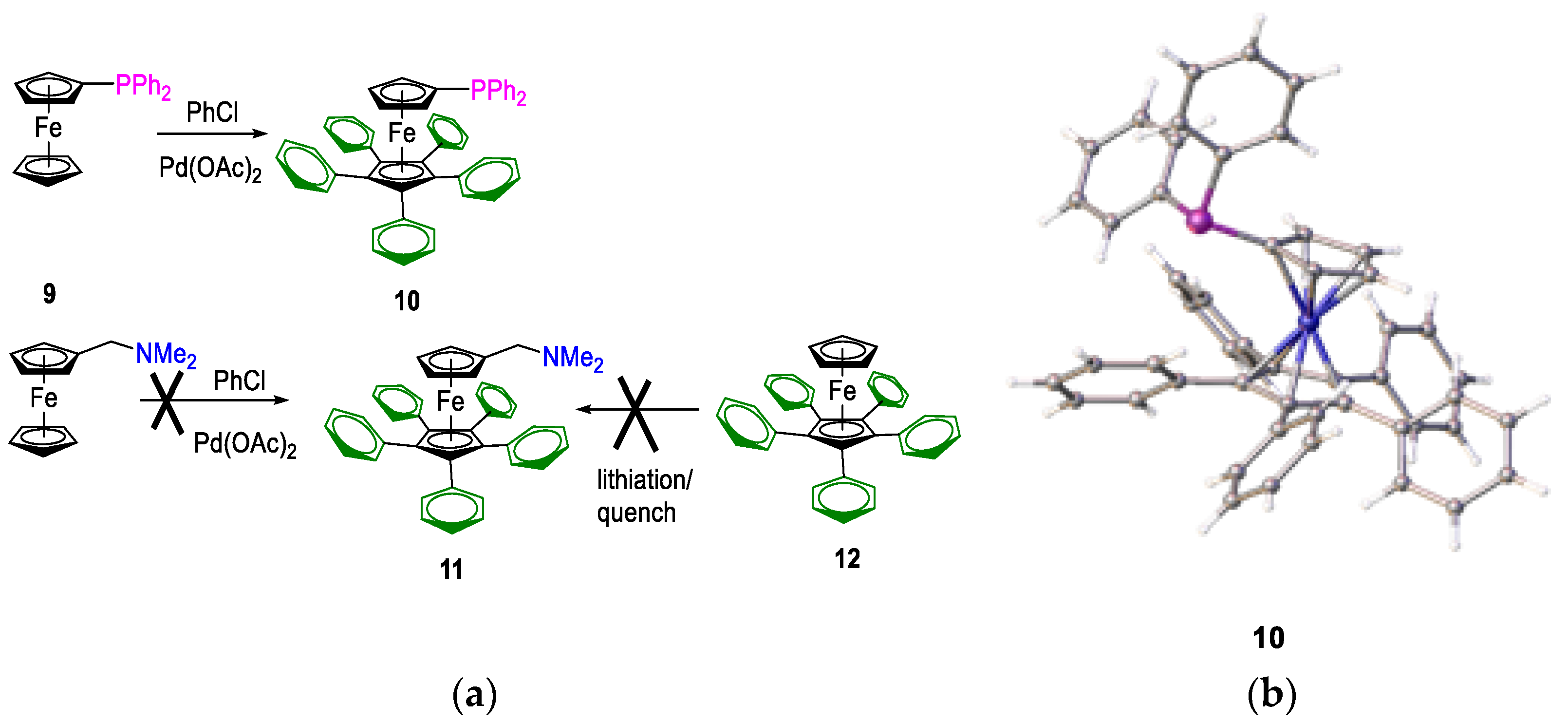
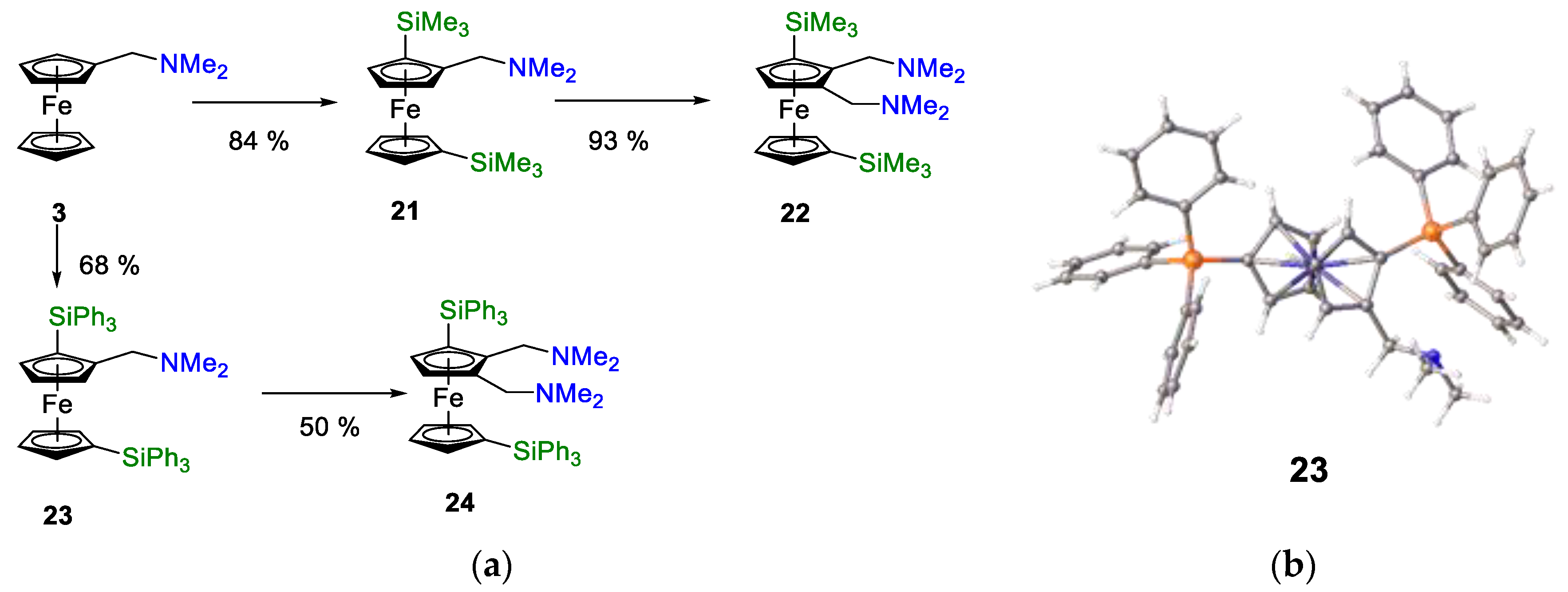
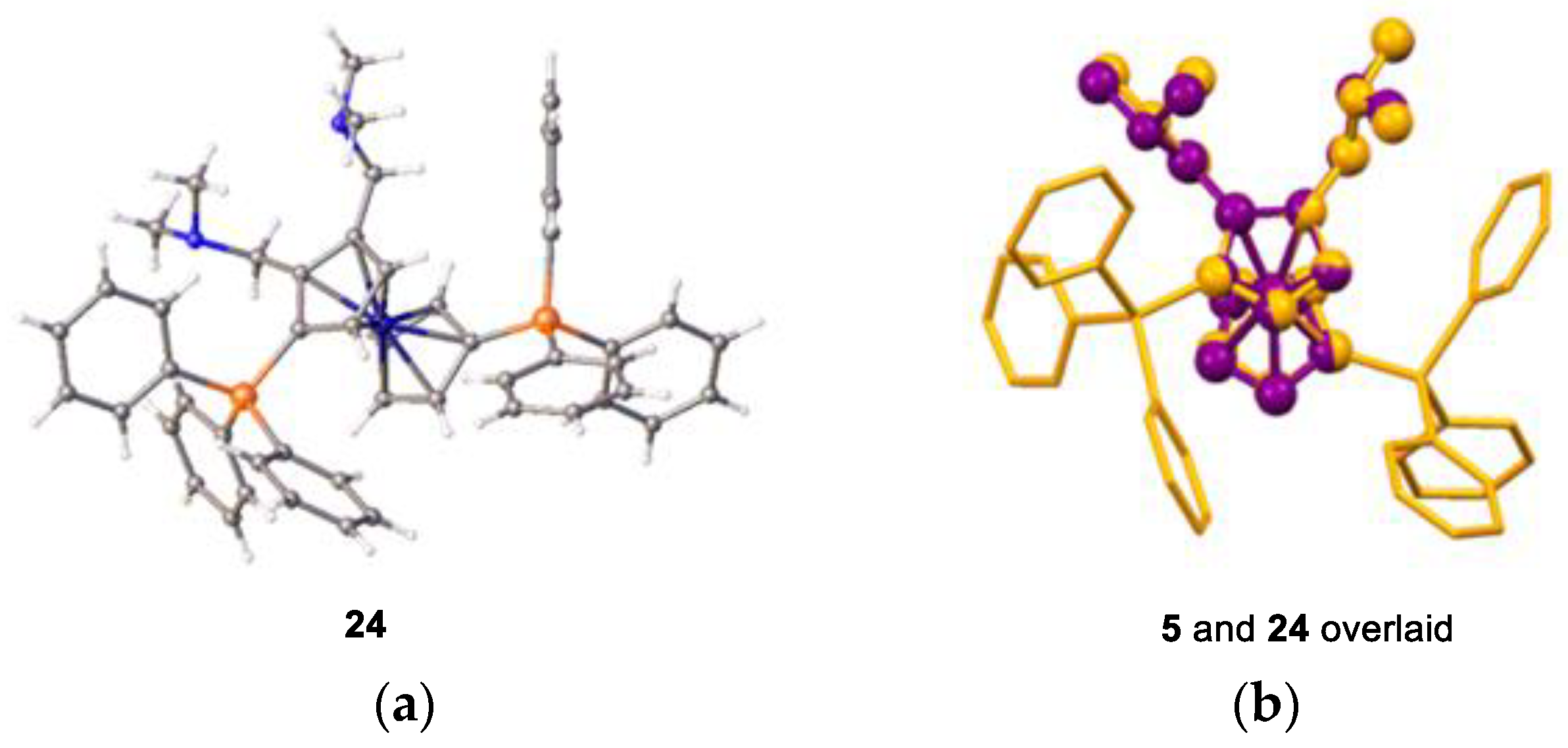
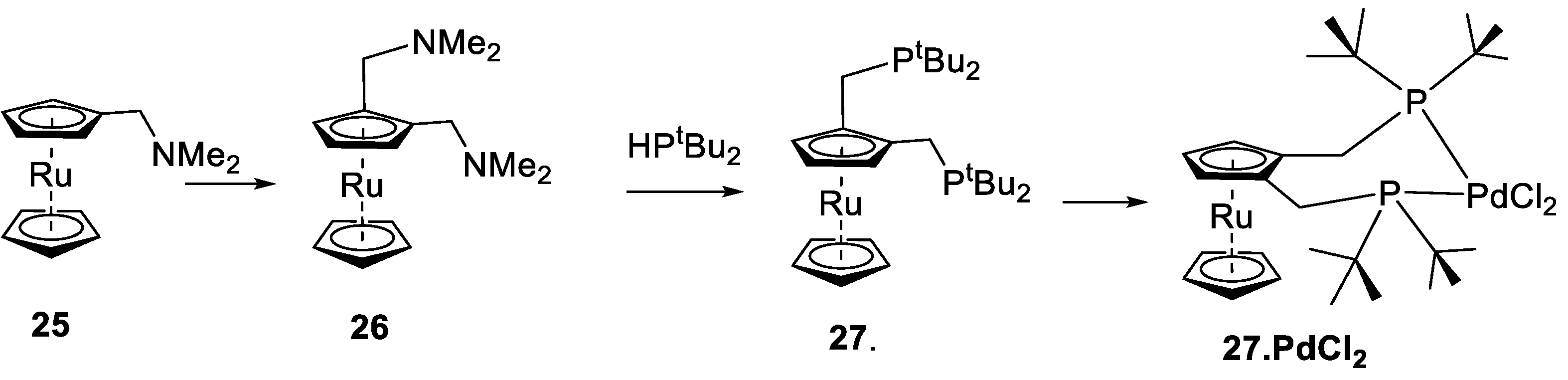
One of the key targets in our ligand design process was the control of substitution of the unsubstituted ferrocenyl cyclopentadienyl ring. Greater control would allow changes in ligand basicity, and it would grant some additional steric control. There are several synthetic methods available to substitute the unsubstituted cyclopentadienyl ring. As reported by Hartwig et al., substitution at all positions on the cp ring is possible for simple mono-phosphino ferrocenes [
58,
59]. These authors modified some related ligands, including di-tert-butylphosphinoferrocene. We adopted this approach for the one-step synthesis of the less basic 6,7,8,9,10-penta-phenyl(diphenylphosphino)ferrocenyl,
10 (
Figure 9); however, the yields were low. We also investigated the possible use of the N, N-dimethylaminomethylferrocene precursors in place of a phosphane,
9. Still, we could not isolate any product because the ortho-metallated palladium complex does not undergo self-catalysis. Attempts to obtain amine
11 were unsuccessful; on the attempted lithiation of
12 with t-butyllithium, a deep green/blue solution formed, which reverted to starting materials upon attempted quenching.
2.1. Catalyst Testing
The structure of the palladium dichloride complex of ligand
2a was originally determined in 2004 from a crystal obtained via slow diffusion of petrol/ether into chloroform (
Supplementary Materials). A more recent structure was crystallised from dichloromethane/petroleum ether (
Figure 16, top and side views).
