
Molecular Pathogenesis of Central and Peripheral Nervous System Complications in Anderson–Fabry Disease

 , ,
, , 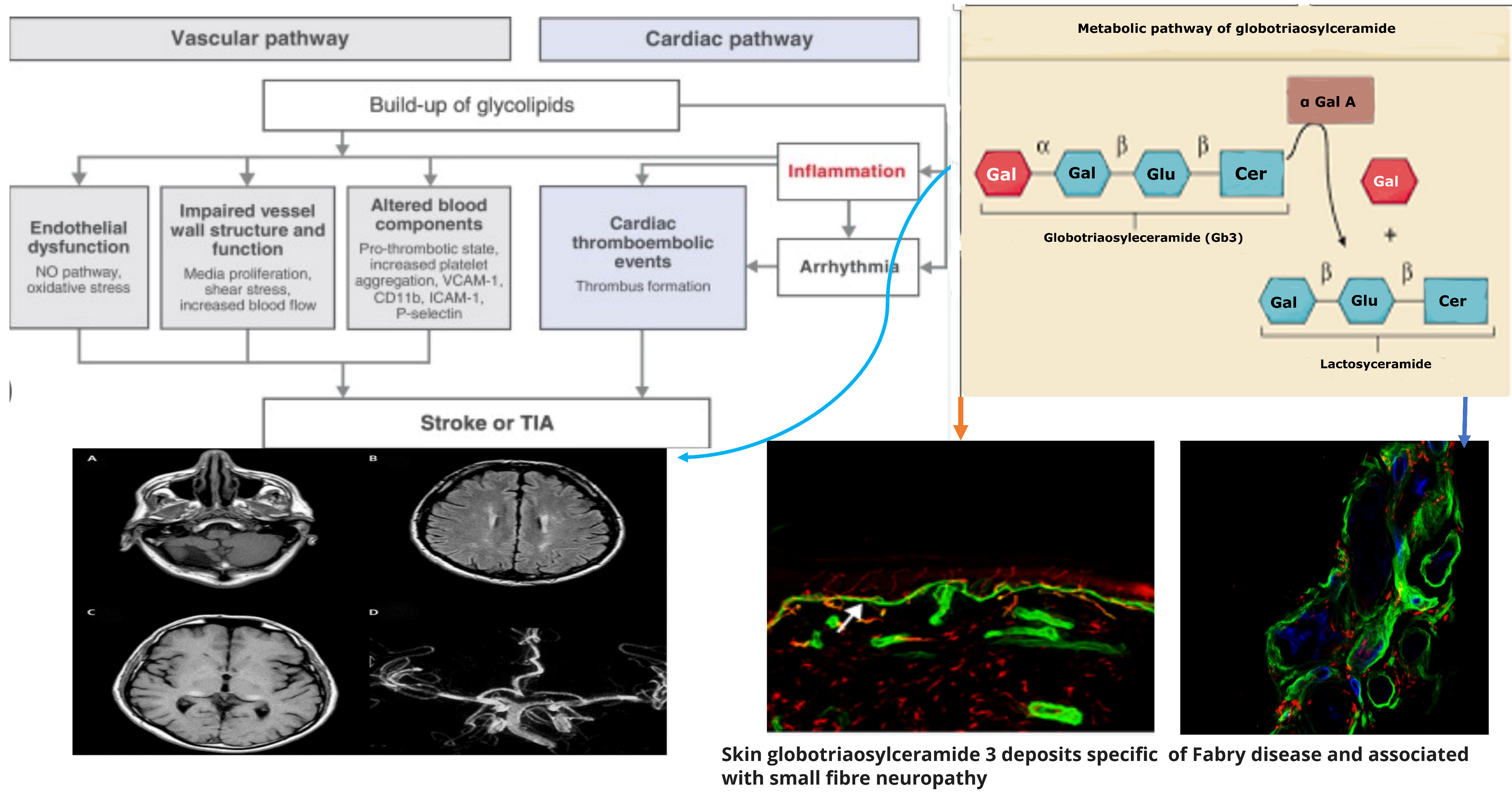{kind=link}
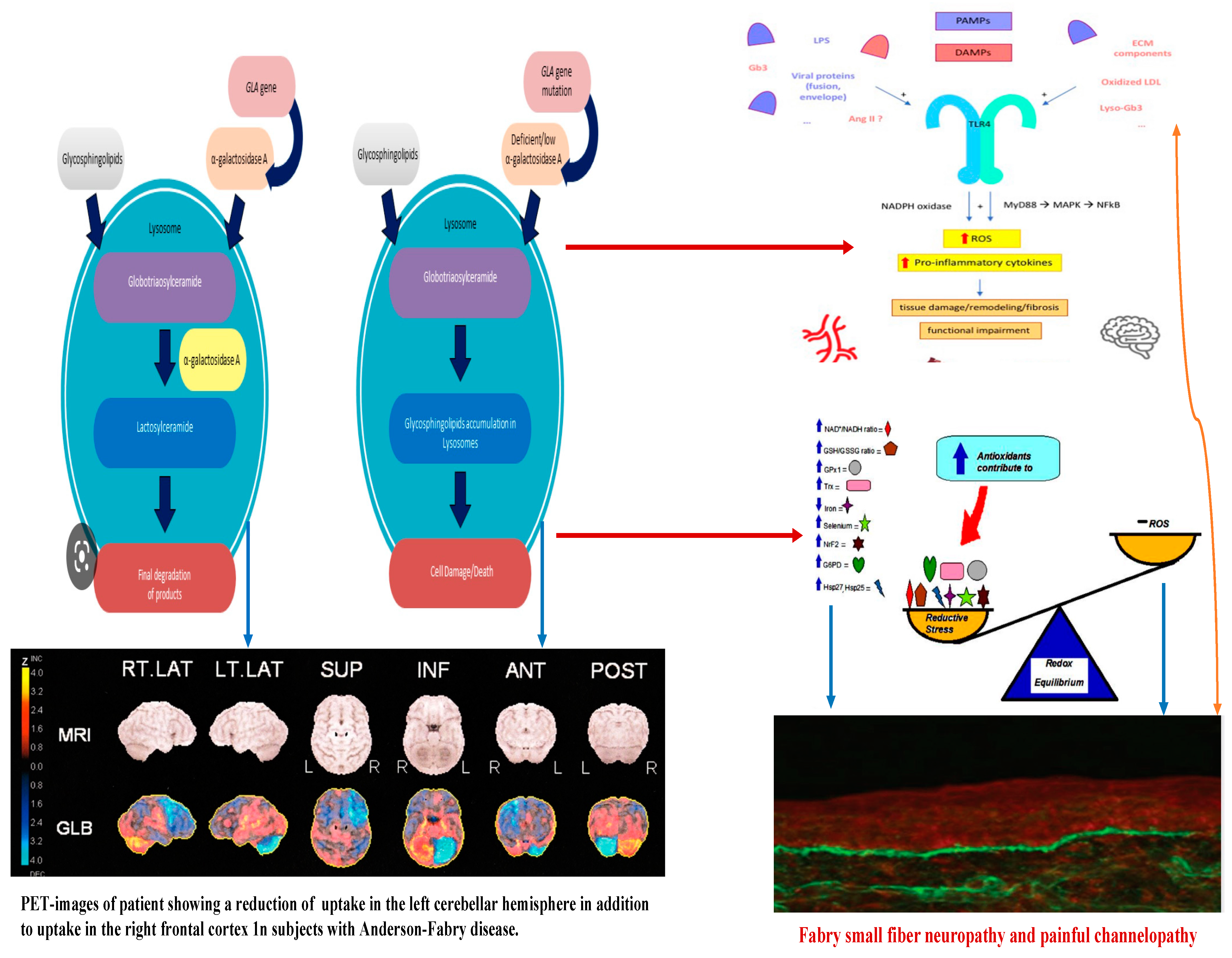{kind=link}
Abstract
:1. Background
2. Molecular Pathogenesis of Anderson–Fabry Disease
3. Molecular Pathogenesis of Central Nervous System Involvement in Anderson–Fabry Disease
4. Molecular Pathogenesis of Peripheral Nerve Involvement in Anderson–Fabry Disease
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Meikle, P.J.; Hopwood, J.J.; Clague, A.E.; Carey, W.F. Prevalence of lysosomal storage disorders. JAMA 1999, 281, 249–254. [Google Scholar] [CrossRef]
- Kahn, P. Anderson-Fabry disease: A histopathological study of three cases with observations on the mechanism of production of pain. J. Neurol. Neurosurg. Psychiatry 1973, 36, 1053–1062. [Google Scholar] [CrossRef]
- Gemignani, F.; Marbini, A.; Bragaglia, M.M.; Govoni, E. Pathological study of the sural nerve in Fabry’s disease. Eur. Neurol. 1984, 23, 173–181. [Google Scholar] [CrossRef]
- Desnick, R.J.; Ionnou, Y.; Eng, C.M. Fabry disease: Alpha galactosidase A deficiency. In The Metabolic and Molecular Bases of Inherited Disease; Scriver, C.H., Beaudet, A.L., Sly, W.S., Valle, D., Eds.; McGraw Hill: New York, NY, USA, 1995; pp. 2741–2784. [Google Scholar]
- Wise, D.; Wallace, H.J.; Jellinek, E.H. Angiokeratoma corporis diVusum. Q. J. Med. 1962, XXXI, 177–212. [Google Scholar]
- Desnick, R.J.; Ioannou, Y.A. α-Galactosidase a Deficiency. Fabry Disease. In The Metabolic and Molecular Bases of Inherited Disease, 8th ed.; Scriver, C.R., Beaudet, A.L., Sly, W.S., Valle, D., Eds.; McGraw-Hill: New York, NY, USA, 2001. [Google Scholar]
- Safyan, R.; Whybra, C.; Beck, M.; Elstein, D.; Altarescu, G. An association study of inflammatory cytokine gene polymorphisms in Fabry disease. Eur. Cytokine Netw. 2006, 17, 271–275. [Google Scholar]
- Thomaidis, T.; Relle, M.; Golbas, M.; Brochhausen, C.; Galle, P.R.; Beck, M.; Schwarting, A. Downregulation of alpha-galactosidase A upreg- ulates CD77: Functional impact for Fabry nephropathy. Kidney Int. 2009, 75, 399–407. [Google Scholar] [CrossRef]
- Tuttolomondo, A.; Pecoraro, R.; Simonetta, I.; Miceli, S.; Arnao, V.; Licata, G.; Pinto, A. Neurological complications of Anderson-Fabry disease. Curr. Pharm. Des. 2013, 19, 6014–6030. [Google Scholar] [CrossRef]
- Tran, N.; Garcia, T.; Aniqa, M.; Ali, S.; Ally, A.; Nauli, S.M. Endothelial Nitric Oxide Synthase (eNOS) and the Cardiovascular System: In Physiology and in Disease States. Am. J. Biomed. Sci. Res. 2022, 15, 153–177. [Google Scholar]
- Sweeley, C.C.; Klionsky, B. Fabry’s Disease: Classification as a sphingolipidosis and partial char-acterization of a novel glycolipid. J. Biol. Chem. 1963, 238, 3148–3150. [Google Scholar] [CrossRef]
- Brady, R.O.; Gal, A.E.; Bradley, R.M.; Martensson, E.; Warshaw, A.L.; Laster, L. Enzymatic Defect in Fabry’s Disease. N. Engl. J. Med. 1967, 276, 1163–1167. [Google Scholar] [CrossRef]
