Room Temperature Ni(II) Catalyzed Hydrophosphination and Cyclotrimerization of Alkynes


Department of Chemistry, University of Bath, Bath BA2 7AY, UK
Inorganics 2018, 6(4), 120; https://0-doi-org.brum.beds.ac.uk/10.3390/inorganics6040120
Submission received: 21 September 2018
/
Revised: 15 October 2018
/
Accepted: 31 October 2018
/
Published: 2 November 2018
(This article belongs to the Special Issue First-Row Transition Metal Complexes)
Abstract
:The catalytic activity of nickel complexes in hydrophosphination involving secondary phosphines is not a commonly studied transformation. Beyond a small number of stand-out examples, many reports in the literature focus on the use of simple nickel salts. β-Diketiminates have been proven to be incredibly effective ligands for catalysis using a range of metal centers. This synthetic study investigates the catalytic ability of a Ni(II) β-diketiminate complex in the hydrophosphination of alkenes and alkynes, with a serendipitous discovery of its ability to effect alkyne cyclotrimerization and phosphine dehydrocoupling.
1. Introduction
Transition metal catalyzed hydrophosphination is an economical route for atoms to access a range of functionalized phosphines from alkenes and alkynes [1,2,3]. We have already demonstrated the efficacy of β-diketiminate iron catalysts in hydrophosphination [4,5], and questioned whether, given the ease with which these complexes can be synthesized, the reactivity could be extended to other first row transition metal β-diketiminate complexes. Nickel is an ideal metal to choose for this purpose: Holland has described an easy, scalable synthetic route to the N(SiMe3)2-ligated complex (1) [6]. Accessing a nickel complex that is a low coordinate with a σ-bound co-ligand should give high levels of reactivity via a σ-bond metathesis-type pathway. This hydrophosphination catalyzed by nickel is surprisingly underexplored [7]. Arguably, the most elegant example came from Togni’s group, where enantioselective hydrophosphination of methacrylonitrile was achieved using [Ni(κ3-Pigiphos)(THF)](ClO4)2 [8]. The reaction provided the highest catalytic turnover using HPtBu2 or HP(1-Ad)2 (where 1-Ad is 1-adamantyl). The authors subsequently explored the effects of counterion, the full substrate scope and performed a detailed mechanistic study [9]. The earliest examples of nickel-catalyzed hydrophosphination came independently from Korolev using a Ni(II) amine complex [10] and Pringle using [Ni{P(CH2OH)3}4] [11] to catalyze the reaction of formaldehyde and PH3. Beletskaya has shown that 5 mol % [NiBr2(PPh2)2] can be used to effect the hydrophosphination of a range of alkenyl–alkyl ethers using HPPh2 in 2 h at 80 °C [12]. Beletskaya has also shown that the simplest nickel salts (i.e., [Ni{P(OEt)3}4], NiBr2 and Ni(acac)2) can be used to hydrophosphinate styrenes [13] and terminal alkynes [14,15]. Taking phenyl acetylene as an example, the authors showed that NiBr2 gives an 86:14 ratio of the Markovnikov:anti-Markovnikov product (the latter forming the E isomer selectively), whereas a change to Ni(acac)2 generates 27:73 Markovnikov:anti-Markovnikov (the latter in a 67:33 mixture of E:Z). Interestingly, when HP(O)(OEt)2 was added to the Ni(acac)2-catalyzed reaction, a further shift in regioselectivity was observed and the almost exclusive formation of the Markovnikov product was obtained. This was the first example of Markovnikov-selective hydrophosphination. Gong and Song have undertaken the hydrophosphination of enones with diarylphosphines (isolating the phosphine oxide product) with a modest yield using a Ni(II) pincer complex [16]. Beyond P(III) phosphines, Montchamp has formed vinyl-H-phosphinates from alkynes and alkyl phosphinates using only 2–3 mol % NiCl2 [17]. Ananikov and Beletskaya have undertaken the Markovnikov-selective phosphorylation of internal and terminal alkynes using a range of phosphites in the presence of catalytic Ni(acac)2 and DPPE (1,2-bis(diphenylphosphino)ethane) [18]. In the interest of preparing products that would potentially be of use in coordination chemistry, the P(III) phosphine, rather than phosphine oxide, was used in this catalysis study. Finally, it is worth noting that catalyst-free, thermal hydrophosphination has been reported [19,20].
2. Results and Discussion
2.1. Reaction Optimization
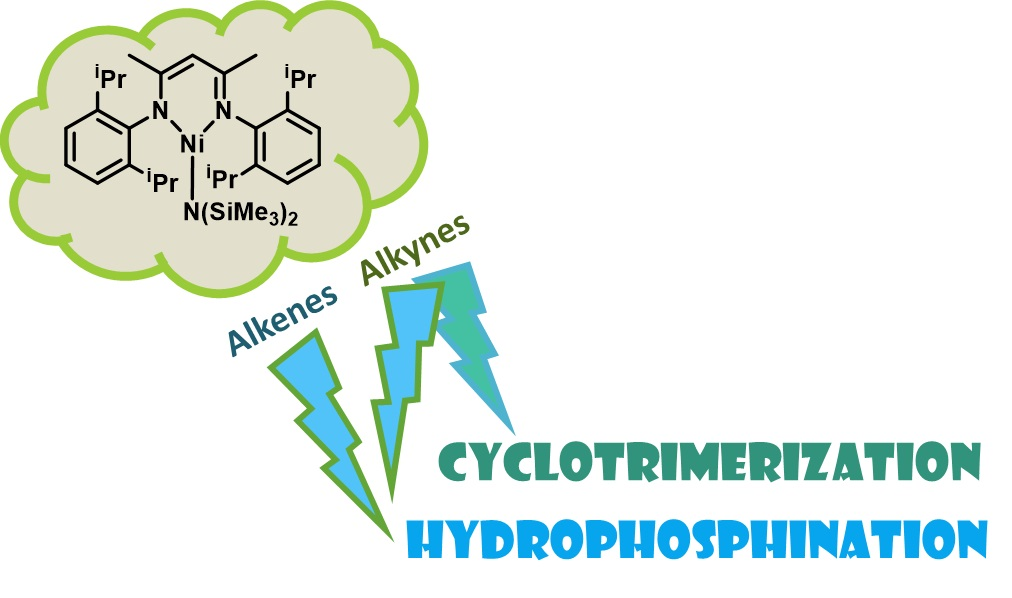
The catalytic hydrophosphination reaction was optimized using 1, phenylacetylene and HPPh2 (Scheme 1, Table 1). When benzene was used as a solvent, dehydrocoupling was observed as a competitive side reaction, therefore leading to a reduced yield of the Z anti-Markovnikov product, 3a (Entry 1). The dehydrocoupling product ((PPh2)2) was observed at a 19% spectroscopic yield and 9% E anti-Markovnikov product (2a) was also obtained. When the solvent was changed to CH2Cl2, no reaction was observed, but a change in the MeCN resulted in an increased product yield (Table 1, Entry 4). When the temperature was dropped to room temperature, a good yield of product was obtained (Entry 5), but lowering the catalyst loading was detrimental to reactivity (Entry 6). A 1:1 ratio of phenylacetylene and HPPh2 still gave a good yield of anti-Markovnikov products with an 18 h reaction time (Entry 8). Increasing the ratio of phenylacetylene to phosphine increased the yield further (Entry 9), but also allowed for good yield after only 4 h at room temperature (RT, Entry 7).
