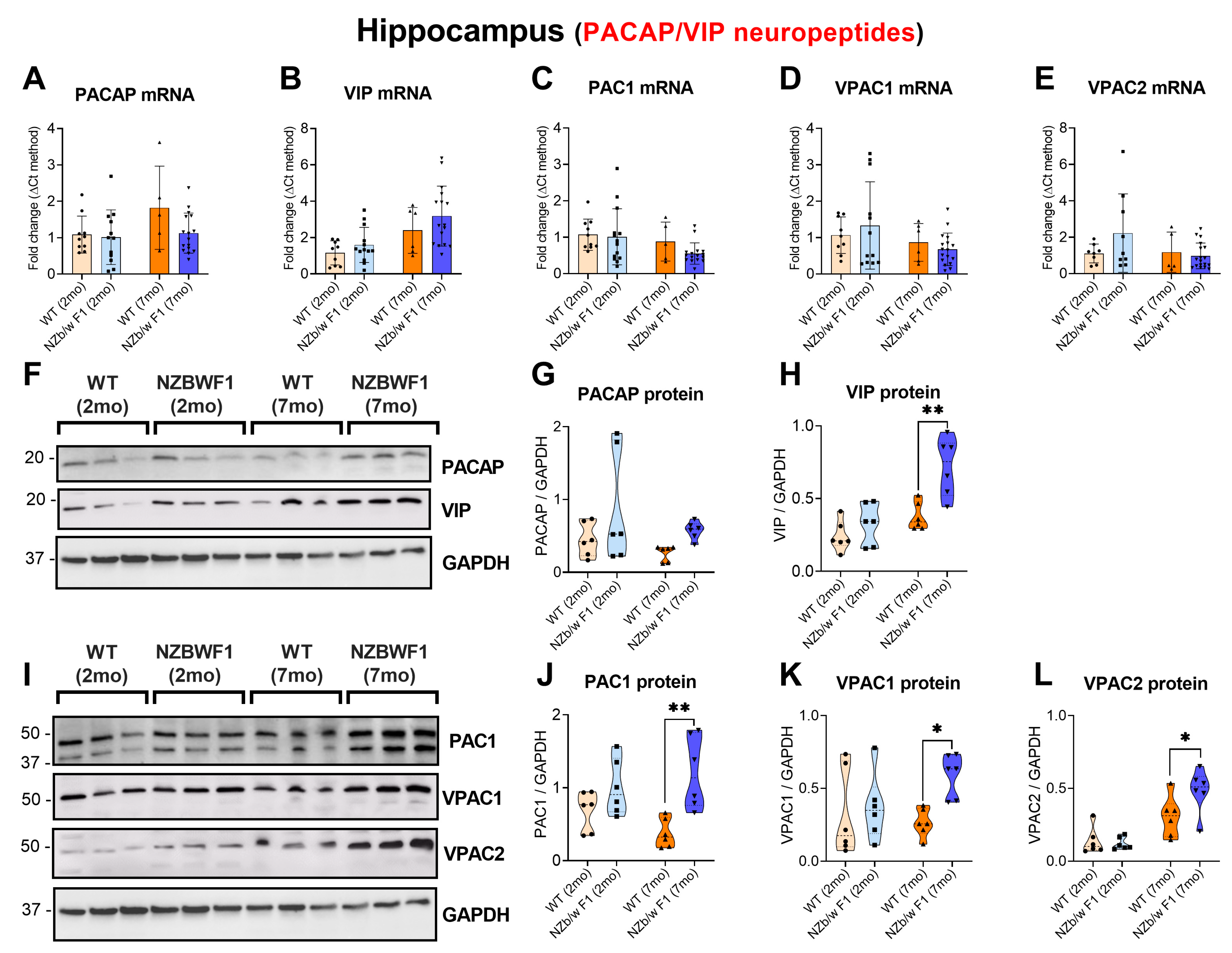
2.2. Increased Hippocampal Expression of VIP/PACAP and Receptors in Older but Not in Younger NZBWF1 Mice
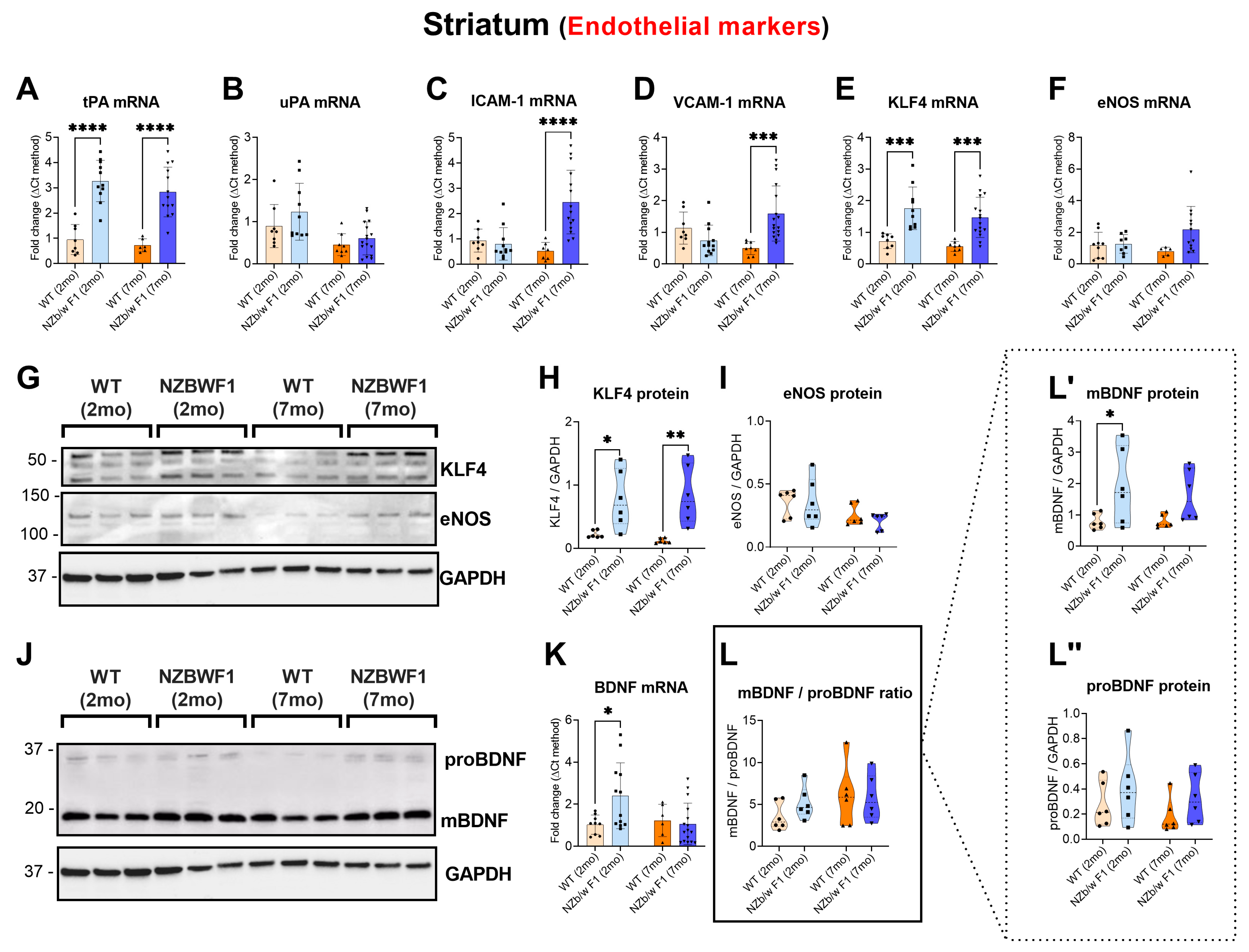
2.3. Robust Transcriptional Alterations of Endothelial Markers in the Striatum of NPSLE Mice
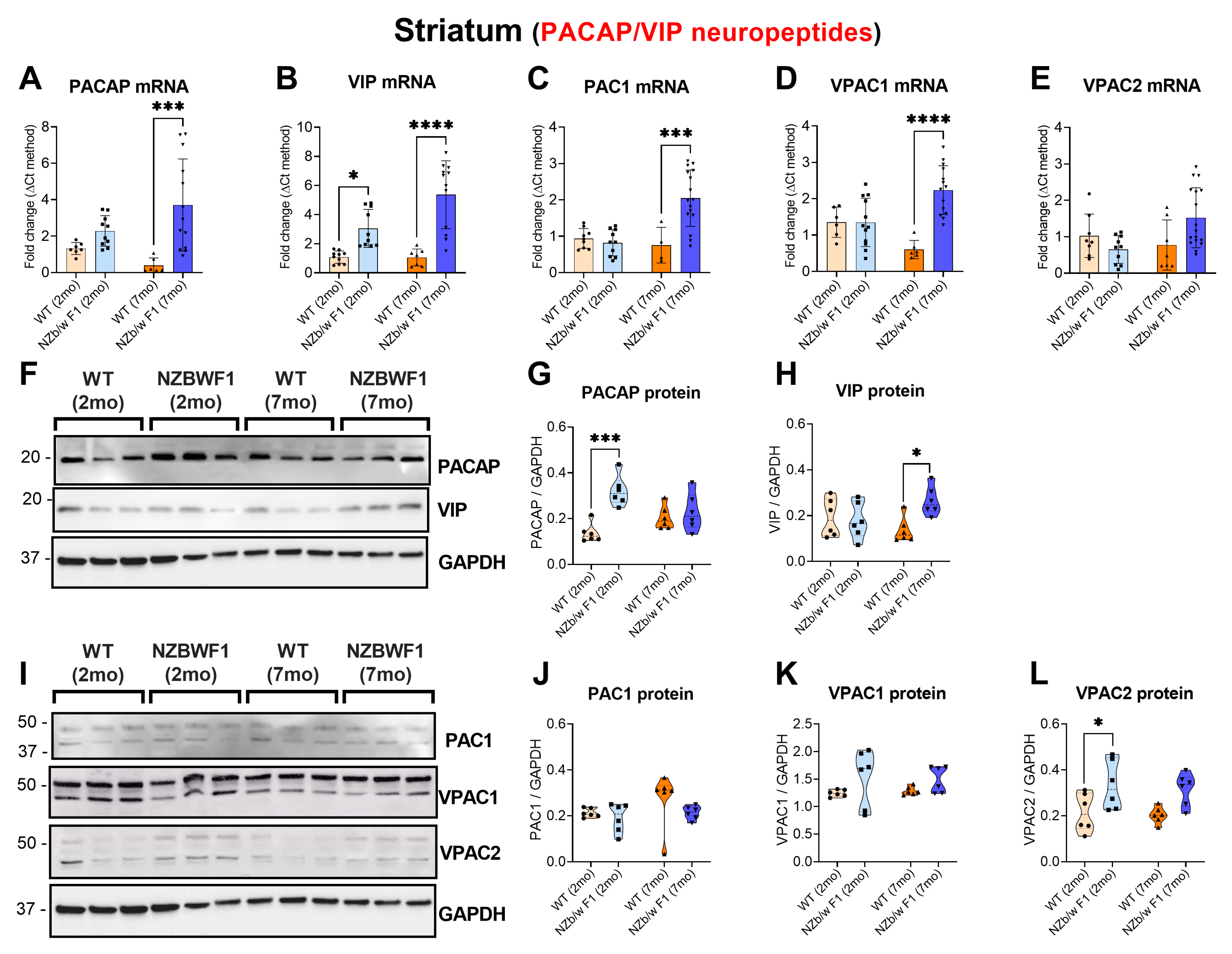
2.4. Striatal Perturbations of VIP/PACAP and Receptors in NZBWF1 Mice
3. Discussion
4. Materials and Methods
4.1. Animals
4.2. Brain Micro-Dissections
4.3. RNA Extraction, cDNA Synthesis and Real-Time Quantitative Polymerase Chain Reaction (RT-qPCR)
4.4. Protein Extraction and Western Blot
4.5. Statistical Analyses
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Mok, C.C.; Lau, C.S. Pathogenesis of systemic lupus erythematosus. J. Clin. Pathol. 2003, 56, 481–490. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carter, E.E.; Barr, S.G.; Clarke, A.E. The global burden of SLE: Prevalence, health disparities and socioeconomic impact. Nat. Rev. Rheumatol. 2016, 12, 605–620. [Google Scholar] [CrossRef] [PubMed]
- Cieślik, P.; Hrycek, A.; Kłuciński, P. Vasculopathy and vasculitis in systemic lupus erythematosus. Pol. Arch. Med. Wewn. 2008, 118, 57–63. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Popescu, A.; Kao, A.H. Neuropsychiatric systemic lupus erythematosus. Curr. Neuropharmacol. 2011, 9, 449–457. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahn, G.Y.; Kim, D.; Won, S.; Song, S.T.; Jeong, H.J.; Sohn, I.W.; Lee, S.; Joo, Y.B.; Bae, S.C. Prevalence, risk factors, and impact on mortality of neuropsychiatric lupus: A prospective, single-center study. Lupus 2018, 27, 1338–1347. [Google Scholar] [CrossRef]
- Muscal, E.; Brey, R.L. Neurologic manifestations of systemic lupus erythematosus in children and adults. Neurol. Clin. 2010, 28, 61–73. [Google Scholar] [CrossRef] [Green Version]
- Sibbitt, W.L., Jr.; Brooks, W.M.; Kornfeld, M.; Hart, B.L.; Bankhurst, A.D.; Roldan, C.A. Magnetic resonance imaging and brain histopathology in neuropsychiatric systemic lupus erythematosus. Semin. Arthritis Rheum. 2010, 40, 32–52. [Google Scholar] [CrossRef] [Green Version]
- Zirkzee, E.J.; Huizinga, T.W.; Bollen, E.L.; van Buchem, M.A.; Middelkoop, H.A.; van der Wee, N.J.; le Cessie, S.; Steup-Beekman, G.M. Mortality in neuropsychiatric systemic lupus erythematosus (NPSLE). Lupus 2014, 23, 31–38. [Google Scholar] [CrossRef]
- Feng, X.; Pan, W.; Liu, L.; Wu, M.; Ding, F.; Hu, H.; Ding, X.; Wei, H.; Zou, Y.; Qian, X.; et al. Prognosis for Hospitalized Patients with Systemic Lupus Erythematosus in China: 5-Year Update of the Jiangsu Cohort. PLoS ONE 2016, 11, e0168619. [Google Scholar] [CrossRef] [Green Version]