- Valbuena, C.; Carvalho, E.; Bustorff, M.; Ganhao, M.; Relvas, S.; Nogueira, R.; Carneiro, F.; Oliveira, J.P. Kidney biopsy findings in heterozygous Fabry disease females with early nephropathy. Virchows Arch. 2008, 453, 329–338. [Google Scholar] [CrossRef]
- Rozenfeld, P.; Agriello, E.; De Francesco, N.; Martinez, P.; Fossati, C. Leukocyte perturbation associated with Fabry disease. J. Inherit. Metab. Dis. 2009, 32 (Suppl. S1), S67–S77. [Google Scholar] [CrossRef]
- Altarescu, G.; Chicco, G.; Whybra, C.; Delgado-Sanchez, S.; Sharon, N.; Beck, M.; Elstein, D. Correlation between interleukin-6 pro- moter and C-reactive protein (CRP) polymorphisms and CRP levels with the Mainz Severity Score Index for Fabry disease. J. Inherit. Metab. Dis. 2008, 31, 117–123. [Google Scholar] [CrossRef]
- Moore, D.F.; Goldin, E.; Gelderman, M.P.; Robinson, C.; Baer, J.; Ries, M.; Elkahloun, A.; Brady, R.O.; Schiffmann, R. Apoptotic abnormalities in differential gene expression in peripheral blood mononuclear cells from children with Fabry disease. Acta Paediatr. Suppl. 2008, 97, 48–52. [Google Scholar] [CrossRef]
- Shen, J.S.; Meng, X.L.; Moore, D.F.; Quirk, J.M.; Shayman, J.A.; Schiffmann, R.; Kaneski, C.R. Globotriaosylceramide induces oxidative stress and up-regulates cell adhesion molecule expression in Fabry disease endothelial cells. Mol. Genet. Metab. 2008, 95, 163–168. [Google Scholar] [CrossRef]
- Rohard, I.; Schaefer, E.; Kampmann, C.; Beck, M.; Gal, A. Association between polymorphisms of endothelial nitric oxide synthase gene (NOS3) and left posterior wall thickness (LPWT) of the heart in Fabry disease. J. Inherit. Metab. Dis. 2008, 31 (Suppl. S2), S349–S356. [Google Scholar] [CrossRef]
- Wang, R.Y.; Abe, J.T.; Cohen, A.H.; Wilcox, W.R. Enzyme replacement therapy stabilizes obstructive pulmonary Fabry disease associated with respiratory globotriaosylceramide storage. J. Inherit. Metab. Dis. 2008, 31 (Suppl. S2), S369–S374. [Google Scholar] [CrossRef]
- Aerts, J.M.; Groener, J.E.; Kuiper, S.; Donker-Koopman, W.E.; Strijland, A.; Ottenhoff, R.; van Roomen, C.; Mirzaian, M.; Wijburg, F.A.; Linthorst, G.E.; et al. Elevated globotriaosylsphingosine is a hallmark of abry disease. Proc. Natl. Acad. Sci. USA 2008, 105, 2812–2817. [Google Scholar] [CrossRef]
- Schiffmann, R. Fabry disease. Pharmacol. Ther. 2009, 122, 65–77. [Google Scholar] [CrossRef]
- Moore, D.F.; Kaneski, C.R.; Askari, H.; Schiffmann, R. The cerebral vasculopathy of Fabry disease. J. Neurol. Sci. 2007, 257, 258–263. [Google Scholar] [CrossRef]
- Rombach, S.M.; Twickler, T.B.; Aerts, J.M.; Linthorst, G.E.; Wijburg, F.A.; Hollak, C.E. Vasculopathy in patients with Fabry disease: Current controversies and research directions. Mol. Genet. Metab. 2010, 99, 99–108. [Google Scholar] [CrossRef]