2.2. Substrate Scope
With these optimized conditions in hand (Table 1, Entry 9), the substrate scope was investigated using a range of sp and sp2 systems (Table 2).
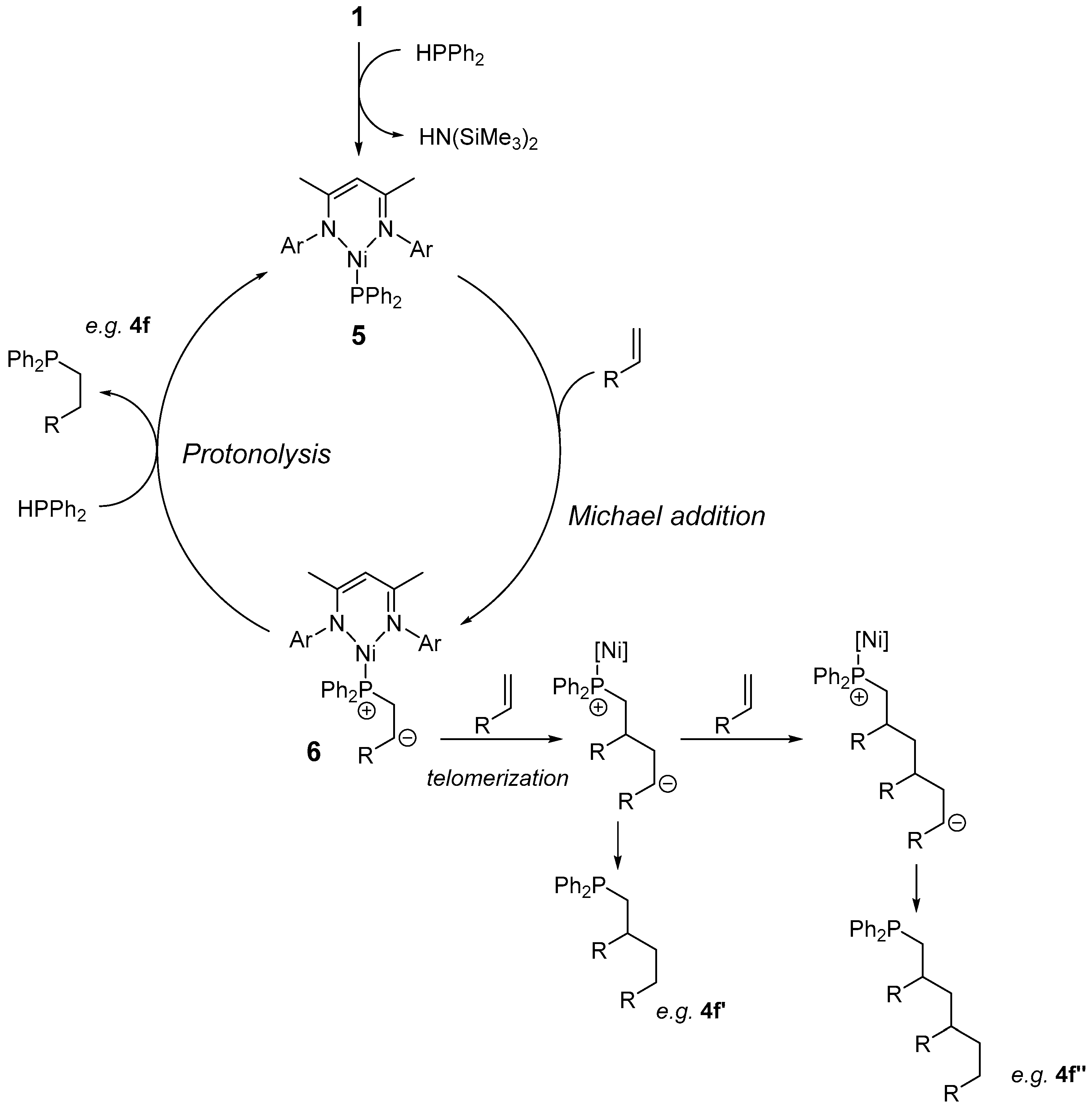
In terms of the substrate scope for this reaction, arylacetylenes were tolerated and underwent hydrophosphination readily at room temperature. It is interesting to note the change in selectivity when the electronics of the acetylene were varied (compare entries 1, 2 and 3). Very high Z selectivity was obtained with phenylacetylene, but when 4-fluorophenylacetylene was employed, the E product dominated. Unfortunately, lower yields of products were also obtained with these electronically diverse substrates. The same levels of selectivity that we obtained using the Fe congener (95:5 Z:E) [5] was not achieved with this Ni system, nor was the same solvent-switching observed (refer to Table 1, Entry 3). The reaction of phenylacetylene with phenylphosphine produced a complex mixture of products, presumably including the products from the addition of one phosphine across two triple bonds and the addition of more than one phosphine to a triple bond. When trimethylsilylacetylene was employed, complete uptake of the starting material was observed and an intractable mixture of products was obtained (Entry 5); this was in vast contrast to the selective reactivity we observed using the iron analogue of the pre-catalyst [5] (albeit with a CH2TMS co-ligand, as opposed to N(SiMe3)2). No reaction was observed when hepta-1,6-diyne and diphenylacetylene were used (Entries 6 and 7). A reaction was not observed when 1-heptyne was used. A reaction with different activated alkenes resulted in the anti-Markovnikov product (Entries 8 to 13). An intractable mixture of products was obtained when cyclohexanone was used as a substrate. The mild reaction conditions required for styrene hydrophosphination in this study were in vast contrast to the forcing conditions needed when we employed our iron β-diketiminate system (70 °C, 24 h) [5]. The fact that more than one alkene was observed for the reaction of acrylonitrile and n-butylacrylate hints at the reaction mechanism [21]. The fact that telomerization was observed, particularly with n-butylacrylate, where three acrylate moieties were observed in one of the products (which was isolable, 4f”), suggests that a Michael-type reaction was taking place [22,23]. A postulated catalytic cycle was shown (Scheme 2), which proceeds via a three-coordinate Ni(II) terminal-phosphido intermediate (5), akin to Stephan’s report of Ni(I) β-diketiminate terminal phosphine complexes [24]. The 5 then undertook a Michael attack of the alkene, forming a zwitterion (6), which can undertake telomerization to produce, for example, 4f’ and 4f”. However, the dominant process would be a protonolysis event, which liberates the hydrophosphination product (e.g., 4f) and regenerates Ni–phosphido. It is presumed that the process was completely redox neutral due to the nature of the pre-catalyst; protonolysis forming the active catalysis through the loss of HN(SiMe3)2 should be favored simply because HN(SiMe3)2 is a good leaving group. Accessing the higher oxidation state Ni via the oxidative addition of HPPh2 to 1 is unlikely and steric congestion around the Ni-center should also preclude higher coordination numbers. With respect to the hydrophosphination of alkynes, we cannot rule out catalyst activation by the alkyne to release SiMe4 and form an on-cycle nickel–alkynyl intermediate that is then attacked by HPPh2 [5]. However, this should then result in the formation of a nickel–hydride intermediate that would necessitate the loss of H2 to regenerate the nickel–alkynyl complex. There is no evidence of gas being evolved during the reaction and thus this particular catalytic cycle seems unlikely. In our previous studies of alkyne hydrophosphination we found that an iron–alkynyl complex was not a good catalyst [5].