- Gulinello, M.; Wen, J.; Putterman, C. Neuropsychiatric Symptoms in Lupus. Psychiatr. Ann. 2012, 42, 322–328. [Google Scholar] [CrossRef] [Green Version]
- Shih, Y.C.; Ou, Y.H.; Chang, S.W.; Lin, C.M. A challenging case of neuropsychiatric systematic lupus erythematosus with recurrent antiphospholipid- related stroke: A case report and literature review. Neurol. Int. 2019, 11, 8182. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Xiang, X.; Sun, J.; Liu, S.; Liu, Y.; Feng, L.; Li, C.; Li, Z. Prevalence, outcome and prognostic factors of neuropsychiatric systemic lupus erythematosus: A real world single center study. Mod. Rheumatol. 2020, 30, 321–326. [Google Scholar] [CrossRef]
- Eisenberg, R. Why can’t we find a new treatment for SLE? J. Autoimmun. 2009, 32, 223–230. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maidhof, W.; Hilas, O. Lupus: An overview of the disease and management options. Pharm. Ther. 2012, 37, 240–249. [Google Scholar]
- Burbach, J.P. What are neuropeptides? Methods Mol. Biol. 2011, 789, 1–36. [Google Scholar] [CrossRef]
- Ganea, D.; Hooper, K.M.; Kong, W. The neuropeptide vasoactive intestinal peptide: Direct effects on immune cells and involvement in inflammatory and autoimmune diseases. Acta Physiol. 2015, 213, 442–452. [Google Scholar] [CrossRef] [PubMed]
- Castorina, A.; Vogiatzis, M.; Kang, J.W.M.; Keay, K.A. PACAP and VIP expression in the periaqueductal grey of the rat following sciatic nerve constriction injury. Neuropeptides 2019, 74, 60–69. [Google Scholar] [CrossRef] [PubMed]
- Hirabayashi, T.; Nakamachi, T.; Shioda, S. Discovery of PACAP and its receptors in the brain. J. Headache Pain 2018, 19, 28. [Google Scholar] [CrossRef]
- Vaudry, D.; Falluel-Morel, A.; Bourgault, S.; Basille, M.; Burel, D.; Wurtz, O.; Fournier, A.; Chow, B.K.; Hashimoto, H.; Galas, L.; et al. Pituitary adenylate cyclase-activating polypeptide and its receptors: 20 years after the discovery. Pharmacol. Rev. 2009, 61, 283–357. [Google Scholar] [CrossRef]
- Karunia, J.; Niaz, A.; Mandwie, M.; Thomas Broome, S.; Keay, K.A.; Waschek, J.A.; Al-Badri, G.; Castorina, A. PACAP and VIP Modulate LPS-Induced Microglial Activation and Trigger Distinct Phenotypic Changes in Murine BV2 Microglial Cells. Int. J. Mol. Sci. 2021, 22, 10947. [Google Scholar] [CrossRef]
- Thomas Broome, S.; Musumeci, G.; Castorina, A. Doxycycline and Minocycline Act as Positive Allosteric Modulators of the PAC1 Receptor and Induce Plasminogen Activators in RT4 Schwann Cells. Appl. Sci. 2021, 11, 7673. [Google Scholar] [CrossRef]
- Jansen, M.I.; Broome, S.T.; Castorina, A. Targeting the neurological comorbidities of multiple sclerosis: The beneficial effects of VIP and PACAP neuropeptides. J. Integr. Neurosci. 2022, 21, 33. [Google Scholar] [CrossRef] [PubMed]