- Namdar, M.; Gebhard, C.; Studiger, R.; Shi, Y.; Mocharla, P.; Schmied, C.; Brugada, P.; Lüscher, T.F.; Camici, G.G. Globotriaosylsphingosine accumulation and not alpha-galactosidase-A deficiency causes endothelial dysfunction in Fabry disease. PLoS ONE 2012, 7, e36373. [Google Scholar] [CrossRef]
- Sestito, S.; Ceravolo, F.; Concolino, D. Anderson-Fabry disease in children. Curr. Pharm. Des. 2013, 19, 6037–6045. [Google Scholar] [CrossRef]
- Sims, K.; Politei, J.; Banikazemi, M.; Lee, P. Stroke in Fabry disease frequently occurs before diagnosis and in the absence of other clinical events: Natural history data from the Fabry Registry. Stroke 2009, 40, 788–794. [Google Scholar] [CrossRef]
- Buechner, S.; Moretti, M.; Burlina, A.P.; Cei, G.; Manara, R.; Ricci, R.; Mignani, R.; Parini, R.; Di Vito, R.; Giordano, G.P.; et al. Central nervous system involvement in Anderson-Fabry disease: A clinical and MRI retrospective study. J. Neurol. Neurosurg. Psychiatry 2008, 79, 1249–1254. [Google Scholar] [CrossRef]
- Saito, S.; Ohno, K.; Sakuraba, H. Fabry-database.org: Database of the clinical phenotypes, genotypes and mutant α-galactosidase A structures in Fabry disease. J. Hum. Genet. 2011, 56, 467–468. [Google Scholar] [CrossRef]
- Cable, W.J.L.; Kolodny, E.H.; Adams, R.D. Fabry disease impaired autonomic function. Neurology 1982, 32, 498. [Google Scholar] [CrossRef]
- Tabira, T.; Goto, I.; Kuroiwa, Y.; Kikuchi, M. Neuropathological and biochemical studies in Fabry’s disease. Acta Neuropathol. 1974, 30, 345–354. [Google Scholar] [CrossRef]
- Scott, L.J.C.; Griffin, J.W.; Luciano, C.; Barton, N.W.; Banerjee, T.; Crawford, T.; McArthur, J.C.; Tournay, A.; Schiffmann, R. Quantitative analysis of epidermal innervation in Fabry disease. Neurology 1999, 52, 1249. [Google Scholar] [CrossRef]
- Schiffmann, R. Neuropathy and Fabry disease: Pathogenesis and enzyme replacement therapy. Acta Neurol. Belg. 2006, 106, 61. [Google Scholar]
- Choi, L.; Vernon, J.; Kopach, O.; Minett, M.S.; Mills, K.; Clayton, P.T.; Meert, T.; Wood, J.N. The Fabry disease-associated lipid Lyso-Gb3 enhances voltage-gated calcium currents in sensory neurons and causes pain. Neurosci. Lett. 2015, 594, 163–168. [Google Scholar] [CrossRef]
- Prado, V.F.; Roy, A.; Kolisnyk, B.; Gros, R.; Prado, M.A.M. Regulation of cholinergic activity by the vesicular acetylcholine transporter. Biochem. J. 2013, 450, 265–274. [Google Scholar] [CrossRef]
- Lücke, T. Fabry disease: Reduced activities of respiratory chain enzymes with decreased levels of energy-rich phosphates in fibroblasts. Mol. Genet. Metab. 2004, 82, 93–97. [Google Scholar] [CrossRef]
- Yamamoto, A.; Abuillan, W.; Burk, A.S.; Körner, A.; Ries, A.; Werz, D.B.; Demé, B.; Tanaka, M. Influence of length and conformation of saccharide head groups on the mechanics of glycolipid membranes: Unraveled by off-specular neutron scattering. J. Chem. Phys. 2015, 142, 154907. [Google Scholar] [CrossRef]