When investigating the hydrophosphination reactivity of methylpropiolate it was interesting to note that a highly exothermic reaction took place. NMR spectroscopic analysis showed that the HPPh2 remained unreacted, but there was complete loss of the alkyne. Isolation of the new organic product revealed that cyclotrimerization had taken place, with high levels of selectivity for the 1,2,4-product (7a, Table 3, Entry 1). We did not observe cyclotrimerization with our iron β-diketiminate congeners. In terms of the nickel-catalyzed cyclotrimerization of alkynes, there are limited examples in the literature. Since the earliest reports from Reppe [25,26,27], Stuhl has described a Ni(II) pre-catalyst ((K·18-crown-6)2[(η2-PhCCPh)Ni(CN)2]) that produces a 40% yield of 1,2,4-triphenylbenzene at room temperature when phenylacetylene is added in a 300 eq. excess [28]. Eisch and co-workers have carried out a series of stoichiometric and catalytic studies that focus on the use of nickelole complexes, which are themselves postulated to be intermediates in nickel catalyzed cyclotrimerization reactions. For example, catalytic reactions of less than 1 mol % nickelole complex with diphenylacetylene in refluxing toluene produced 78% hexaphenylbenzene [29]. Eisch followed up this original study with a wide-ranging investigation into cyclotrimerization using a range of nickel species [30]. Bennett investigated Ni(0)–benzyne complexes at the stoichiometric level, but also provided catalytic cyclotrimerization results with a highly reactive 14-electron nickel species [31,32]. More recently, Lord and Groysman undertook a detailed study into cyclotrimerization catalyzed by a dinuclear iminopyridine complex. Catalysis proceeded for a range of terminal alkynes including ethyl propiolate, phenylacetylene and methyl propargyl ether. The former two reagents showed good conversion at 1 to 5 mol % catalyst loading at room temperature or 50 °C. There was also a moderate preference for the 1,2,4-regioisomer. Somewhat lower reactivity was observed in the ether substrate where cyclotetraenes were also formed [33]. Beyond discrete homogeneous Ni catalysts, Blümel has described silica supported complexes, which are efficient for cyclotrimerisation [34].
In the case of pre-catalyst 1, good reactivity and selectivity was also observed with ethylpropiolate (Table 3, Entry 2), whereas heating to 80 °C was necessary to convert phenylacetylene. It should be noted that when other phosphines such as HPCy2 and H2PCy were reacted with phenylacetylene, only the cyclotrimerized product was observed.
Mechanistically, a nickelole intermediate cannot be ruled out. However, if the β-diketiminate remains as an anionic ligand, this would mean the nickel center is in the +3 oxidation state. Although not impossible [35,36], the non-innocent nature of the ligand [37] or the possibility of hemi-lability or the role of non-ligated Ni(0) cannot be ignored, and this aspect of reactivity, along the wider area of first row transition metal catalyzed cyclotrimerization, is undergoing further investigation in our laboratory.