- Jozwiak-Bebenista, M.; Kowalczyk, E. Neuroleptic Drugs and PACAP Differentially Affect the mRNA Expression of Genes Encoding PAC1/VPAC Type Receptors. Neurochem. Res. 2017, 42, 943–952. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, W.; Wang, H.; Li, Y.S.; Luo, W. Role of vasoactive intestinal peptide in osteoarthritis. J. Biomed. Sci. 2016, 23, 63. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martinez, C.; Abad, C.; Delgado, M.; Arranz, A.; Juarranz, M.G.; Rodriguez-Henche, N.; Brabet, P.; Leceta, J.; Gomariz, R.P. Anti-inflammatory role in septic shock of pituitary adenylate cyclase-activating polypeptide receptor. Proc. Natl. Acad. Sci. USA 2002, 99, 1053–1058. [Google Scholar] [CrossRef] [PubMed]
- Delgado, M.; Abad, C.; Martinez, C.; Juarranz, M.G.; Arranz, A.; Gomariz, R.P.; Leceta, J. Vasoactive intestinal peptide in the immune system: Potential therapeutic role in inflammatory and autoimmune diseases. J. Mol. Med. 2002, 80, 16–24. [Google Scholar] [CrossRef]
- Castorina, A.; Giunta, S.; Mazzone, V.; Cardile, V.; D’Agata, V. Effects of PACAP and VIP on hyperglycemia-induced proliferation in murine microvascular endothelial cells. Peptides 2010, 31, 2276–2283. [Google Scholar] [CrossRef]
- Lund, A.M.; Hannibal, J. Localization of the neuropeptides pituitary adenylate cyclase-activating polypeptide, vasoactive intestinal peptide, and their receptors in the basal brain blood vessels and trigeminal ganglion of the mouse CNS; an immunohistochemical study. Front. Neuroanat. 2022, 16, 991403. [Google Scholar] [CrossRef]
- Goncharov, N.V.; Nadeev, A.D.; Jenkins, R.O.; Avdonin, P.V. Markers and Biomarkers of Endothelium: When Something Is Rotten in the State. Oxid. Med. Cell Longev. 2017, 2017, 9759735. [Google Scholar] [CrossRef] [Green Version]
- Bobik, A.; Tkachuk, V. Metalloproteinases and plasminogen activators in vessel remodeling. Curr. Hypertens. Rep. 2003, 5, 466–472. [Google Scholar] [CrossRef]
- Yang, J.; Shi, Q.-D.; Song, T.-B.; Feng, G.-F.; Zang, W.-J.; Zong, C.-H.; Chang, L. Vasoactive intestinal peptide increases VEGF expression to promote proliferation of brain vascular endothelial cells via the cAMP/PKA pathway after ischemic insult in vitro. Peptides 2013, 42, 105–111. [Google Scholar] [CrossRef]
- Frank, P.G.; Lisanti, M.P. ICAM-1: Role in inflammation and in the regulation of vascular permeability. Am. J. Physiol. Heart Circ. Physiol. 2008, 295, H926–H927. [Google Scholar] [CrossRef] [PubMed]