- Park, S.; Kim, J.A.; Joo, K.Y.; Choi, S.; Choi, E.N.; Shin, J.A.; Han, K.H.; Jung, S.C.; Suh, S.H. Globotriaosylceramide leads to KCa3.1 channel dysfunction: A new insight into endothelial dysfunction in Fabry disease. Cardiovasc. Res. 2011, 89, 290–299. [Google Scholar] [CrossRef]
- Schäfer, M.K.-H.; Eiden, L.E.; Weihe, E. Cholinergic neurons and terminal fields revealed by immunohistochemistry for the vesicular acetylcholine transporter. II. The peripheral nervous system. Neuroscience 1998, 84, 361–376. [Google Scholar] [CrossRef]
- Moore, A.M.; Wood, M.D.; Chenard, K.; Hunter, D.A.; Mackinnon, S.E.; Sakiyama-Elbert, S.E.; Borschel, G.H. Controlled delivery of glial cell line-derived neurotrophic factor enhances motor nerve regeneration. J. Hand Surg. Am. 2010, 35, 2008–2017. [Google Scholar] [CrossRef]
- Politei, J.M.; Bouhassira, D.; Germain, D.P.; Goizet, C.; Guerrero-Sola, A.; Hilz, M.J.; Hutton, E.J.; Karaa, A.; Liguori, R.; Üçeyler, N.; et al. Pain in Fabry Disease: Practical Recommendations for Diagnosis and Treatment. CNS Neurosci. Ther. 2016, 22, 568–576. [Google Scholar] [CrossRef]
- Miller, J.J.; Aoki, K.; Mascari, C.A.; Beltrame, A.K.; Sokumbi, O.; North, P.E.; Tiemeyer, M.; Kriegel, A.J.; Dahms, N.M. α-Galactosidase A-deficient rats accumulate glycosphingolipids and develop cardiorenal phenotypes of Fabry disease. FASEB J. 2018, 33, 418–429. [Google Scholar] [CrossRef]
- Burand, A.J., Jr.; Stucky, C.L. Fabry disease pain: Patient and preclinical parallels. Pain 2021, 162, 1305–1321. [Google Scholar] [CrossRef]
- Kaneski, C.R.; Brady, R.O.; Hanover, J.A.; Schueler, U.H. Development of a model system for neuronal dysfunction in Fabry disease. Mol. Genet. Metab. 2016, 119, 144–150. [Google Scholar] [CrossRef]
- Sluka, K.A.; Winter, O.C.; Wemmie, J.A. Acid-sensing ion channels: A new target for pain and CNS diseases. Curr. Opin. Drug Discov. Devel. 2009, 12, 693–704. [Google Scholar]
- DeGraba, T.; Azhar, S.; Dignat-George, F.; Brown, E.; Boutière, B.; Altarescu, G.; McCarron, R.; Schiffmann, R. Profile of endothelial and leukocyte activation in Fabry patients. Ann. Neurol. 2000, 47, 229–233. [Google Scholar] [CrossRef]
- van Breemen, M.J.; Rombach, S.M.; Dekker, N.; Poorthuis, B.J.; Linthorst, G.E.; Zwinderman, A.H.; Breunig, F.; Wanner, C.; Aerts, J.M.; Hollak, C.E. Reduction of elevated plasma globotriaosylsphingosine in patients with classic Fabry disease following enzyme replacement therapy. Biochim. Biophys. Acta 2011, 1812, 70–76. [Google Scholar] [CrossRef]
- Zampetti, A.; Gnarra, M.; Borsini, W.; Giurdanella, F.; Antuzzi, D.; Piras, A.; Smaldone, C.; Pieroni, M.; Cadeddu, C.; de Waure, C.; et al. Vascular endothelial growth factor (VEGF-a) in Fabry disease: Association with cutaneous and systemic manifestations with vascular involvement. Cytokine 2013, 61, 933–939. [Google Scholar] [CrossRef]
- Klug, K.; Spitzel, M.; Hans, C.; Klein, A.; Schottmann, N.M.; Erbacher, C.; Üçeyler, N. Endothelial Cell Dysfunction and Hypoxia as Potential Mediators of Pain in Fabry Disease: A Human-Murine Translational Approach. Int. J. Mol. Sci. 2023, 24, 15422. [Google Scholar] [CrossRef]