In addition to our report, a further study was also published later by Claver et al. [
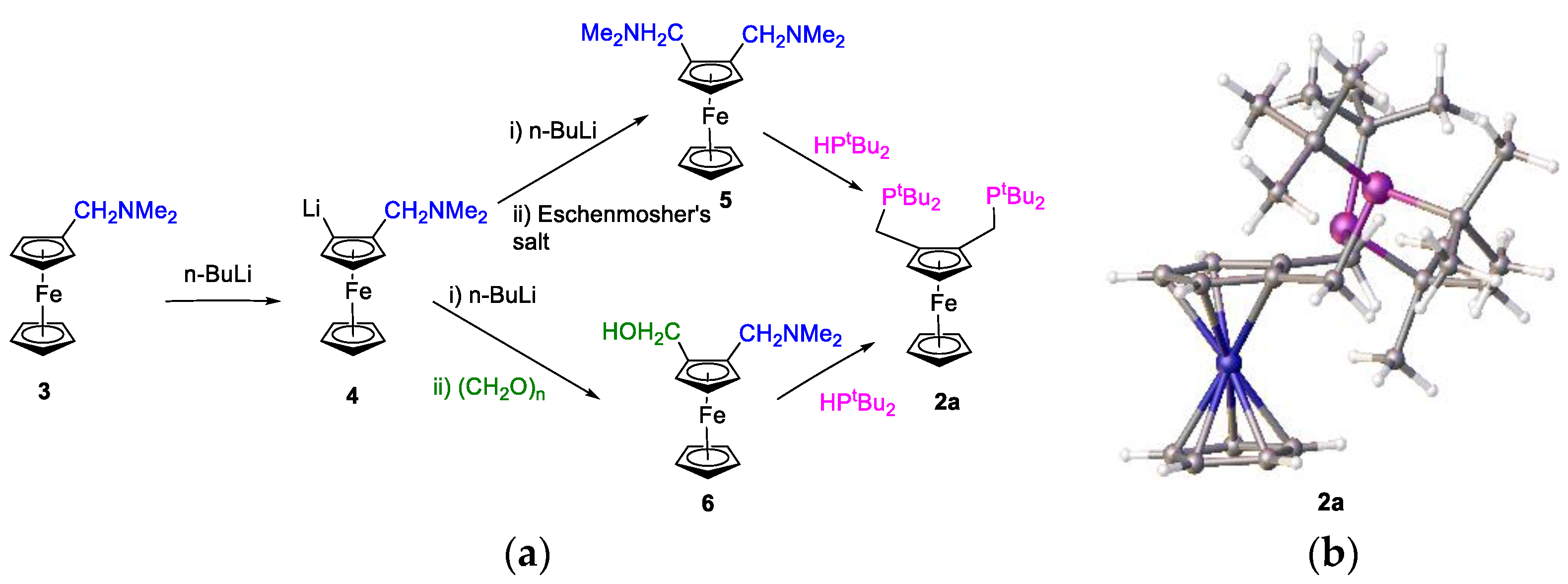
63]. These workers reported the preparation of the complex from a sample of the ligand donated without our knowledge. The catalyst testing was performed concurrently with benzene-based ligands at Lucite International Ltd., Cleveland, OH, USA. The synthetic procedure was as follows: reactions of 1,2-bis-(N,N-dimethylaminomethyl)ferrocene,
5, with a secondary phosphane were performed in acetic acid/acetic anhydride at 70 °C for 60 h. This methodology initially resulted in low (20–30%) yields of the bidentate phosphanes and, therefore, we developed an improved method. The improved method involved the reaction of a secondary phosphane with compound
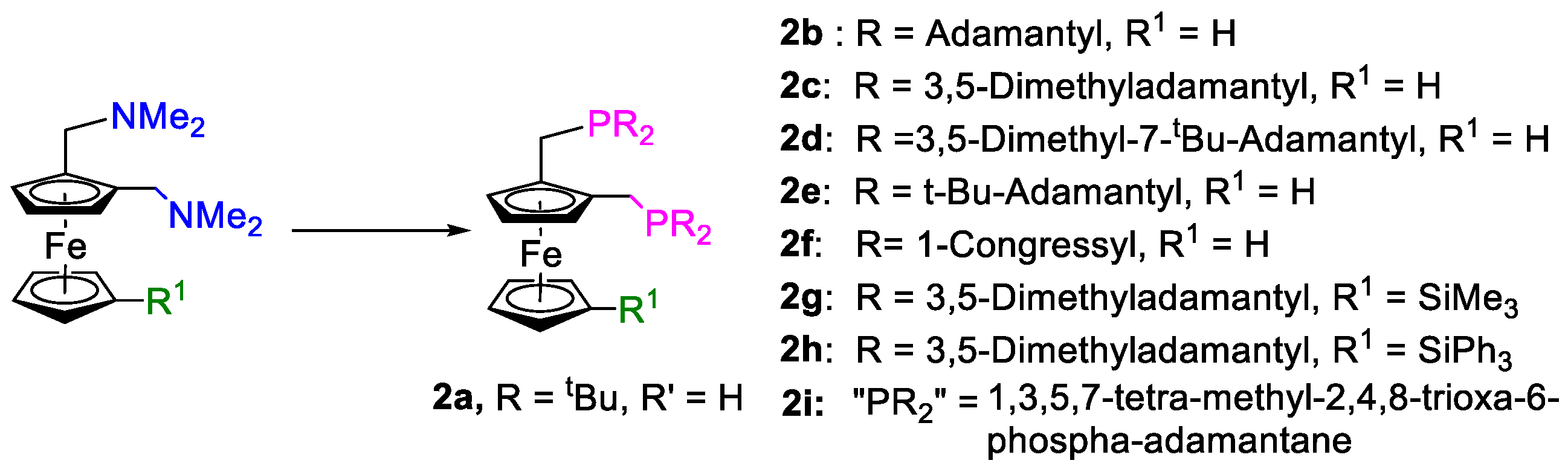
5 at ca. 130 °C in a mixture of degassed acetic acid and acetic anhydride (9:1). The solvent was removed in vacuo, and the residue was stirred in methanol for 30 min. After removal of the methanol in vacuo, the residue was washed with ethanol. This preparation furnished a free-flowing solid of the adamantylphosphine-substituted derivative, with up to 83% yield. In
Figure 17, drawings of bulky ferrocenylphosphanes, prepared in situ, are shown. These complexes were prepared and used under industrial conditions, and so complete characterisation data are unavailable. However, the synthetic methodology is available in the
Supplementary Materials. The commercially available phosphane 1,3,5,7-tetramethyl-2,4,8-trioxa-6-phospha-adamantane was also reacted with
5 to provide the corresponding bidentate ferrocenylphosphane ligand,
2i (
Figure 17).