- Kong, D.H.; Kim, Y.K.; Kim, M.R.; Jang, J.H.; Lee, S. Emerging Roles of Vascular Cell Adhesion Molecule-1 (VCAM-1) in Immunological Disorders and Cancer. Int. J. Mol. Sci. 2018, 19, 1057. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghaleb, A.M.; Yang, V.W. Krüppel-like factor 4 (KLF4): What we currently know. Gene 2017, 611, 27–37. [Google Scholar] [CrossRef] [PubMed]
- Heiss, C.; Rodriguez-Mateos, A.; Kelm, M. Central role of eNOS in the maintenance of endothelial homeostasis. Antioxid. Redox Signal. 2015, 22, 1230–1242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Napolitano, F.; Montuori, N. Role of Plasminogen Activation System in Platelet Pathophysiology: Emerging Concepts for Translational Applications. Int. J. Mol. Sci. 2022, 23, 6065. [Google Scholar] [CrossRef]
- Stepanova, V.; Jayaraman, P.S.; Zaitsev, S.V.; Lebedeva, T.; Bdeir, K.; Kershaw, R.; Holman, K.R.; Parfyonova, Y.V.; Semina, E.V.; Beloglazova, I.B.; et al. Urokinase-type Plasminogen Activator (uPA) Promotes Angiogenesis by Attenuating Proline-rich Homeodomain Protein (PRH) Transcription Factor Activity and De-repressing Vascular Endothelial Growth Factor (VEGF) Receptor Expression. J. Biol. Chem. 2016, 291, 15029–15045. [Google Scholar] [CrossRef] [Green Version]
- Crookston, K.P.; Sibbitt, W.L., Jr.; Chandler, W.L.; Qualls, C.R.; Roldan, C.A. Circulating microparticles in neuropsychiatric systemic lupus erythematosus. Int. J. Rheum. Dis. 2013, 16, 72–80. [Google Scholar] [CrossRef]
- Pang, P.T.; Teng, H.K.; Zaitsev, E.; Woo, N.T.; Sakata, K.; Zhen, S.; Teng, K.K.; Yung, W.H.; Hempstead, B.L.; Lu, B. Cleavage of proBDNF by tPA/plasmin is essential for long-term hippocampal plasticity. Science 2004, 306, 487–491. [Google Scholar] [CrossRef]
- Castorina, A.; D’Amico, A.; Scuderi, S.; Leggio, G.; Drago, F.; D’Agata, V. Dopamine D3 receptor deletion increases tissue plasminogen activator (tPA) activity in prefrontal cortex and hippocampus. Neuroscience 2013, 250, 546–556. [Google Scholar] [CrossRef]
- Chen, A.; Xiong, L.J.; Tong, Y.; Mao, M. The neuroprotective roles of BDNF in hypoxic ischemic brain injury. Biomed. Rep. 2013, 1, 167–176. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meng, Y.; Chopp, M.; Zhang, Y.; Liu, Z.; An, A.; Mahmood, A.; Xiong, Y. Subacute intranasal administration of tissue plasminogen activator promotes neuroplasticity and improves functional recovery following traumatic brain injury in rats. PLoS ONE 2014, 9, e106238. [Google Scholar] [CrossRef] [PubMed]
- Iacobas, D.A.; Wen, J.; Iacobas, S.; Putterman, C.; Schwartz, N. TWEAKing the Hippocampus: The Effects of TWEAK on the Genomic Fabric of the Hippocampus in a Neuropsychiatric Lupus Mouse Model. Genes 2021, 12, 1172. [Google Scholar] [CrossRef] [PubMed]