- Biegstraaten, M.; Hollak, C.E.; Bakkers, M.; Faber, C.G.; Aerts, J.M.; van Schaik, I.N. Small fiber neuropathy in Fabry disease. Mol. Genet. Metab. 2012, 106, 135–141. [Google Scholar] [CrossRef]
- Schuller, Y.; Linthorst, G.E.; Hollak, C.E.; Van Schaik, I.N.; Biegstraaten, M. Pain management strategies for neuropathic pain in Fabry disease--a systematic review. BMC Neurol. 2016, 16, 25. [Google Scholar]
- Hilz, M.J.; Kolodny, E.H.; Brys, M.; Stemper, B.; Haendl, T.; Marthol, H. Reduced cerebral blood flow velocity and impaired cerebral autoregulation in patients with Fabry disease. J. Neurol. 2004, 251, 564–570. [Google Scholar] [CrossRef]
- Moore, D.F.; Ye, F.; Brennan, M.; Gupta, S.; Barshop, B.A.; Steiner, R.D.; Rhead, W.J.; Brady, R.O.; Hazen, S.L.; Schiffmann, R. Ascorbate decreases fabry cerebral hyperperfusion suggesting a reactive oxygen species abnormality: An arterial spin tagging study. J. Magn. Reson. Imaging 2004, 20, 674–683. [Google Scholar] [CrossRef]
- Geevasinga, N.; Tchan, M.; Sillence, D.; Vucic, S. Upregulation of inward rectifying currents and Fabry disease neuropathy. J. Peripher. Nerv. Syst. 2012, 17, 399–406. [Google Scholar] [CrossRef]
- Castellanos, L.C.S.; Rozenfeld, P.; Gatto, R.G.; Reisin, R.C.; Uchitel, O.D.; Weissmann, C. Upregulation of ASIC1a channels in an in vitro model of Fabry disease. Neurochem. Int. 2020, 140, 104824. [Google Scholar] [CrossRef]
- Hofmann, L.; Hose, D.; Grießhammer, A.; Blum, R.; Döring, F.; Dib-Hajj, S.; Waxman, S.; Sommer, C.; Wischmeyer, E.; Üçeyler, N. Characterization of small fiber pathology in a mouse model of Fabry disease. Elife 2018, 7, e39300. [Google Scholar] [CrossRef]
- Waltz, T.B.; Burand, A.J., Jr.; Sadler, K.E.; Stucky, C.L. Sensory-specific peripheral nerve pathology in a rat model of Fabry disease. Neurobiol. Pain 2021, 10, 100074. [Google Scholar] [CrossRef]
- Kocen, R.S.; Thomas, P.K. Peripheral Nerve Involvement in Fabry’s Disease. Arch. Neurol. 1970, 22, 81–88. [Google Scholar] [CrossRef]
- Torvin Møller, A.; Winther Bach, F.; Feldt-Rasmussen, U.; Rasmussen, A.; Hasholt, L.; Lan, H.; Sommer, C.; Kølvraa, S.; Ballegaard, M.; Staehelin Jensen, T. Functional and structural nerve fiber findings in heterozygote patients with Fabry disease. Pain 2009, 145, 237–245. [Google Scholar] [CrossRef]
- Uceyler, N.; Ganendiran, S.; Kramer, D.; Sommer, C. Characterization of pain in fabry disease. Clin. J. Pain 2014, 30, 915–920. [Google Scholar] [CrossRef]
- Burlina, A.P.; Sims, K.B.; Politei, J.M.; Bennett, G.J.; Baron, R.; Sommer, C.; Møller, A.T.; Hilz, M.J. Early diagnosis of peripheral nervous system involvement in Fabry disease and treatment of neuropathic pain: The report of an expert panel. BMC Neurol. 2011, 11, 61. [Google Scholar] [CrossRef]