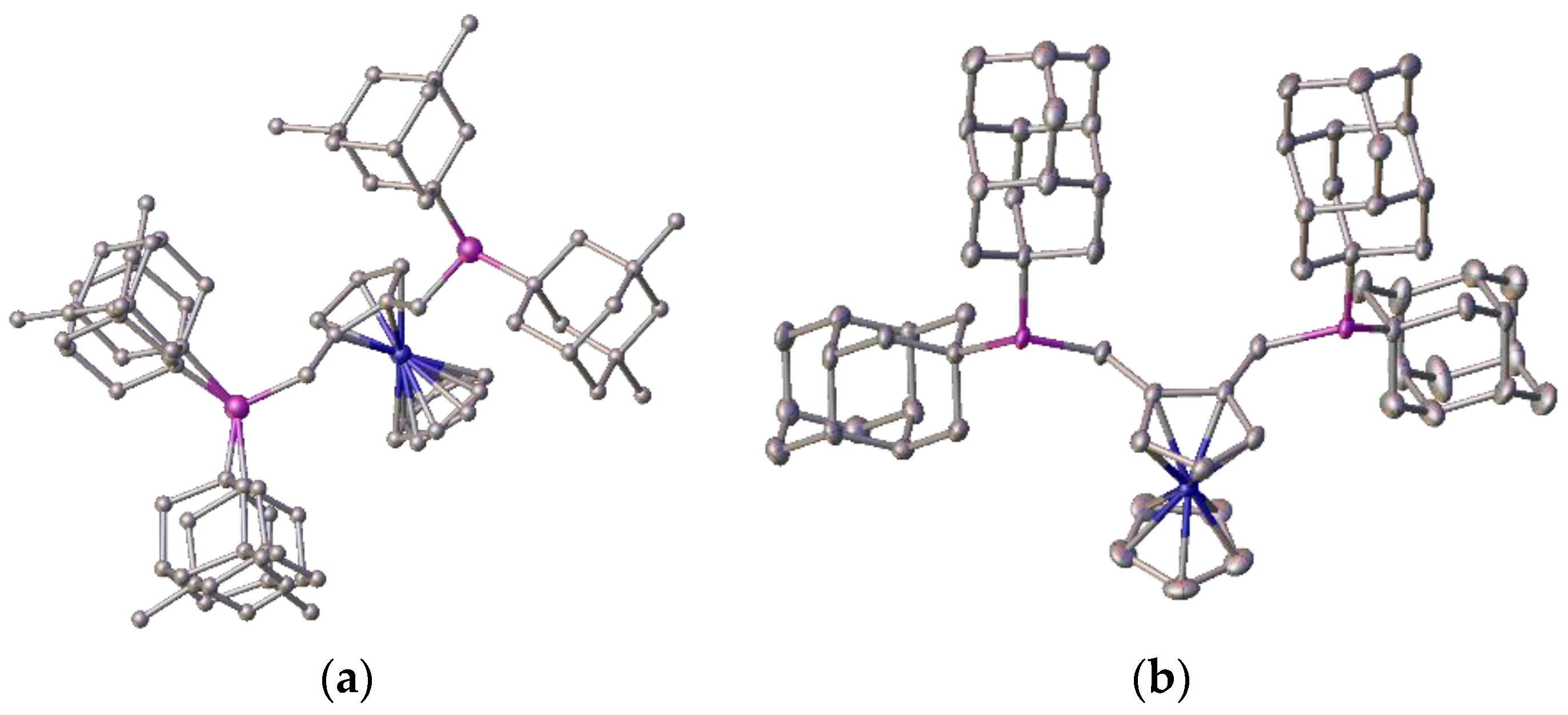
As examples, two of these bulky ligands
2c and
2f were characterised by crystallography, and are shown in
Figure 18.
2.2. Catalyst Testing: Initial Evaluation of Ferrocene-Based Ligands vs. Alpha Ligand in the Palladium-Catalysed Formation of Methyl Propanoate from Ethene, CO, and Methanol
We continued catalysis development using the standard Alpha ligand palladium complexes in tandem with this work during the catalytic trials. We altered the experimental conditions throughout the study in order to accommodate improvements. Consequently, it is difficult to make direct comparisons except within individual datasets. The catalyst precursors generally were prepared in situ to reflect industrial conditions. We carried out the coordination of the ferrocenyl phosphine ligand to palladium to form the active catalyst according to the method reported for diphenylphosphinopropane [
64,
65].
In a typical run, [Pd(OAc)
2] (1.44
× 10
−5 mol) was added to an autoclave, together with ligand 2 (7.61 × 10
−5 mol) and 70%
w/w methyl propanoate/methanol (300 mL) and methanesulphonic acid (140 × Pd equivalent). The autoclave was heated to 100 °C and maintained at this operating temperature for the duration of the experiment. Then, we repeated the procedure for each ligand,
1,
2a, and
8.
Table 1 shows the results from these initial trials over the first three hours.
In all cases, the initial reaction rates for the benzene-based ligand (
1) complex and the ferrocene-based complex (
2a) are similar (circa 30,000 (mol cat.) (mol prod.)
−1 h
−1). However, turnover numbers (TONs) for the ferrocene-based complex are significantly higher than those for the benzene-based analogues. The selectivity for the formation of methyl propanoate for the catalyst derived from ligand
2a was always >99%; thus, these ligands are vastly superior to other ferrocene-based ligands, such as dppf (see [
66] for an overview). The high turnover numbers for the ferrocenylphosphane-based catalysts resulted from the greater basicity of the ferrocenyl ligand and the difference in its bite angle. The fall in reaction rate, which occurred after three hours, was attributed to catalyst decomposition, possibly because of de-ligation of one of the phosphines from the metal centre. We used the palladium complex of ligand
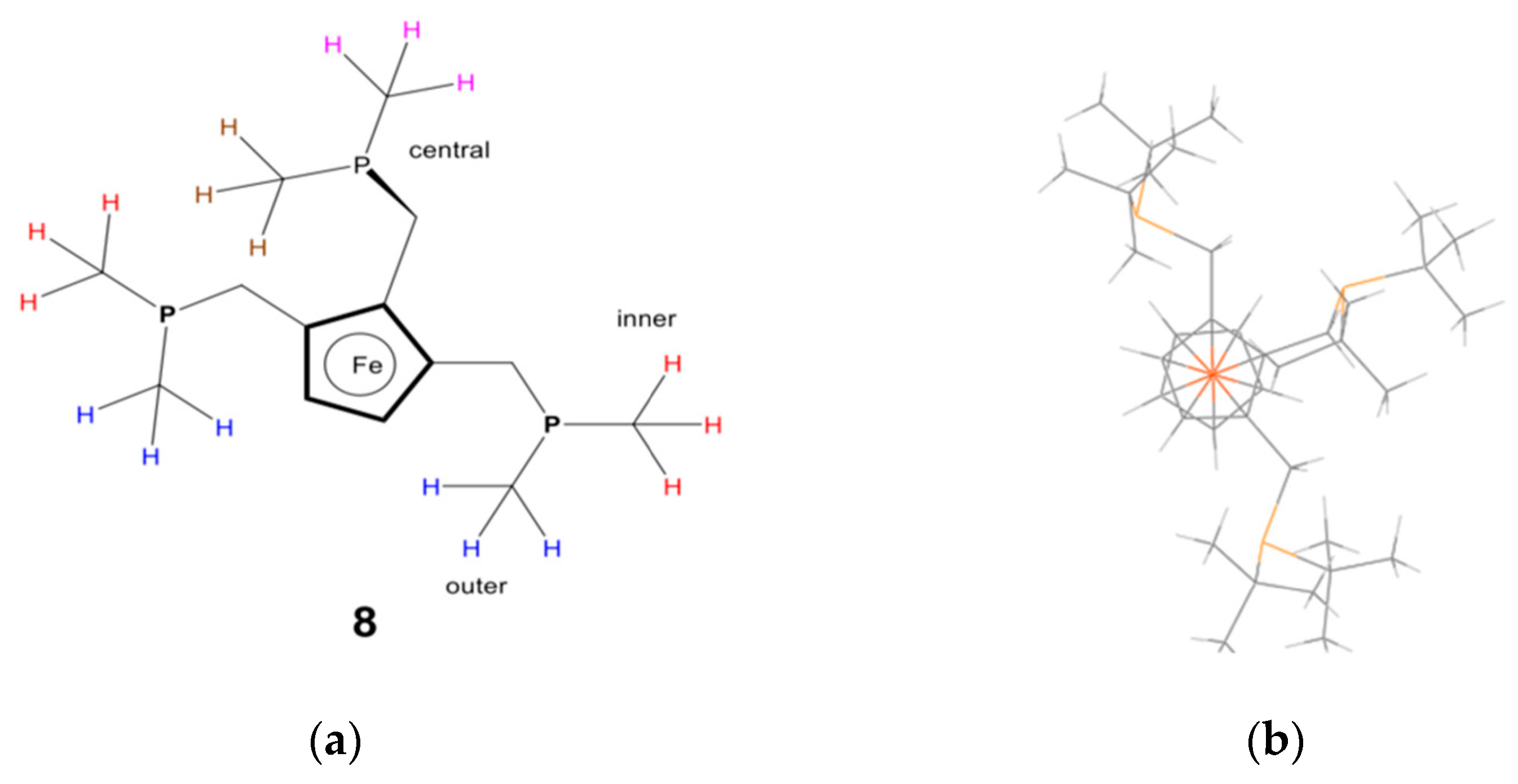
8 to counter this; however, the turnover number was reduced, even though the maximum reaction rate was reached more quickly (8–10 min compared to 25 min). More comprehensive tests were carried out following these initial results, again, under the industrial operating conditions.