- Budhram, A.; Butendieck, R.R.; Duarte-Garcia, A.; Brinjikji, W.; Zalewski, N.L. Striatal Encephalitis: Potential Inflammatory Vasculopathy in Systemic Lupus Erythematosus. Can. J. Neurol. Sci. 2021, 48, 415–416. [Google Scholar] [CrossRef]
- Schwartz, N.; Stock, A.D.; Putterman, C. Neuropsychiatric lupus: New mechanistic insights and future treatment directions. Nat. Rev. Rheumatol. 2019, 15, 137–152. [Google Scholar] [CrossRef] [PubMed]
- Lin, L.; Hu, K. Tissue plasminogen activator and inflammation: From phenotype to signaling mechanisms. Am. J. Clin. Exp. Immunol. 2014, 3, 30–36. [Google Scholar] [PubMed]
- Xu, D.; Lian, D.; Wu, J.; Liu, Y.; Zhu, M.; Sun, J.; He, D.; Li, L. Brain-derived neurotrophic factor reduces inflammation and hippocampal apoptosis in experimental Streptococcus pneumoniae meningitis. J. Neuroinflamm. 2017, 14, 156. [Google Scholar] [CrossRef] [Green Version]
- Sangwung, P.; Zhou, G.; Nayak, L.; Chan, E.R.; Kumar, S.; Kang, D.W.; Zhang, R.; Liao, X.; Lu, Y.; Sugi, K.; et al. KLF2 and KLF4 control endothelial identity and vascular integrity. JCI Insight 2017, 2, e91700. [Google Scholar] [CrossRef]
- Jin, H.; Zhu, Y.; Li, Y.; Ding, X.; Ma, W.; Han, X.; Wang, B. BDNF-mediated mitophagy alleviates high-glucose-induced brain microvascular endothelial cell injury. Apoptosis 2019, 24, 511–528. [Google Scholar] [CrossRef]
- Williamson, L.L.; Bilbo, S.D. Chemokines and the hippocampus: A new perspective on hippocampal plasticity and vulnerability. Brain Behav. Immun. 2013, 30, 186–194. [Google Scholar] [CrossRef]
- Small, S.A. Isolating pathogenic mechanisms embedded within the hippocampal circuit through regional vulnerability. Neuron 2014, 84, 32–39. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kelley, B.P.; Corrigan, J.J.; Patel, S.C.; Griffith, B.D. Neuropsychiatric Lupus with Antibody-Mediated Striatal Encephalitis. AJNR Am. J. Neuroradiol. 2018, 39, 2263–2269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raventhiranathan, N.; Hussien, A.R.; Mirchia, K.; Swarnkar, A.; Mangla, R. Striatal dominant lupus encephalitis-Is it vasculitis or an autoimmune process? Literature review & new case report with vessel wall imaging. Radiol. Case Rep. 2022, 17, 1205–1210. [Google Scholar] [CrossRef] [PubMed]
- Uchida, H.; Matsushita, Y.; Ueda, H. Epigenetic regulation of BDNF expression in the primary sensory neurons after peripheral nerve injury: Implications in the development of neuropathic pain. Neuroscience 2013, 240, 147–154. [Google Scholar] [CrossRef]
- Bayas, A.; Hummel, V.; Kallmann, B.A.; Karch, C.; Toyka, K.V.; Rieckmann, P. Human cerebral endothelial cells are a potential source for bioactive BDNF. Cytokine 2002, 19, 55–58. [Google Scholar] [CrossRef]