- Üçeyler, N.; Kahn, A.K.; Kramer, D.; Zeller, D.; Casanova-Molla, J.; Wanner, C.; Weidemann, F.; Katsarava, Z.; Sommer, C. Impaired small fiber conduction in patients with Fabry disease: A neurophysiological case-control study. BMC Neurol. 2013, 13, 47. [Google Scholar] [CrossRef]
- Dütsch, M.; Marthol, H.; Stemper, B.; Brys, M.; Haendl, T.; Hilz, M.J. Small fiber dysfunction predominates in Fabry neuropathy. J. Clin. Neurophysiol. 2002, 19, 575–586. [Google Scholar] [CrossRef]
- Üçeyler, N.; He, L.; Schönfeld, D.; Kahn, A.K.; Reiners, K.; Hilz, M.J.; Breunig, F.; Sommer, C. Small fibers in Fabry disease: Baseline and follow-up data under enzyme replacement therapy. J. Peripher. Nerv. Syst. 2011, 16, 304–314. [Google Scholar] [CrossRef]
- Politei, J.M.; Pagano, M.A. Peripheral neuropathy in AndersonFabry disease: Its physiology, evaluation and treatment. Rev. Neurol. 2004, 38, 979–983. [Google Scholar]
- Siedler, G.; Káhn, A.K.; Weidemann, F.; Wanner, C.; Sommer, C.; Üçeyler, N. Dyshidrosis is associated with reduced amplitudes in electrically evoked pain-related potentials in women with Fabry disease. Clin. Neurophysiol. 2019, 130, 528–536. [Google Scholar] [CrossRef]
- Schiffmann, R.; Scott, L.J. Pathophysiology and assessment of neuropathic pain in Fabry disease. Acta Paediatr. Suppl. 2002, 91, 48–52. [Google Scholar] [CrossRef]
- Hilz, M.J.; Koehn, J.; Kolodny, E.H.; Brys, M.; Moeller, S.; Stemper, B. Metronomic breathing shows altered parasympathetic baroreflex function in untreated Fabry patients and baroreflex improvement after enzyme replacement therapy. J. Hypertens. 2011, 29, 2387–2394. [Google Scholar] [CrossRef]
- deVeber, G.A.; Schwarting, G.A.; Kolodny, E.H.; Kowall, N.W. Fabry disease: Immunocytochemical characterization of neuronal involvement. Ann. Neurol. 1992, 31, 409–415. [Google Scholar] [CrossRef]
- Lao, L.-M.; Kumakiri, M.; Mima, H.; Kuwahara, H.; Ishida, H.; Ishiguro, K.; Fujita, T.; Ueda, K. The ultrastructural characteristics of eccrine sweat glands in a Fabry disease patient with hypohidrosis. J. Dermatol. Sci. 1998, 18, 109–117. [Google Scholar] [CrossRef]
- Lim, S.N.; Huang, C.C.; Kuo, H.C.; Hsieh, Y.C.; Chu, C.C. Subtle Changes in Cutaneous Nerves and Sural Nerve Biopsy in a Patient With Fabry’s Disease. J. Clin. Neuromuscul. Dis. 2005, 7, 19–24. [Google Scholar] [CrossRef]
- Gayathri, N.; Yasha, T.; Kanjalkar, M.; Agarwal, S.; Sagar, B.C.; Santosh, V.; Shankar, S. Fabry’s disease: An ultrastructural study of nerve biopsy. Ann. Indian Acad. Neurol. 2008, 11, 182–184. [Google Scholar] [CrossRef]
- Onishi, A.; Dyck, P.J. Loss of small peripheral sensory neurons in Fabry disease. Histologic and morphometric evaluation of cutaneous nerves, spinal ganglia, and posterior columns. Arch. Neurol. 1974, 31, 120–127. [Google Scholar] [CrossRef]
- Toyooka, K.; Said, G. Nerve biopsy findings in hemizygous and heterozygous patients with Fabry’s disease. J. Neurol. 1997, 244, 464–468. [Google Scholar] [CrossRef] [PubMed]