Table 2 provides the results data as average weight gains for the complexes for standard Alpha
1 and
butphos 2a. There was no evidence of metal deposition in any of these runs, and all reaction solutions from the autoclave were pale green/yellow.
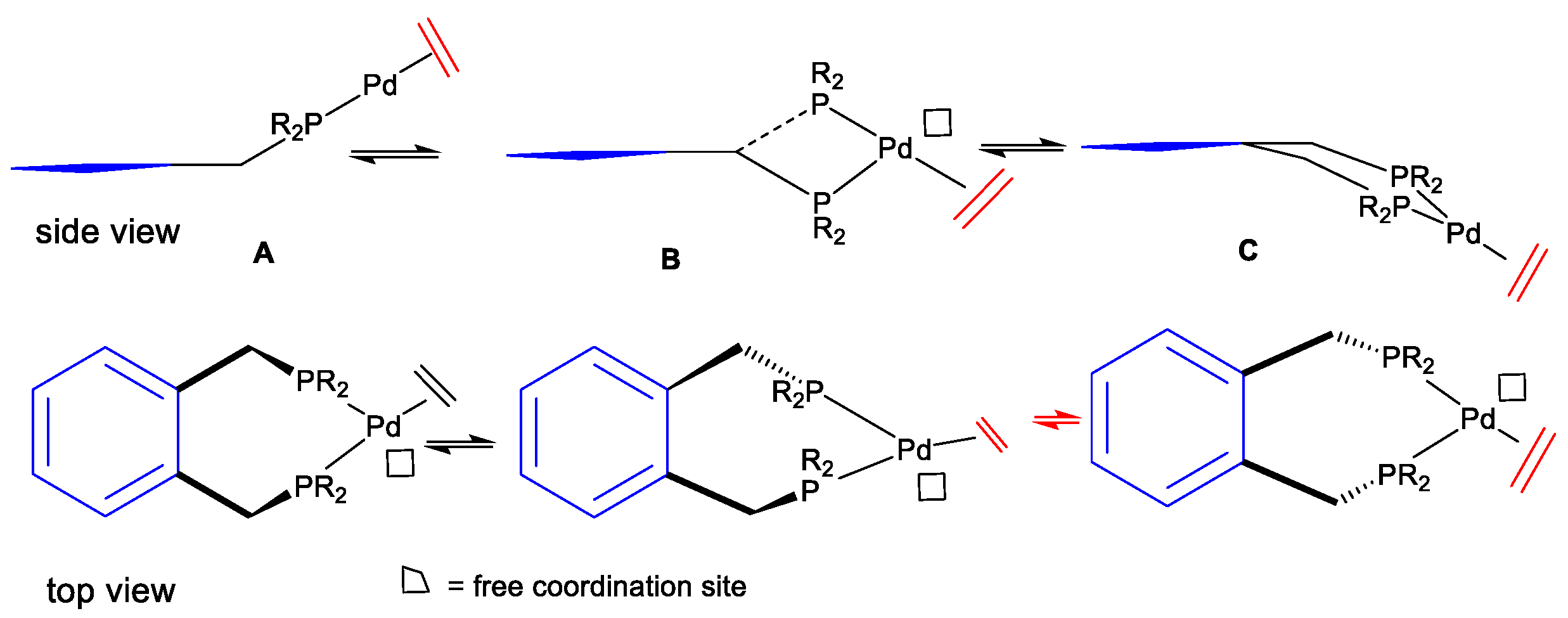
In this set of experiments, 0.1mmol of the palladium complex was used in neat methanol. The gas composition was 50:50 CO:ethene for all experiments. The use of neat methanol and relatively high palladium concentrations is generally optimal for high initial rates but accelerated catalyst decay can occur under these conditions. The palladium complex of the Alpha
1 standard gave rise to an average productivity after 3 h of approximately 257.3 g—or an average TON of 45,932. Independent research carried out by Lucite International has shown that average productivity is enhanced when bulky groups, such as tert-butyl, are attached to the 4-position, and increase the steric bulk of alpha ligands [
64]. The disadvantage in benzene-derived Alpha ligand design (
Figure 10), identified from crystallographic analysis of their palladium complexes, is that the Pd, P, and methylene atoms in the ligands lie approximately in the same plane of a square planar palladium(II) complex [
65]. The benzene ring then tilts at 60–80 degrees to this plane, suggesting that the benzene ring can flip between three different positions: up, down, or one up and one down. This ring flipping can, in some cases, lead to ligand dissociation and, hence, catalyst decay. A study of complexes like [(P-P)PdCl
2] supports this, and the exchange of the CH
2 proton has been observed [
65]. For the Alpha ligand, by adding steric bulk to the benzene ring, it is possible to influence the rate of the ring flipping and, consequently, retard the rate of catalyst decay. In the
butphos ligand family
2a, the average productivity after 3 h outperforms the Alpha standard, with an average weight gain of 300.9 g, or an average TON of 53,731.