- Schmidt-Kastner, R.; Freund, T.F. Selective vulnerability of the hippocampus in brain ischemia. Neuroscience 1991, 40, 599–636. [Google Scholar] [CrossRef]
- Schmidt-Kastner, R. Genomic approach to selective vulnerability of the hippocampus in brain ischemia-hypoxia. Neuroscience 2015, 309, 259–279. [Google Scholar] [CrossRef]
- Mizrachi, M.; Anderson, E.; Carroll, K.R.; Tehrani, N.; Volpe, B.T.; Diamond, B. Cognitive dysfunction in SLE: An understudied clinical manifestation. J. Autoimmun. 2022, 132, 102911. [Google Scholar] [CrossRef]
- Förstermann, U.; Sessa, W.C. Nitric oxide synthases: Regulation and function. Eur. Heart J. 2012, 33, 829–837. [Google Scholar] [CrossRef] [Green Version]
- Banoujaafar, H.; Monnier, A.; Pernet, N.; Quirié, A.; Garnier, P.; Prigent-Tessier, A.; Marie, C. Brain BDNF levels are dependent on cerebrovascular endothelium-derived nitric oxide. Eur. J. Neurosci. 2016, 44, 2226–2235. [Google Scholar] [CrossRef]
- Li, S.T.; Pan, J.; Hua, X.M.; Liu, H.; Shen, S.; Liu, J.F.; Li, B.; Tao, B.B.; Ge, X.L.; Wang, X.H.; et al. Endothelial nitric oxide synthase protects neurons against ischemic injury through regulation of brain-derived neurotrophic factor expression. CNS Neurosci. Ther. 2014, 20, 154–164. [Google Scholar] [CrossRef] [PubMed]
- Cohen, D.; Rijnink, E.C.; Nabuurs, R.J.; Steup-Beekman, G.M.; Versluis, M.J.; Emmer, B.J.; Zandbergen, M.; van Buchem, M.A.; Allaart, C.F.; Wolterbeek, R.; et al. Brain histopathology in patients with systemic lupus erythematosus: Identification of lesions associated with clinical neuropsychiatric lupus syndromes and the role of complement. Rheumatology 2017, 56, 77–86. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tan, Y.V.; Abad, C.; Wang, Y.; Lopez, R.; Waschek, J. VPAC2 (vasoactive intestinal peptide receptor type 2) receptor deficient mice develop exacerbated experimental autoimmune encephalomyelitis with increased Th1/Th17 and reduced Th2/Treg responses. Brain Behav. Immun. 2015, 44, 167–175. [Google Scholar] [CrossRef]
- Wang, M.; Wang, Z.; Zhang, S.; Wu, Y.; Zhang, L.; Zhao, J.; Wang, Q.; Tian, X.; Li, M.; Zeng, X. Progress in the Pathogenesis and Treatment of Neuropsychiatric Systemic Lupus Erythematosus. J. Clin. Med. 2022, 11, 4955. [Google Scholar] [CrossRef] [PubMed]
- Mandwie, M.; Karunia, J.; Niaz, A.; Keay, K.A.; Musumeci, G.; Rennie, C.; McGrath, K.; Al-Badri, G.; Castorina, A. Metformin Treatment Attenuates Brain Inflammation and Rescues PACAP/VIP Neuropeptide Alterations in Mice Fed a High-Fat Diet. Int. J. Mol. Sci. 2021, 22, 13660. [Google Scholar] [CrossRef]