- Brakch, N.; Dormond, O.; Bekri, S.; Golshayan, D.; Correvon, M.; Mazzolai, L.; Steinmann, B.; Barbey, F. Evidence for a role of sphingosine-1 phosphate in cardiovascular remodelling in Fabry disease. Eur. Heart J. 2010, 31, 67–76. [Google Scholar] [CrossRef] [PubMed]
- Tomé, F.M.; Fardeau, M.; Lenoir, G. Ultrastructure of muscle and sensory nerve in Fabry’s disease. Acta Neuropathol. 1977, 38, 187–194. [Google Scholar] [CrossRef] [PubMed]
- Azevedo, E.; Mendes, A.; Seixas, D.; Santos, R.; Castro, P.; Ayres-Basto, M.; Rosengarten, B.; Oliveira, J.P. Functional transcranial Doppler: Presymptomatic changes in Fabry disease. Eur. Neurol. 2012, 67, 331–337. [Google Scholar] [CrossRef] [PubMed]
- Tuttolomondo, A.; Pedone, C.; Pinto, A.; Di Raimondo, D.; Fernandez, P.; Di Sciacca, R.; Licata, G.; Gruppo Italiano di Farmacoepidemiologia dell’Anziano (GIFA) researchers. Predictors of outcome in acute ischemic cerebrovascular syndromes: The GIFA study. Int. J. Cardiol. 2008, 125, 391–396. [Google Scholar] [CrossRef] [PubMed]
- Davì, G.; Tuttolomondo, A.; Santilli, F.; Basili, S.; Ferrante, E.; Di Raimondo, D.; Pinto, A.; Licata, G. CD40 ligand and MCP-1 as predictors of cardiovascular events in diabetic patients with stroke. J. Atheroscler. Thromb. 2009, 16, 707–713. [Google Scholar] [CrossRef] [PubMed]
- Siragusa, S.; Malato, A.; Saccullo, G.; Iorio, A.; Di Ianni, M.; Caracciolo, C.; Coco, L.L.; Raso, S.; Santoro, M.; Guarneri, F.P.; et al. Residual vein thrombosis for assessing duration of anticoagulation after unprovoked deep vein thrombosis of the lower limbs: The extended DACUS study. Am. J. Hematol. 2011, 86, 914–917. [Google Scholar] [CrossRef]
- Daidone, M.; Ferrantelli, S.; Tuttolomondo, A. Machine learning applications in stroke medicine: Advancements, challenges, and future prospectives. Neural Regen Res. 2024, 19, 769–773. [Google Scholar] [CrossRef]
- Tuttolomondo, A.; Di Raimondo, D.; Pecoraro, R.; Maida, C.; Arnao, V.; Della Corte, V.; Simonetta, I.; Corpora, F.; Di Bona, D.; Maugeri, R.; et al. Early High-dosage Atorvastatin Treatment Improved Serum Immune-inflammatory Markers and Functional Outcome in Acute Ischemic Strokes Classified as Large Artery Atherosclerotic Stroke: A Randomized Trial. Medicine 2016, 95, e3186. [Google Scholar] [CrossRef]
- Della Corte, V.; Tuttolomondo, A.; Pecoraro, R.; Di Raimondo, D.; Vassallo, V.; Pinto, A. Inflammation, Endothelial Dysfunction and Arterial Stiffness as Therapeutic Targets in Cardiovascular Medicine. Curr. Pharm. Des. 2016, 22, 4658–4668. [Google Scholar] [CrossRef]
- Petta, S.; Marrone, O.; Torres, D.; Buttacavoli, M.; Cammà, C.; Di Marco, V.; Licata, A.; Lo Bue, A.; Parrinello, G.; Pinto, A.; et al. Obstructive Sleep Apnea Is Associated with Liver Damage and Atherosclerosis in Patients with Non-Alcoholic Fatty Liver Disease. PLoS ONE 2015, 10, e0142210. [Google Scholar] [CrossRef] [PubMed]