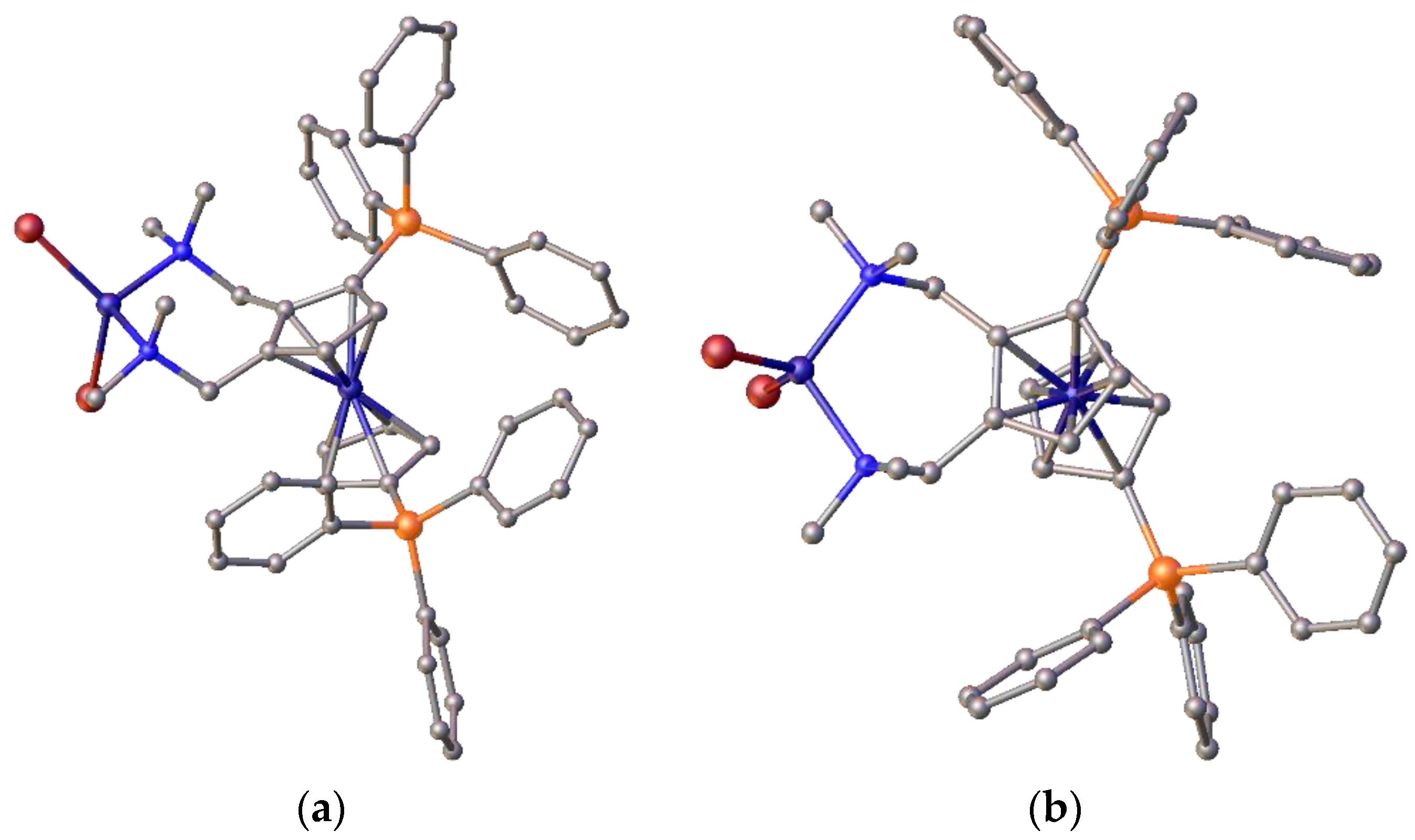
Additionally, variable-temperature NMR analysis of 1,2-di-tert-butylphosphinomethyl ferrocene palladium complexes shows that the chemical shifts of CH
2 protons are essentially static at room temperature (see
supporting information). Thus, the ferrocenyl ring is not flipping on the NMR timescale at room temperature. Finally, the conformation of the complexed ligand is confirmed by crystal structural data obtained from complexes of methanesulphonic acid salts of the ferrocenylphosphines (isolated during catalyst trials), as shown in
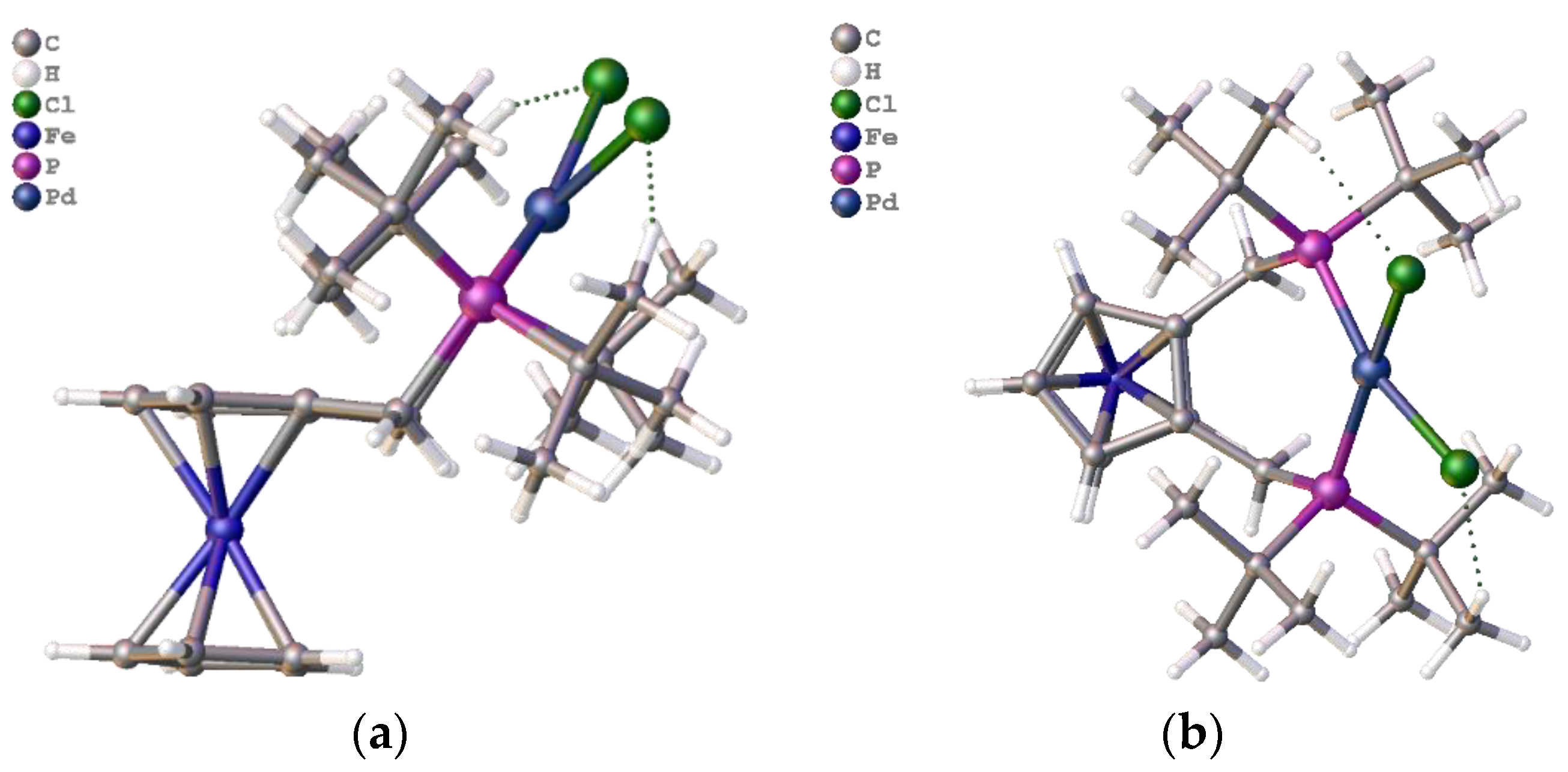
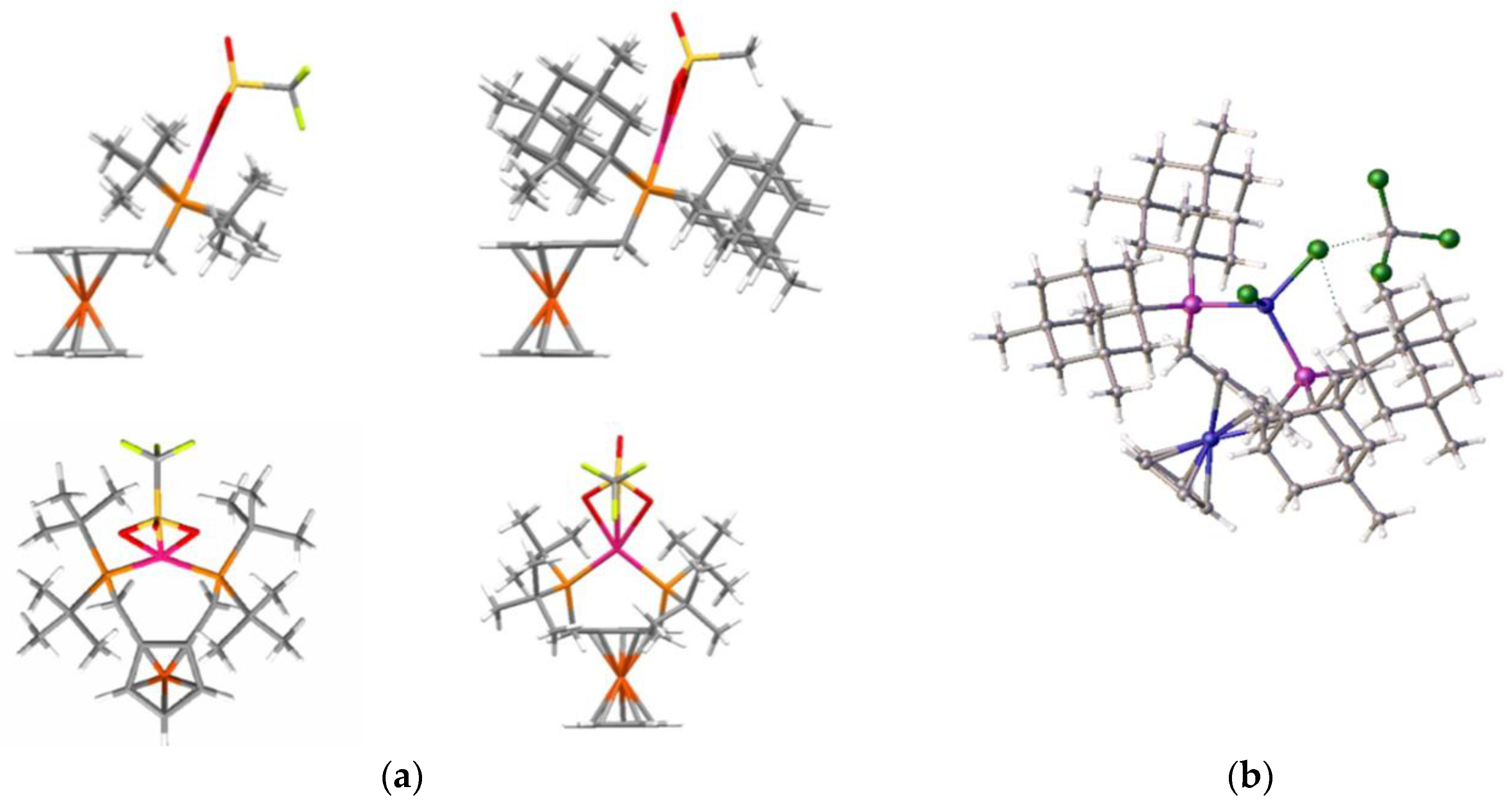
Figure 19.