- Pardo, L.M.; Rizzu, P.; Francescatto, M.; Vitezic, M.; Leday, G.G.R.; Sanchez, J.S.; Khamis, A.; Takahashi, H.; van de Berg, W.D.J.; Medvedeva, Y.A.; et al. Regional differences in gene expression and promoter usage in aged human brains. Neurobiol. Aging 2013, 34, 1825–1836. [Google Scholar] [CrossRef] [Green Version]
- Hawkes, C.A.; Gatherer, M.; Sharp, M.M.; Dorr, A.; Yuen, H.M.; Kalaria, R.; Weller, R.O.; Carare, R.O. Regional differences in the morphological and functional effects of aging on cerebral basement membranes and perivascular drainage of amyloid-β from the mouse brain. Aging Cell 2013, 12, 224–236. [Google Scholar] [CrossRef]
- Tzang, B.S.; Hsu, T.C.; Chen, T.Y.; Huang, C.Y.; Li, S.L.; Kao, S.H. Cystamine ameliorates ventricular hypertrophy associated with modulation of IL-6-mediated signaling in lupus-prone mice. Life Sci. 2013, 92, 719–726. [Google Scholar] [CrossRef]
- Paxinos, G.; Franklin, K.B. Paxinos and Franklin’s the Mouse Brain in Stereotaxic Coordinates; Academic Press: Cambridge, MA, USA, 2019. [Google Scholar]
- Schmittgen, T.D.; Livak, K.J. Analyzing real-time PCR data by the comparative C(T) method. Nat. Protoc. 2008, 3, 1101–1108. [Google Scholar] [CrossRef]
- Roy, J.G.; McElhaney, J.E.; Verschoor, C.P. Reliable reference genes for the quantification of mRNA in human T-cells and PBMCs stimulated with live influenza virus. BMC Immunol. 2020, 21, 4. [Google Scholar] [CrossRef]
- Bucolo, C.; Leggio, G.M.; Maltese, A.; Castorina, A.; D’Agata, V.; Drago, F. Dopamine-(3) receptor modulates intraocular pressure: Implications for glaucoma. Biochem. Pharmacol. 2012, 83, 680–686. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Accession No. | Gene Name (in Parenthesis) | Primer Sequences (5′-3′) | Length (bp) |
|---|---|---|---|
| NM_009625.2 | Pituitary adenylate cyclase-activating polypeptide (Adcyap1) | Fwd CTGCGTGACGCTTACGCCCT Rev CCTAGGTTCTCCCCCGCGCC | 152 |
| NM_011702.2 | Vasoactive intestinal peptide (Vip) | Fwd TGGCAAACGAATCAGCAGCAGCA Rev AGCCATTTGCTTTCTGAGGCGGG | 106 |
| NM_007407.3 | PAC1 receptor (Adcyap1r1) | Fwd CAGTCCCCAGACATGGGAGGCA Rev AGCGGGCCAGCCGTAGAGTA | 139 |
| NM_011703.4 | VPAC1 receptor (Vipr1) | Fwd TCAATGGCGAGGTGCAGGCAG Rev TGTGTGCTGCACGAGACGCC | 127 |
| NM_009511.2 | VPAC2 receptor (Vipr2) | Fwd GCGTCGGTGGTGCTGACCTG Rev ACACCGCTGCAGGCTCTCTGAT | 155 |
| NM_008872.2 | Tissue plasminogen activator (Plat) | Fwd GCCTGTCCGAAGTTGCAGCGA Rev TGCTGTGCTCCACGTGCCTC | 184 |
| NM_008873.3 | Urokinase plasminogen activator (Plau) | Fwd TTCGCAGCCATCTACCAGAA Rev TGGGAGTTGAATGAAGCAGTG | 117 |
| NM_010493.3 | Intercellular adhesion molecule-1 (Icam 1) | Fwd CCTCCGGACTTTCGATCTTC Rev TCACTGCTGTTTGTGCTCTC | 180 |