- Tuttolomondo, A.; Petta, S.; Casuccio, A.; Maida, C.; Corte, V.D.; Daidone, M.; Di Raimondo, D.; Pecoraro, R.; Fonte, R.; Cirrincione, A.; et al. Reactive hyperemia index (RHI) and cognitive performance indexes are associated with histologic markers of liver disease in subjects with non-alcoholic fatty liver disease (NAFLD): A case control study. Cardiovasc. Diabetol. 2018, 17, 28. [Google Scholar] [CrossRef] [PubMed]
- Petta, S.; Tuttolomondo, A.; Gagliardo, C.; Zafonte, R.; Brancatelli, G.; Cabibi, D.; Cammà, C.; Di Marco, V.; Galvano, L.; La Tona, G.; et al. The Presence of White Matter Lesions Is Associated With the Fibrosis Severity of Nonalcoholic Fatty Liver Disease. Medicine 2016, 95, e3446. [Google Scholar] [CrossRef] [PubMed]
- Tuttolomondo, A.; Colomba, C.; Di Bona, D.; Casuccio, A.; Di Raimondo, D.; Clemente, G.; Arnao, V.; Pecoraro, R.; Ragonese, P.; Aiello, A.; et al. HLA and killer cell immunoglobulin-like receptor (KIRs) genotyping in patients with acute viral encephalitis. Oncotarget 2018, 9, 17523–17532. [Google Scholar] [CrossRef] [PubMed]
- Tuttolomondo, A.; Casuccio, A.; Della Corte, V.; Maida, C.; Pecoraro, R.; Di Raimondo, D.; Vassallo, V.; Simonetta, I.; Arnao, V.; Pinto, A. Endothelial function and arterial stiffness indexes in subjects with acute ischemic stroke: Relationship with TOAST subtype. Atherosclerosis 2017, 256, 94–99. [Google Scholar] [CrossRef]
- Tuttolomondo, A.; Di Raimondo, D.; Pecoraro, R.; Casuccio, A.; Di Bona, D.; Aiello, A.; Accardi, G.; Arnao, V.; Clemente, G.; Corte, V.D.; et al. HLA and killer cell immunoglobulin-like receptor (KIRs) genotyping in patients with acute ischemic stroke. J. Neuroinflamm. 2019, 16, 88. [Google Scholar] [CrossRef]
- Zanoli, L.; Ozturk, K.; Cappello, M.; Inserra, G.; Geraci, G.; Tuttolomondo, A.; Torres, D.; Pinto, A.; Duminuco, A.; Riguccio, G.; et al. Inflammation and Aortic Pulse Wave Velocity: A Multicenter Longitudinal Study in Patients With Inflammatory Bowel Disease. J. Am. Heart Assoc. 2019, 8, e010942. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tuttolomondo, A.; Baglio, I.; Riolo, R.; Todaro, F.; Parrinello, G.; Miceli, S.; Simonetta, I. Molecular Pathogenesis of Central and Peripheral Nervous System Complications in Anderson–Fabry Disease. Int. J. Mol. Sci. 2024, 25, 61. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms25010061
Tuttolomondo A, Baglio I, Riolo R, Todaro F, Parrinello G, Miceli S, Simonetta I. Molecular Pathogenesis of Central and Peripheral Nervous System Complications in Anderson–Fabry Disease. International Journal of Molecular Sciences. 2024; 25(1):61. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms25010061
Chicago/Turabian StyleTuttolomondo, Antonino, Irene Baglio, Renata Riolo, Federica Todaro, Gaspare Parrinello, Salvatore Miceli, and Irene Simonetta. 2024. "Molecular Pathogenesis of Central and Peripheral Nervous System Complications in Anderson–Fabry Disease" International Journal of Molecular Sciences 25, no. 1: 61. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms25010061