The reaction of HPPh2 with catalytic 1 at 80 °C, without any olefin added, produced the dehydrocoupled product 8 in good yield. When dehydrocoupling was attempted at room temperature, only a trace amount of product was obtained, but a new peak at 8.8 ppm was observed in the 31P NMR. This is postulated to be the three-coordinate nickel(II)–phosphido compound, because the chemical shift observed was consistent with other nickel(II) phosphides reported in the literature [38]. Unfortunately, this phosphido intermediate could not be crystallized or fully characterized.
3. Materials and Methods
3.1. General Method for Hydrophosphination
1 (1.8 mg, 5 mol %) was weighed into a J-Young NMR tube. CD3CN (300 μL) was added to this, followed by alkene/alkyne (1 mmol) and diphenylphosphine (87 μL, 0.5 mmol, 1 eq.). The reaction was monitored by 1H and 31P NMR (Bruker (Avance), Coventry, UK). The same procedure was followed for cyclotrimerization and dehydrocoupling, but in the absence of phosphine and alkyne, respectively. The same procedure was followed for cyclotrimerization and dehydrocoupling, but without the addition of phosphine or alkyne to the respective reactions.
For the spectroscopic yield, the reaction solutions were exposed to air and 45 μL of 1,2-dichloroethane was added as an integration standard. All reaction products were compared to isolated compounds in the literature. Products were then isolated by silica gel column chromatography (see individual products for column conditions). It is important to note that the products oxidize slowly over time.
3.2. Analysis Data for Isolated Products
2a/3a (Isolated using 5% EtOAc/pentane. White solid, 116 mg, 81%): 1H NMR (300 MHz, 298 K, CDCl3) δ 7.37–6.98 (m, 2a + 3a), 6.80 (d, J = 10.7 Hz, C=CH, 2a), 2.39–2.33 (dd, J = 12.6, 2.8 Hz, C=CH, 3a); 31P{1H} NMR (121.5 MHz, 298 K, CDCl3) δ −10.6 (2a), −23.9 (3a). Data comparable to that reported elsewhere [39,40].
4a (Isolated using 5% EtOAc/pentane, Rf = 0.30. White solid, 133 mg, 92%): 1H NMR (300 MHz, 298 K, CDCl3) δ 7.47–7.15 (m, 14H), 2.73–2.67 (m, 2H), 2.39–2.33 (m, 2H); 13C{1H} NMR (75 MHz, 298 K, CDCl3) δ 142.7 (d, J = 12.7 Hz), 138.6 (d, J = 19.2 Hz), 132.8 (d, J = 13.2 Hz), 128.9, 128.6 (d, J = 6.2 Hz), 128.2, 128.0, 126.1, 32.3 (d, J = 3.3 Hz), 30.2 (d, J = 7.7 Hz); 31P{1H} NMR (121.5 MHz, 298 K, CDCl3) δ −15.0. Data comparable to that reported elsewhere [41].
4b (Isolated using 5% EtOAc/pentane, Rf = 0.24. white solid, 124 mg, 68%): 1H NMR (300 MHz, 298 K, CDCl3) δ 7.70–7.33 (m, 19H), 2.90–2.85 (m, 2H), 2.55–2.50 (m, 2H); 13C{1H} NMR (75 MHz, 298 K, CDCl3) δ 141.7 (d, J = 13.0 Hz), 141.1, 139.0, 138.5 (d, J = 12.4 Hz), 132.7 (d, J = 18.6 Hz), 128.7, 128.62, 128.56, 128.50 (d, J = 6.5 Hz), 127.2, 127.03, 126.98, 31.9 (d, J = 17.7 Hz), 30.2 (d, J = 12.4 Hz); 31P{1H} NMR (121.5 MHz, 298 K, CDCl3) δ −14.9. Data comparable to that reported elsewhere [41].
4c (Isolated using 5% EtOAc/pentane, Rf = 0.26. White solid, 85 mg, 49%): 1H NMR (300 MHz, 298 K, CDCl3) δ 7.54–7.42 (m, 4H), 7.41–7.39 (m, 6H), 6.41 (d, 2H, J = 2.3 Hz), 6.37 (t, 1H, J = 2.3 Hz), 3.81 (s, 6H) 2.78–2.70 (m, 2H) 2.47–2.41 (m, 2H); 13C{1H} NMR (75 MHz, 298 K, CDCl3) δ 160.9, 145.1 (d, J = 13.4 Hz), 138.5, 133.0 (d, J = 18.6 Hz), 128.8, 128.6 (d, J = 6.5 Hz), 106.3, 98.0, 55.3, 32.6 (d, J = 18.0 Hz), 30.0 (d, J = 12.4 Hz); 31P{1H} NMR (121.5 MHz, 298 K, CDCl3) δ −14.9. Data comparable to that reported elsewhere [41].
4e (Isolated using 20% EtOAc/pentane, Rf = 0.74. Colourless oil, 62 mg, 52%): 1H NMR (300 MHz, 298 K, CDCl3) δ 7.46–7.38 (m, 10H), 2.42–2.37 (m, 4H); 13C{1H} NMR (75 MHz, 298 K, CDCl3) δ 136.3 (d, J = 12.4 Hz), 132.6 (d, J = 19.2 Hz), 129.3, 128.8 (d, J = 6.8 Hz), 119.4 (d, J = 14.6 Hz), 24.1 (d, J = 15.5 Hz), 14.1 (d, J = 23.9 Hz); 31P{1H} NMR (121.5 MHz, 298 K, CDCl3) δ −15.2. HRMS (EI) 239.0864 calc.; 239.0861 found. Data comparable to that reported elsewhere [23].