The graphics in
Figure 19a, which were prepared from crystal structural data, clearly show the palladium atom positioned above the plane of the ferrocene cyclopentadienyl rings. This positioning may be one reason for the improved activity over the standard Alpha ligand system and allows better steric control of the catalyst system. Even in the nickel complex of ligand
2c, the nickel lies above the cp plane (
Figure 19b). However, in this complex, it was also clear that the steric crowding required for optimal catalytic performance was not present in the tetrahedral coordination mode. Finally, data were collected from the silyl-substituted ligand complexes and run again, compared with the Alpha standard.
Table 3 presents the results from these experiments in tabular form.
The average weight gains achieved with the trimethylsilyl-substituted ligand
2g were low and did not significantly improve catalytic performance. However, the triphenyl-substituted ligand,
2h, did show the expected improvement. In addition, the effect of increasing the steric bulk of the phosphine substituents on the catalysis rate did occur for the diadamantyl-substituted ligand
2b and the bulky dimethyladamantylphosphinoferrocene
2c (
Table 4).
These data show that the steric bulk on the ferrocenylphosphine increases the reaction rate and turnover numbers. In addition, the catalysts are recyclable, and give highly cumulative turnover numbers. In the case of ligand
2c, the fall in reaction rate from the initial rate of 52,700 to 34,125 (approx. 58% of original activity) occurred on its fourth run with the same catalyst, making this a very stable catalyst. The palladium catalyst derived from the standard Alpha ligand had lost approximately 95% of its activity after two recycles. The catalyst derived from ligand
2b also retained about 50% of its activity after two cycles. The adamantylphosphinoferrocene complexes had a much higher TON (approximately 40% increase) when compared to Alpha standards. We also compared some of the more sterically bulky ligands under identical catalytic conditions (
Table 5).
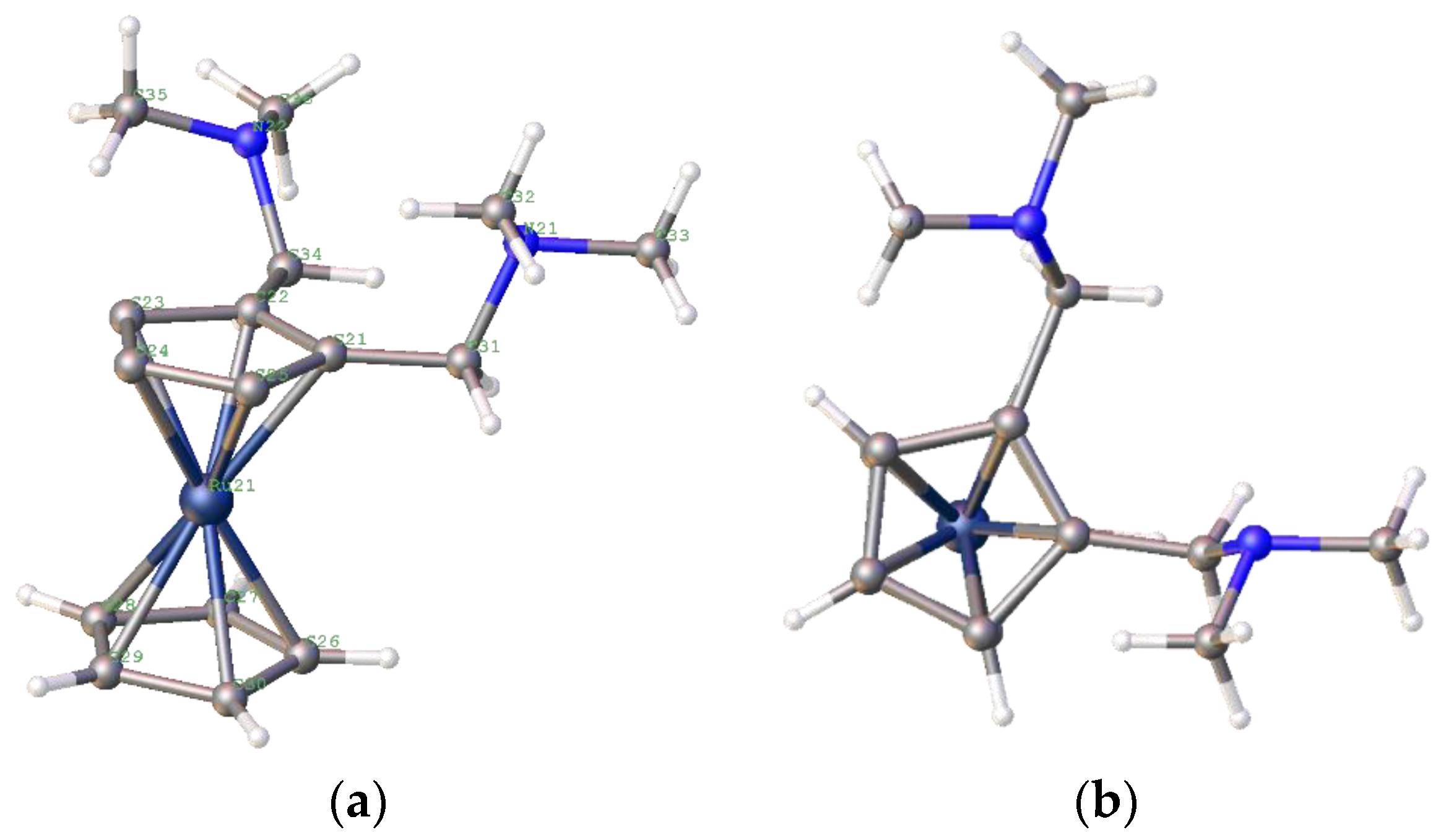
From these data, the catalysts prepared from less bulky phosphines show high initial rates; however, the weight gain data show that the metal complexes with more bulky ligands perform better after 3 h. Finally, for completeness, the ruthenocenyl bis-amine analogue of compound
5—compound
26—was prepared and crystallographically characterised (
Figure 20).
Although the basic structure of compound
26 is remarkably similar to that of compound
5, with the amines both sitting above the cp ring, the actual macrostructures of the crystals used had rather different characteristics. Compound
25 was the starting compound [
66,
67,
68,
69]; it was only possible to obtain crystals of
26 as fine needles, generally clumped together. This clumping made the crystallography difficult, and the structure obtained indicated two molecules—the second with 15% occupancy—at a 53° angle to the first, and the second Ru atom sitting 1.4 Å away from the first (
Supplementary Materials). However, the ligand precursor is isomorphic; we would expect similar reactivity of the metal complexes, with any differences caused by the relative stabilities of the complexes and the different electron donation properties of ruthenocene compared to ferrocene. The complex
27.PdCl2 (
Figure 21) was prepared in situ, and we compare its performance in the catalytic preparation of methyl propanoate to its ferrocenyl analogue
2a.PdCl2.