| NM_011693.3 | Vascular cell adhesion molecule-1 (Vcam 1) | Fwd GGATACTGTTTGCAGTCTCTCA Rev GCGTTTAGTGGGCTGTCTAT | 160 |
| NM_007540.4 | Brain-derived neurotrophic factor (Bdnf) | Fwd CGAGTGGGTCACAGCGGCAG Rev GCCCCTGCAGCCTTCCTTGG | 160 |
| NM_008713.4 | Endothelial nitric oxide synthase (Nos3) | Fwd AGGTATTTGATGCTCGGGAC Rev CTGTGATGGCTGAACGAAGA | 108 |
| NM_010637.3 | Krüppel-like factor 4 (Klf4) | Fwd CCCACACTTGTGACTATGCAG Rev GTTTCTCGCCTGTGTGAGTT | 90 |
| NM_011296.2 | 18s ribosomal subunit (s18) | Fwd CCCTGAGAAGTTCCAGCACA Rev GGTGAGGTCGATGTCTGCTT | 145 |
| Primary Antibodies (All Raised in Rabbit) | Dilution | Source (Cat. No.) |
|---|---|---|
| Pituitary adenylate cyclase-activating polypeptide (PACAP) | 1:1000 | GeneTex (Irvine, CA, USA, GTX37576) |
| Vasoactive intestinal peptide (VIP) | 1:1000 | GeneTex (Irvine, CA, USA, GTX129461) |
| PAC1 receptor | 1:1000 | GeneTex (Irvine, CA, USA, GTX30026) |
| VPAC1 receptor | 1:500 | Sigma-Aldrich (Castle Hill, NSW, Australia, SAB4503084) |
| VPAC2 receptor | 1:500 | Sigma-Aldrich (Castle Hill, NSW, Australia, AB2266) |
| Brain-derived neurotrophic factor (BDNF) | 1:1000 | GeneTex (Irvine, CA, USA, GTX132621) |
| Endothelial nitric oxide synthase (eNOS) | 1:1000 | GeneTex (Irvine, CA, USA, GTX129058) |
| Krüppel-like factor 4 (KLF4) | 1:1000 | GeneTex (Irvine, CA, USA, GTX101508) |
| Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) | 1:2000 | Bio-Rad (Gladesville, NSW, Australia, VPA00187) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, J.; Thomas Broome, S.; Jansen, M.I.; Mandwie, M.; Logan, G.J.; Marzagalli, R.; Musumeci, G.; Castorina, A. Altered Hippocampal and Striatal Expression of Endothelial Markers and VIP/PACAP Neuropeptides in a Mouse Model of Systemic Lupus Erythematosus. Int. J. Mol. Sci. 2023, 24, 11118. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms241311118
Lee J, Thomas Broome S, Jansen MI, Mandwie M, Logan GJ, Marzagalli R, Musumeci G, Castorina A. Altered Hippocampal and Striatal Expression of Endothelial Markers and VIP/PACAP Neuropeptides in a Mouse Model of Systemic Lupus Erythematosus. International Journal of Molecular Sciences. 2023; 24(13):11118. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms241311118
Chicago/Turabian StyleLee, Jayden, Sarah Thomas Broome, Margo Iris Jansen, Mawj Mandwie, Grant J. Logan, Rubina Marzagalli, Giuseppe Musumeci, and Alessandro Castorina. 2023. "Altered Hippocampal and Striatal Expression of Endothelial Markers and VIP/PACAP Neuropeptides in a Mouse Model of Systemic Lupus Erythematosus" International Journal of Molecular Sciences 24, no. 13: 11118. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms241311118