4e’ (Isolated using 20% EtOAc/pentane, Rf = 0.48. Colourless oil, 29 mg, 20%): 1H NMR (300 MHz, 298 K, CDCl3) δ 7.48–7.38 (m, 10H), 2.76–2.64 (m, 1H), 2.62–2.45 (m, 3H), 2.36–2.29 (m, 1H), 2.25–2.13 (m, 1H), 2.13–1.98 (m, 1H); 13C{1H} NMR (75 MHz, 298 K, CDCl3) δ 136.2 (d, J = 7.1 Hz), 136.1 (d, J = 7.1 Hz), 132.8 (d, J = 2.2 Hz), 132.6 (d, J = 2.2 Hz), 129.5, 128.9 (d, J = 7.1 Hz), 119.9 (d, J = 6.5 Hz), 117.7, 31.3 (d, J = 17.4 Hz), 29.0 (d, J = 9.9 Hz), 28.4 (d, J = 21.1 Hz), 15.2; 31P{1H} NMR (121.5 MHz, 298 K, CDCl3) δ −20.3. HRMS (EI) 292.1129 calc.; 292.1131 found.
4f (Isolated using 5% EtOAc/pentane, Rf = 0.46. White solid, 67 mg, 43%): 1H NMR (300 MHz, 298 K, CDCl3) δ 7.49–7.46 (m, 4H), 7.37–7.35 (m, 6H), 4.08 (t, 2H, J = 6.7 Hz), 2.44–2.39 (m, 4H), 1.61 (app. tt, 2H, J = 15.5, 6.7 Hz), 1.37 (app. tq, 2H, J = 15.5, 7.4 Hz), 0.95 (t, 3H, J = 7.4 Hz); 13C{1H} NMR (75 MHz, 298 K, CDCl3) δ 173.3 (d, J = 15.2 Hz), 137.8 (d, J = 18.6 Hz), 132.8 (d, J = 18.6 Hz), 128.9, 128.6 (d, J = 6.8 Hz), 64.6, 30.9, 30.7, 23.0 (d, J = 11.5 Hz), 19.2, 13.8; 31P{1H} NMR (121.5 MHz, 298 K, CDCl3) δ −14.9. Data comparable to that reported elsewhere [41].
4f’ (Isolated using 5% EtOAc/pentane, Rf = 0.26. White solid, 38 mg, 17%): 1H NMR (500 MHz, 298 K, CDCl3) δ 7.45–7.41 (m, 4H), 7.34–7.33 (m, 6H), 4.08–3.98 (m, 4H), 2.49–2.44 (m, 2H), 2.37–2.24 (m, 2H), 2.21–2.16 (m, 1H), 2.06–2.01 (m, 2H), 1.58 (tt, 4H, J = 14.5, 7.3 Hz), 1.36 (dq, 4H, J = 15.1, 7.4 Hz), 0.94 (m, 6H); 13C{1H} NMR (63 MHz, 298 K, CDCl3) δ 174.8 (d, J = 4.6 Hz), 172.7, 138.1 (d, J = 13.3 Hz), 137.9 (d, J = 12.9 Hz), 132.9 (d, J = 8.7 Hz), 132.6 (d, J = 8.3 Hz), 128.7 (d, J = 6.0 Hz), 128.5 (d, J = 3.2 Hz), 128.4 (d, J = 3.2 Hz), 64.5, 64.3, 42.2 (d, J = 17.0 Hz), 31.8, 31.2 (d, J = 14.3 Hz), 30.6 (d, J = 1.4 Hz), 28.6 (d, J = 10.6 Hz), 19.1 (d, J = 3.2 Hz), 13.7; 31P{1H} NMR (202 MHz, 298 K, CDCl3) δ −20.0. HRMS (EI) 442.2273 calc.; 442.2278 found.
4f” (Isolated using 5% EtOAc/pentane, Rf = 0.15. White solid, 10 mg, 4%): 1H NMR (500 MHz, 298 K, CDCl3) δ 7.49–7.39 (m, 4H), 7.34–7.33 (m, 6H), 4.04–3.94 (m, 6H), 2.48–2.35 (m, 3H), 2.30–2.20 (m, 3H), 2.16–2.09 (m, 1H), 1.87–1.75 (m, 3H), 1.62–1.54 (m, 6H), 1.39–1.32 (m, 6H), 0.95–0.89 (m, 9H); 31P{1H} NMR (202 MHz, 298 K, CDCl3) δ −20.5. HRMS (EI) 570.3111 calc.; 570.3118 found.
7a (Isolated using 20% EtOAc/pentane. White solid, 166 mg, 66%): 1H NMR (300 MHz, 298 K, CDCl3) δ 8.41 (d, 1H, J = 1.5 Hz), 8.19 (dd, 1H, J = 8.1, 1.5 Hz), 7.74 (d, 1H, J = 8.1 Hz), 3.95 (s, 3H), 3.92 (s, 6H). Data comparable to that reported elsewhere [42].
7c (Isolated using 20% EtOAc/pentane. White solid, 125 mg, 41%): 1H NMR (300 MHz, 298 K, CDCl3) δ 7.90–7.10 (m, 18H); 13C{1H} NMR (63 MHz, 298 K, CDCl3) δ 142.1, 141.6, 141.1, 139.3, 130.6, 130.3, 129.2, 129.1, 128.2, 127.8, 127.3, 127.1, 126.7, 125.8. Data comparable to that reported elsewhere [43].
4. Conclusions
In summary, the hydrophosphination of a range of alkenes and alkynes was described using a three-coordinate Ni(II) β-diketiminate complex. The Ni complex is a proficient catalyst but, in comparison to the previously reported Fe(II) β-diketiminate pre-catalyst, this group 10 congener does not show the same levels of reaction selectivity. It appears that Fe is a more competent catalyst, however, the effect of the co-ligand (N(SiMe3)2 for Ni and CH2TMS for Fe) is yet to be determined. The Ni catalyst is effective for cyclotrimerization and this, along with other potential catalytic reactions, warrant investigation in more detail.
Funding
This research was funded by the EPSRC.
Acknowledgments
The EPSRC UK National Mass Spectrometry Facility at Swansea University is thanked for mass spectrometry analyses (Waters (Xevo G2-S), Elstree, UK).