These data (
Table 6) indicate that the average catalyst activity of the ruthenocene analogue of the catalyst again outperforms both catalysts
1.PdCl
2 and
2a.PdCl
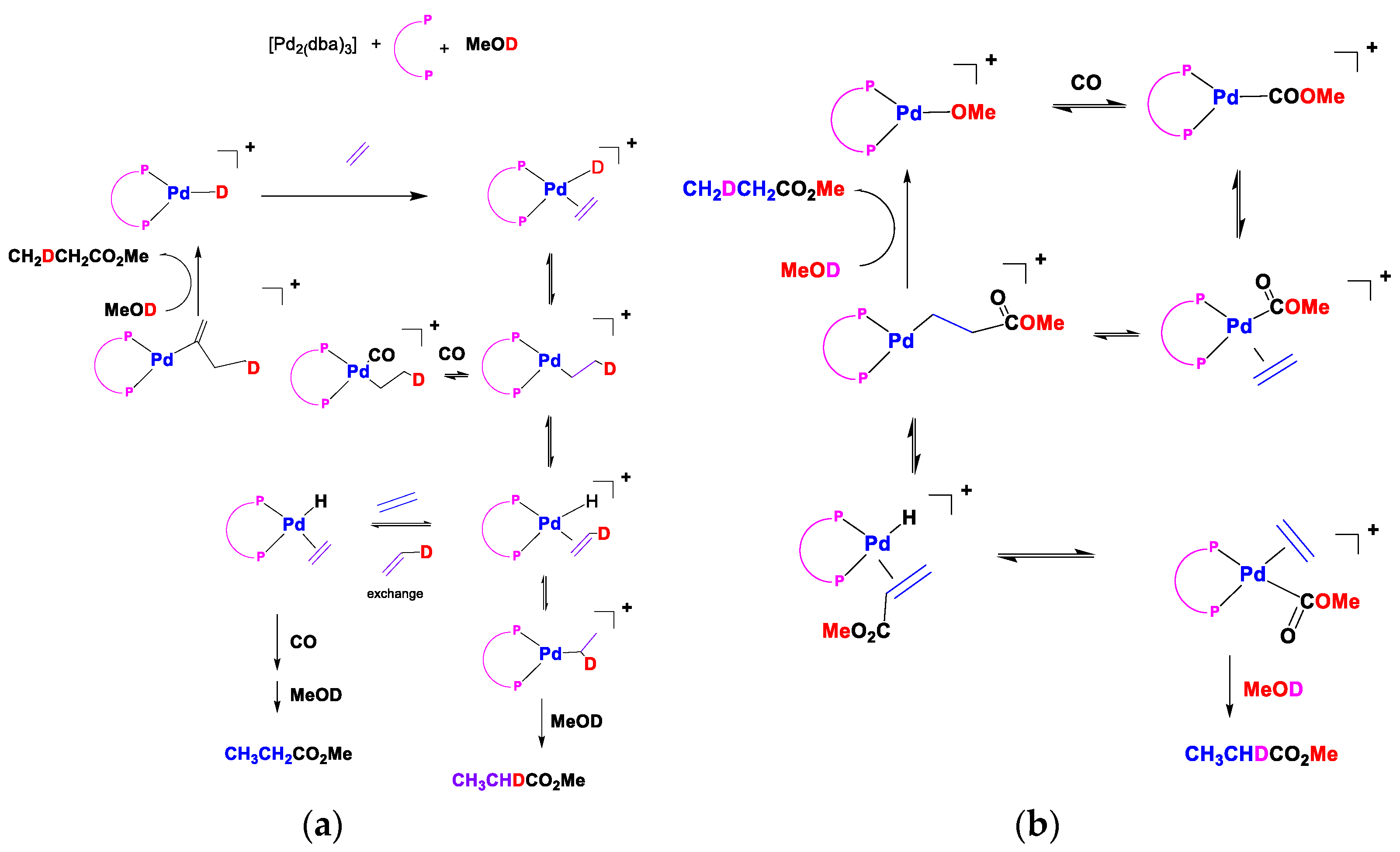
2 under the reaction conditions used. The rationale for this is the improved stability of the ligand and its palladium complex under the operating conditions. These data were all produced pre-2006, and it is necessary to update the reader on more recent progress with ferrocene-based ligands. We briefly discuss the reaction mechanism studies of the hydride and methoxy routes (
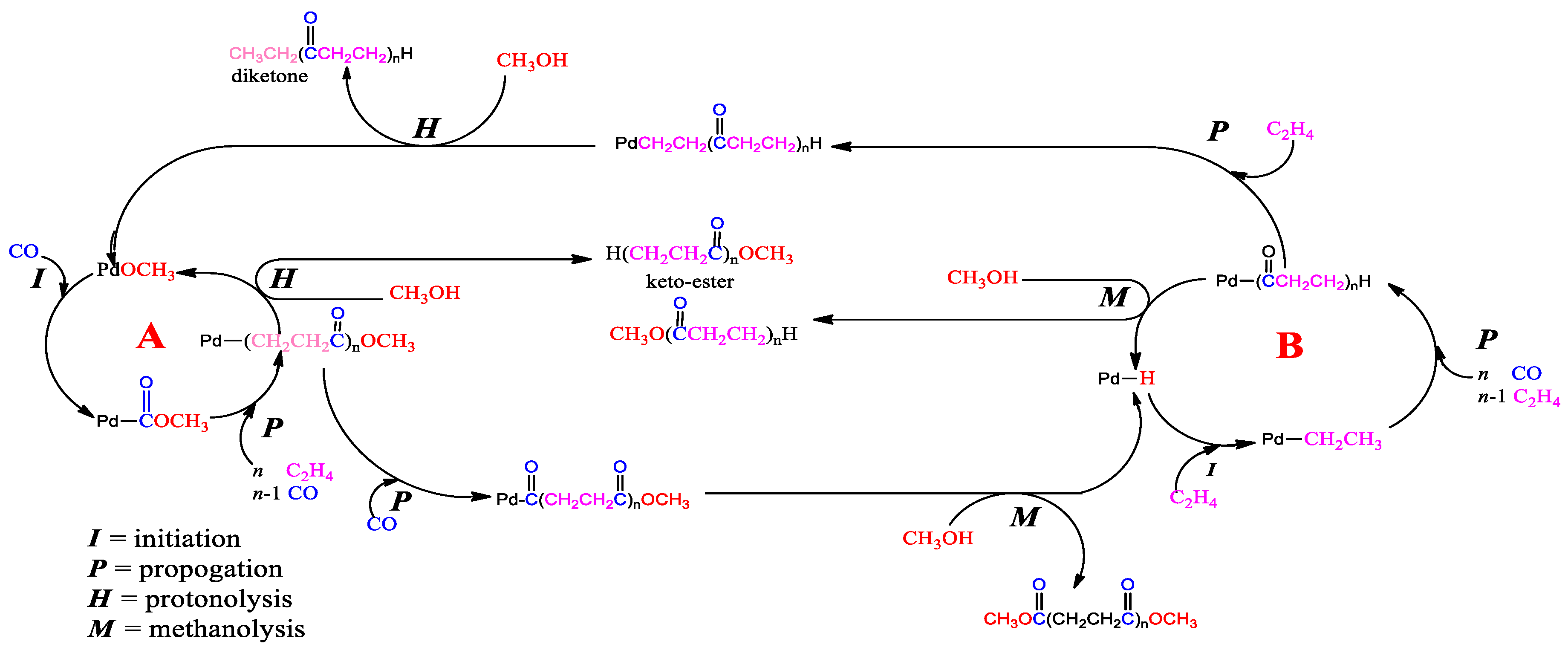
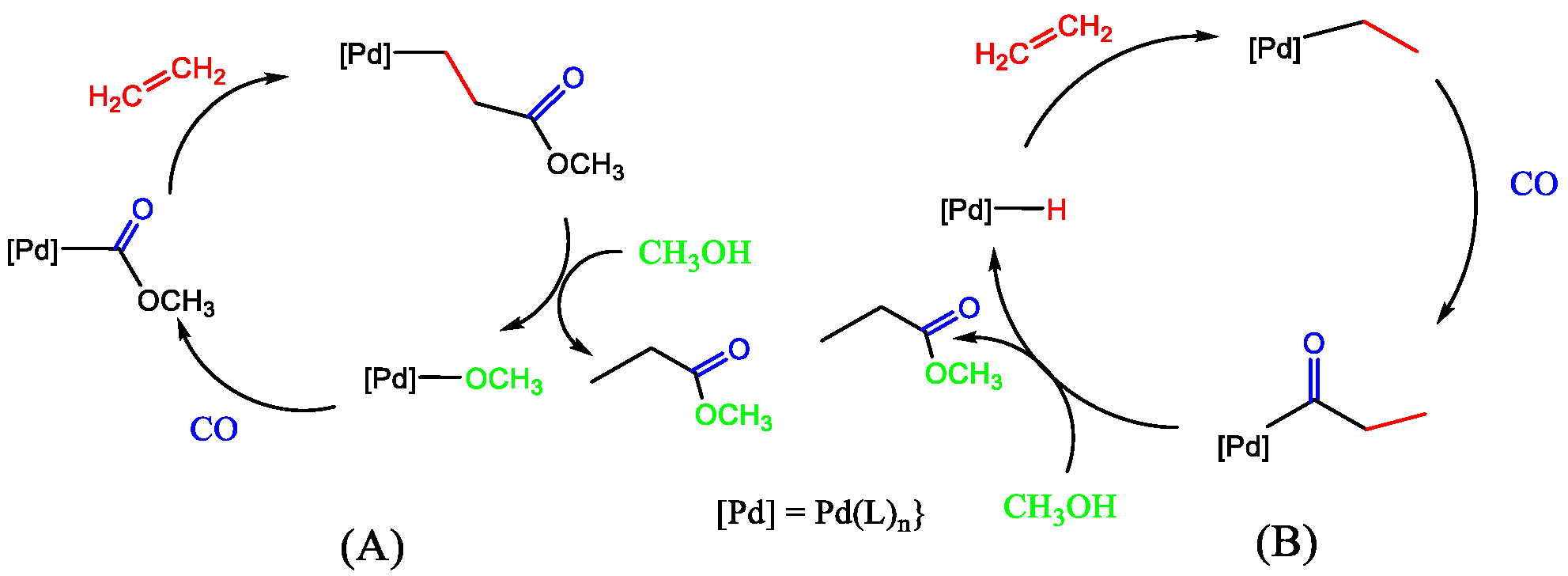
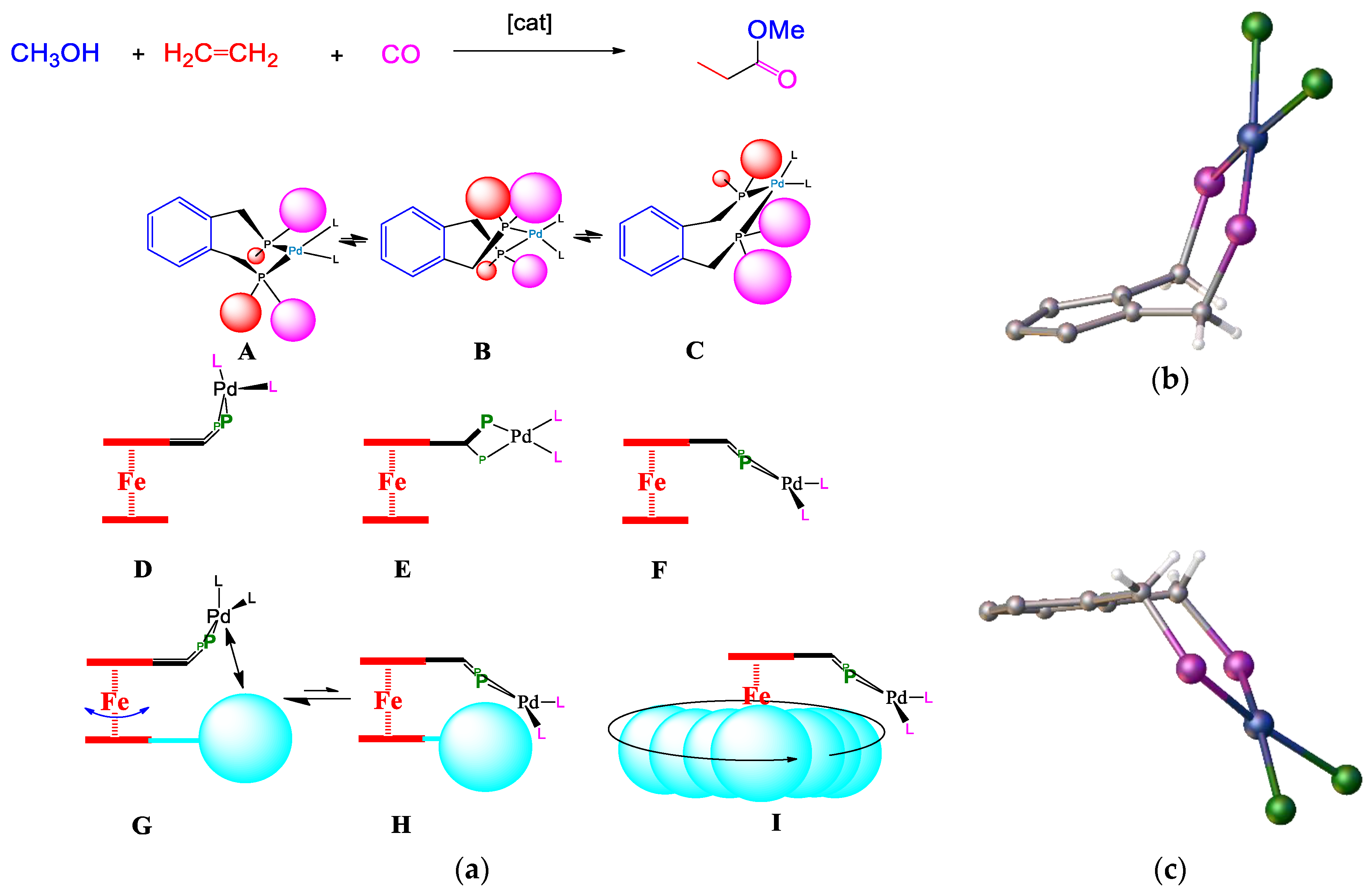
Figure 22). Several reversible steps account for the incorporation of deuterium into the methyl propanoate part when d-methanol is a substrate for both cycles. Maintaining a strict square planar coordination leads to a congested environment around palladium when bulky ligands are present. In these mechanisms, there are many intermediates where there is a free coordination site where stabilisation by solvent will play a significant role. It would be interesting to see whether any deuterium ends up in the ligand because of the possible stabilisation of the metal centre by protons on the bulky ligands. The mechanism for our catalyst is more likely to follow the protonation route, given the strongly acidic conditions of our reaction environment.
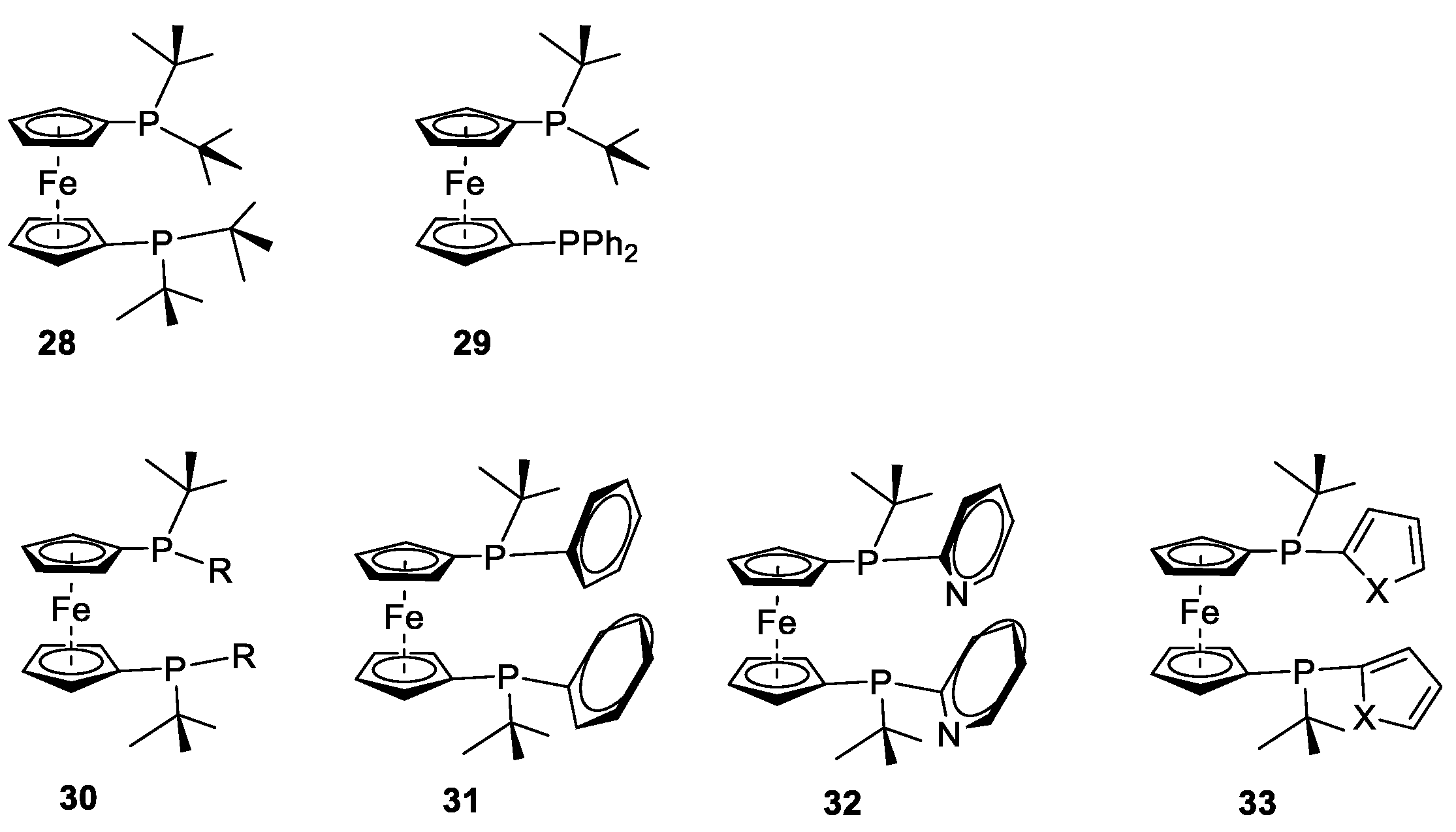
Finally, it is prudent to update the reader on the work of Beller et al. [
70,
71,
72,
73,
74,