Conflicts of Interest
The author declares no conflict of interest.
References
- Koshti, V.; Gaikwad, S.; Chikkali, S.H. Contemporary avenues in catalytic PH bond addition reaction: A case study of hydrophosphination. Coord. Chem. Rev. 2014, 265, 52–73. [Google Scholar] [CrossRef]
- Wauters, I.; Debrouwer, W.; Stevens, C.V. Preparation of phosphines through C–P bond formation. Beilstein J. Org. Chem. 2014, 10, 1064–1096. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bange, C.A.; Waterman, R. Challenges in Catalytic Hydrophosphination. Chem. Eur. J. 2016, 22, 12598–12605. [Google Scholar] [CrossRef] [PubMed]
- Espinal-Viguri, M.; King, A.K.; Lowe, J.P.; Mahon, M.F.; Webster, R.L. Hydrophosphination of Unactivated Alkenes and Alkynes Using Iron(II): Catalysis and Mechanistic Insight. ACS Catal. 2016, 6, 7892–7897. [Google Scholar] [CrossRef] [Green Version]
- King, A.K.; Gallagher, K.J.; Mahon, M.F.; Webster, R.L. Markovnikov versus anti-Markovnikov Hydrophosphination: Divergent Reactivity Using an Iron(II) β-Diketiminate Pre-Catalyst. Chem. Eur. J. 2017, 23, 9039–9043. [Google Scholar] [CrossRef] [PubMed]
- Eckert, N.A.; Bones, E.M.; Lachicotte, R.J.; Holland, P.L. Nickel Complexes of a Bulky β-Diketiminate Ligand. Inorg. Chem. 2003, 42, 1720–1725. [Google Scholar] [CrossRef] [PubMed]
- Bezzenine-Lafollée, S.; Gil, R.; Prim, D.; Hannedouche, J. First-Row Late Transition Metals for Catalytic Alkene Hydrofunctionalisation: Recent Advances in C–N, C–O and C–P Bond Formation. Molecules 2017, 22, 1901. [Google Scholar] [CrossRef] [PubMed]
- Sadow, A.D.; Haller, I.; Fadini, L.; Togni, A. Nickel(II)-Catalyzed Highly Enantioselective Hydrophosphination of Methacrylonitrile. J. Am. Chem. Soc. 2004, 126, 14704–14705. [Google Scholar] [CrossRef] [PubMed]
- Sadow, A.D.; Togni, A. Enantioselective Addition of Secondary Phosphines to Methacrylonitrile: Catalysis and Mechanism. J. Am. Chem. Soc. 2005, 127, 17012–17024. [Google Scholar] [CrossRef] [PubMed]
- Dorfman, Y.A.; Levina, L.V.; Grekov, L.I.; Korolev, A.V. Hydroxymethylation of phosphine in the presence of Ni(II) amines. Kinet. Catal. 1989, 30, 578–583. [Google Scholar]
- Hoye, P.A.T.; Pringle, P.G.; Smith, M.B.; Worboys, K. Hydrophosphination of formaldehyde catalysed by tris-(hydroxymethyl)phosphine complexes of platinum, palladium or nickel. J. Chem. Soc. Dalton Trans. 1993, 269–274. [Google Scholar] [CrossRef]
- Kazankova, M.A.; Shulyupin, M.O.; Beletskaya, I.P. Catalytic Hydrophosphination of Alkenylalkyl Ethers. Synlett 2003, 2003, 2155–2158. [Google Scholar] [CrossRef]
- Kazankova, M.A.; Shulyupin, M.O.; Borisenko, A.A.; Beletskaya, I.P. Synthesis of Alkyl(diphenyl)phosphines by Hydrophosphination of Vinylarenes Catalyzed by Transition Metal Complexes. Russ. J. Org. Chem. 2002, 38, 1479–1484. [Google Scholar] [CrossRef]
- Kazankova, M.A.; Efimova, I.V.; Kochetkov, A.N.; Afanasev, V.V.; Beletskaya, I.P.; Dixneuf, P.H. New Approach to Vinylphosphines Based on Pd- and Ni-Catalyzed Diphenylphosphine Addition to Alkynes. Synlett 2001, 2001, 497–500. [Google Scholar] [CrossRef]
- Kazankova, M.A.; Efimova, I.V.; Kochetkov, A.N.; Afanas’ev, V.V.; Beletskaya, I.P. Synthesis of Vinylphosphines by Hydrophosphination of Alkynes in the Presence of Transition Metal Complexes. Russ. J. Org. Chem. 2002, 38, 1465–1474. [Google Scholar] [CrossRef]
- Yang, M.-J.; Liu, Y.-J.; Gong, J.-F.; Song, M.-P. Unsymmetrical Chiral PCN Pincer Palladium(II) and Nickel(II) Complexes with Aryl-Based Aminophosphine-Imidazoline Ligands: Synthesis via Aryl C–H Activation and Asymmetric Addition of Diarylphosphines to Enones. Organometallics 2011, 30, 3793–3803. [Google Scholar] [CrossRef]
- Ribière, P.; Bravo-Altamirano, K.; Antczak, M.I.; Hawkins, J.D.; Montchamp, J.-L. NiCl2-Catalyzed Hydrophosphinylation. J. Org. Chem. 2005, 70, 4064–4072. [Google Scholar] [CrossRef] [PubMed]
- Ananikov, V.P.; Khemchyan, L.L.; Beletskaya, I.P.; Starikova, Z.A. Acid-Free Nickel Catalyst for Stereo- and Regioselective Hydrophosphorylation of Alkynes: Synthetic Procedure and Combined Experimental and Theoretical Mechanistic Study. Adv. Synth. Catal. 2010, 352, 2979–2992. [Google Scholar] [CrossRef]
- Alonso, F.; Moglie, Y.; Radivoy, G.; Yus, M. Solvent- and catalyst-free regioselective hydrophosphanation of alkenes. Green Chem. 2012, 14, 2699–2702. [Google Scholar] [CrossRef]
- Moglie, Y.; Gonzalez-Soria, M.J.; Martin-Garcia, I.; Radivoy, G.; Alonso, F. Catalyst- and solvent-free hydrophosphination and multicomponent hydrothiophosphination of alkenes and alkynes. Green Chem. 2016, 18, 4896–4907. [Google Scholar] [CrossRef] [Green Version]
- Rosenberg, L. Mechanisms of Metal-Catalyzed Hydrophosphination of Alkenes and Alkynes. ACS Catal. 2013, 2845–2855. [Google Scholar] [CrossRef]
- Scriban, C.; Kovacik, I.; Glueck, D.S. A protic additive suppresses formation of byproducts in platinum-catalyzed hydrophosphination of activated olefins. Evidence for P–C and C–C bond formation by Michael addition. Organometallics 2005, 24, 4871–4874. [Google Scholar] [CrossRef]
- Scriban, C.; Glueck, D.S.; Zakharov, L.N.; Kassel, W.S.; DiPasquale, A.G.; Golen, J.A.; Rheingold, A.L. P–C and C–C bond formation by Michael addition in platinum-catalyzed hydrophosphination and in the stoichiometric reactions of platinum phosphido complexes with activated alkenes. Organometallics 2006, 25, 5757–5767. [Google Scholar] [CrossRef]
- Bai, G.; Wei, P.; Das, A.K.; Stephan, D.W. P–H and P–P bond activation by Ni(I) and Fe(I) β-diketiminato-complexes. Dalton Trans. 2006, 1141–1146. [Google Scholar] [CrossRef] [PubMed]
- Reppe, W.; Schlichting, O.; Klager, K.; Toepel, T. Cyclisierende Polymerisation von Acetylen 1. Uber Cyclooctatetraen. Ann. Chem. Justus Liebig 1948, 560, 1–92. [Google Scholar] [CrossRef]
- Reppe, W.; Schlicting, O.; Meister, H. Cyclisierende Polymerisation von Acetylen 2. Uber Die Kohlenwasserstoffe C10H10, C12H12 und Azulen. Ann. Chem. Justus Liebig 1948, 560, 93–104. [Google Scholar] [CrossRef]
- Reppe, W.; Schweckendiek, W.J. Cyclisierende Polymerisation von Acetylen 3. Benzol, Benzolderivate und Hydroaromatische Verbindungen. Ann. Chem. Justus Liebig 1948, 560, 104–116. [Google Scholar]
- Del Rosario, R.; Stuhl, L.S. Organonickel cyanide chemistry. Reactions of [(PhC.tplbond.CPh)Ni(CN)2]2−. An improved synthesis of [Ni(CN)2(CO)2]2−. Organometallics 1986, 5, 1260–1262. [Google Scholar] [CrossRef]
- Eisch, J.J.; Galle, J.E.; Aradi, A.A.; Bolesa̵wski, M.P. Organic chemistry of subvalent transition metal complexes: XI. Oxidative additions of nickel(0) complexes to carbon–carbon bonds in alkynes: Nickelirenes and nickeloles as catalytic carriers in the oligomerization of alkynes. J. Organomet. Chem. 1986, 312, 399–416. [Google Scholar] [CrossRef]
- Eisch, J.J.; Ma, X.; Han, K.I.; Gitua, J.N.; Krüger, C. Mechanistic Comparison of the Nickel(0)-Catalyzed Homo-Oligomerization and Co-Oligomerization of Alkynes and Nitriles. Eur. J. Inorg. Chem. 2001, 2001, 77–88. [Google Scholar] [CrossRef]
- Bennett, M.A.; Hockless, D.C.R.; Wenger, E. Generation of (2,3-η)-Naphthalyne–Nickel(0) Complexes and Their Reactions with Unsaturated Molecules. Organometallics 1995, 14, 2091–2101. [Google Scholar] [CrossRef]
- Bennett, M.A.; Wenger, E. Insertion Reactions of Benzyne–Nickel(0) Complexes with Acetylenes. Organometallics 1995, 14, 1267–1277. [Google Scholar] [CrossRef]
- Hollingsworth, R.L.; Bheemaraju, A.; Lenca, N.; Lord, R.L.; Groysman, S. Divergent reactivity of a new dinuclear xanthene-bridged bis(iminopyridine) di-nickel complex with alkynes. Dalton Trans. 2017, 46, 5605–5616. [Google Scholar] [CrossRef] [PubMed]
- Reinhard, S.; Šoba, P.; Rominger, F.; Blümel, J. New Silica-Immobilized Nickel Catalysts for Cyclotrimerizations of Acetylenes. Adv. Synth. Catal. 2003, 345, 589–602. [Google Scholar] [CrossRef]
- Haines, R.I.; McAuley, A. Synthesis and reactions of nickel(III) complexes. Coord. Chem. Rev. 1981, 39, 77–119. [Google Scholar] [CrossRef]
- Bhattacharya, S.; Saha, B.; Dutta, A.; Banerjee, P. Electron transfer reactions of nickel(III) and nickel(IV) complexes. Coord. Chem. Rev. 1998, 170, 47–74. [Google Scholar] [CrossRef]
- Camp, C.; Arnold, J. On the non-innocence of “Nacnacs”: Ligand-based reactivity in β-diketiminate supported coordination compounds. Dalton Trans. 2016, 45, 14462–14498. [Google Scholar] [CrossRef] [PubMed]
- Sabater, S.; Page, M.J.; Mahon, M.F.; Whittlesey, M.K. Stoichiometric and Catalytic Reactivity of Ni(6-Mes)(PPh3)2. Organometallics 2017, 36, 1776–1783. [Google Scholar] [CrossRef]
- Duncan, M.; Gallagher, M.J. The 1H, 13C and 31P NMR spectra of EZ pairs of some phosphorus substituted alkenes. Org. Magn. Reson. 1981, 15, 37–42. [Google Scholar] [CrossRef]
- Hayashi, M.; Matsuura, Y.; Watanabe, Y. Regio- and Stereoselective Synthesis of Alkenylphosphines: A Rhodium-Catalyzed Hydrophosphination of Alkynes Using a Silylphosphine. J. Org. Chem. 2006, 71, 9248–9251. [Google Scholar] [CrossRef] [PubMed]
- Gallagher, K.J.; Webster, R.L. Room temperature hydrophosphination using a simple iron salen pre-catalyst. Chem. Commun. 2014, 50, 12109–12111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hilt, G.; Vogler, T.; Hess, W.; Galbiati, F. A simple cobalt catalyst system for the efficient and regioselective cyclotrimerisation of alkynes. Chem. Commun. 2005, 1474–1475. [Google Scholar] [CrossRef] [PubMed]
- Yoshikai, N.; Mashima, H.; Nakamura, E. Nickel-Catalyzed Cross-Coupling Reaction of Aryl Fluorides and Chlorides with Grignard Reagents under Nickel/Magnesium Bimetallic Cooperation. J. Am. Chem. Soc. 2005, 127, 17978–17979. [Google Scholar] [CrossRef] [PubMed]
Scheme 1.