75] in this field, which is much more recent and detailed. These researchers have examined methoxycarbonylation using metal complexes of several adapted ferrocene-based ligands. These ligands generally contain phosphines substituted with t-butyl groups, such as compound
29 shown in
Figure 23. These ligand types are essentially variants of those developed by Cullen et al. [
76,
77]. We had trialled some of these in early our work but discounted them because of their poorer selectivity. These tend to be sterically less congested around the metal centre in comparison to our ligands; however, they incorporate additional features due to the nature of additional functional groups, such as coordinating and protonation sites. However, clearly Beller et al. have championed these ligands and have reported excellent product yields. They have also incorporated adamantyl groups on the phosphorus similar to those in our ligand family thus adding additional steric bulk.
The inclusion of pyridyl substituents in the ligand design is a useful innovation. However, these would probably become protonated during catalysis, decreasing their solubility. It would be interesting to use the palladium complex of these ligands to isolate critical intermediates. In summary, they have shown that ferrocene-based ligands are extremely valuable in this research area, and numerous patents have come from their work. Thus, we have even more confidence that the ligands described in this paper are worthy of much deeper investigation, as are the new ligands under development within our group. A recent DFT study may help advance the mechanistic insights of this process [
78]. Further synthetic details of in situ methods for phosphane preparation can be found in the
Supplementary Materials.


 ,
, 























{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}