Hydrophosphination optimization process using phenylacetylene, HPPh2 and Ni(II) pre-catalyst 1.
Scheme 1.
Hydrophosphination optimization process using phenylacetylene, HPPh2 and Ni(II) pre-catalyst 1.

Scheme 2.
Postulated catalytic cycle for the hydrophosphination of activated alkenes.


{kind=link}
{kind=link}
{kind=link}
Table 1.
Optimization of hydrophosphination.
| Entry | 1 (mol %) | Solvent | Conditions | Spectroscopic Yield 2a + 3a (%) a |
|---|---|---|---|---|
| 1 | 5 | C6H6 | 80 °C | 53 |
| 2 | 5 | C6H6 | RT | 5 |
| 3 | 5 | CH2Cl2 | 80 °C | No reaction |
| 4 | 5 | CH3CN | 80 °C | 90 |
| 5 | 5 | CH3CN | RT, 18 h | 77 |
| 6 | 2 | CH3CN | RT, 18 h | 33 |
| 7 b | 5 | CH3CN | RT, 4 h | 82 |
| 8 c | 5 | CH3CN | RT, 18 h | 74 |
| 9 b,d | 5 | CH3CN | RT, 18 h | 94 (81) {1:12 E:Z} |
Reaction conditions: phenylacetylene (0.6 mmol), HPPh2 (0.5 mmol), 1, solvent (300 μL). a Determined by 1H NMR using 1,2-DCE as an internal standard. b 1 mmol phenyl acetylene. c 0.5 mmol phenylacetylene. d (Isolated yield, %).
Table 2.
Hydrophosphination substrate scope.
| Entry | Olefin | Product | Ratio E:Z (2:3) | Spectroscopic Yield, % a (Isolated Yield, %) | |
|---|---|---|---|---|---|
| 1 |  |  | 2a 3a | 1:12 | 94 (81) |
| 2 |  |  | 2b 3b | 1:1 | 45 |
| 3 |  |  | 2c 3c | 2:1 | 52 |
| 4 |  |  | 2d 3d | 6:5 | 67 |
| 5 |  |  | 2e 3e | - | Complex mixture |
| 6 b,c |  |  | 2f 3f | - | No reaction |
| 7 b |  |  | 2g 3g | - | No reaction |
| 8 |  |  | 4a | - | 100 (92) |
| 9 |  |  | 4b | - | 100 (68) |
| 10 |  |  | 4c | - | 100 (49) |
| 11 |  |  | 4d | - | 11 |
| 12 |  |  | 4e | - | 85 (52/20) |
 | 4e’ | ||||
| 13 |  |  | 4f | - | 100 (43/17/4) |
 | 4f’ | ||||
 | 4f” | ||||
| 14 |  |  | 4g | - | Complex mixture |
Reaction conditions: alkyne/alkene (1 mmol), HPPh2 (0.5 mmol), 1 (5 mol %), MeCN (300 μL), RT, 18 h. a Yield derived from the limiting reagent (0.5 mmol HPPh2) and determined by 1H NMR using 0.5 mmol 1,2-DCE as an internal standard. b 100 °C, 24 h. c 0.5 mmol acetylene.
Table 3.
Cyclotrimerization and the dehydrocoupling substrate scope.
| Entry | Olefin | Product | Ratio 1,2,4:1,3,5 | Spectroscopic Yield, % a (Isolated Yield, %) |
|---|---|---|---|---|
| 1 |  |  7a 7a | 93:7 | 100 (66) |
| 2 |  |  7b 7b | 85:15 | 100 |
| 3 b |  |  7c 7c | Not determined | 66 (41) |
| 4 b |  |  8 8 | - | 56 |
Reaction conditions: alkyne or HPPh2 (1 mmol), 1 (5 mol %), MeCN (500 μL), RT, 1 h. a Determined by 1H NMR using 1,2-DCE as an internal standard. b 80 °C, 18 h.
© 2018 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Webster, R.L. Room Temperature Ni(II) Catalyzed Hydrophosphination and Cyclotrimerization of Alkynes. Inorganics 2018, 6, 120. https://0-doi-org.brum.beds.ac.uk/10.3390/inorganics6040120
AMA Style
Webster RL. Room Temperature Ni(II) Catalyzed Hydrophosphination and Cyclotrimerization of Alkynes. Inorganics. 2018; 6(4):120. https://0-doi-org.brum.beds.ac.uk/10.3390/inorganics6040120
Chicago/Turabian StyleWebster, Ruth L. 2018. "Room Temperature Ni(II) Catalyzed Hydrophosphination and Cyclotrimerization of Alkynes" Inorganics 6, no. 4: 120. https://0-doi-org.brum.beds.ac.uk/10.3390/inorganics6040120
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.