Targeted Gene Delivery through the Respiratory System: Rationale for Intratracheal Gene Transfer

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Cellular Organization in the Normal Lung
3. Target Cells for Gene Therapy in Different Lung Diseases with Genetic Backgrounds
3.1. Pulmonary Arterial Hypertension
3.2. Cystic Fibrosis
3.3. α-1 Antitrypsin Deficiency
3.4. Surfactant Protein-B Deficiency
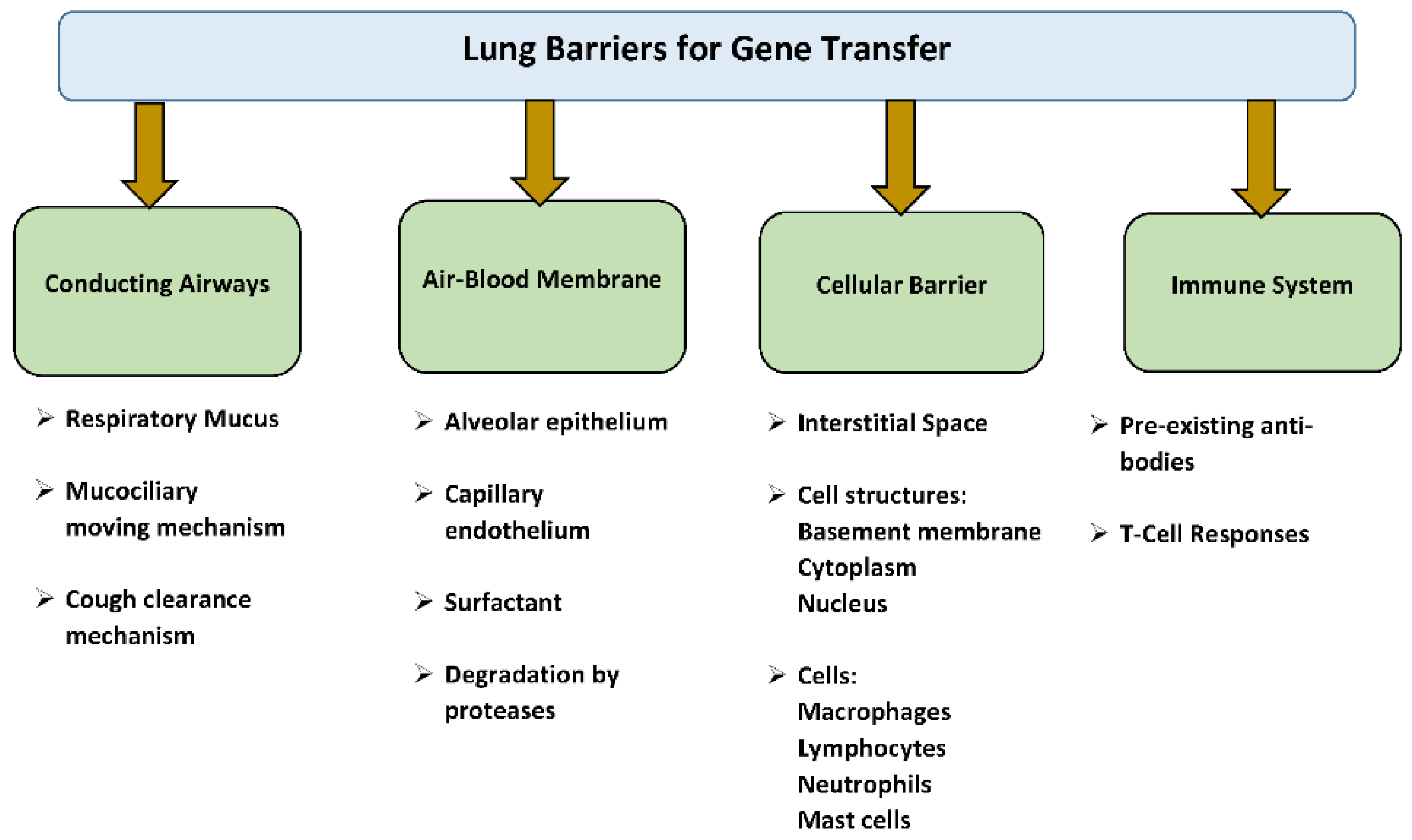
4. Pulmonary Barriers to Gene Transfer
4.1. Respiratory Mucus, Mucociliary, and Cough Clearance Mechanism
4.2. Air–Blood Barrier
4.3. Cellular and Immune Barriers
- Pre-existing antibodies and T- cell response against viral vectors
- Neutralizing antibodies generated against capsid proteins of viral vector
- T-cell-mediated response against viral vector capsid
- Neutralizing antibodies and T cell-mediated response against transgenes
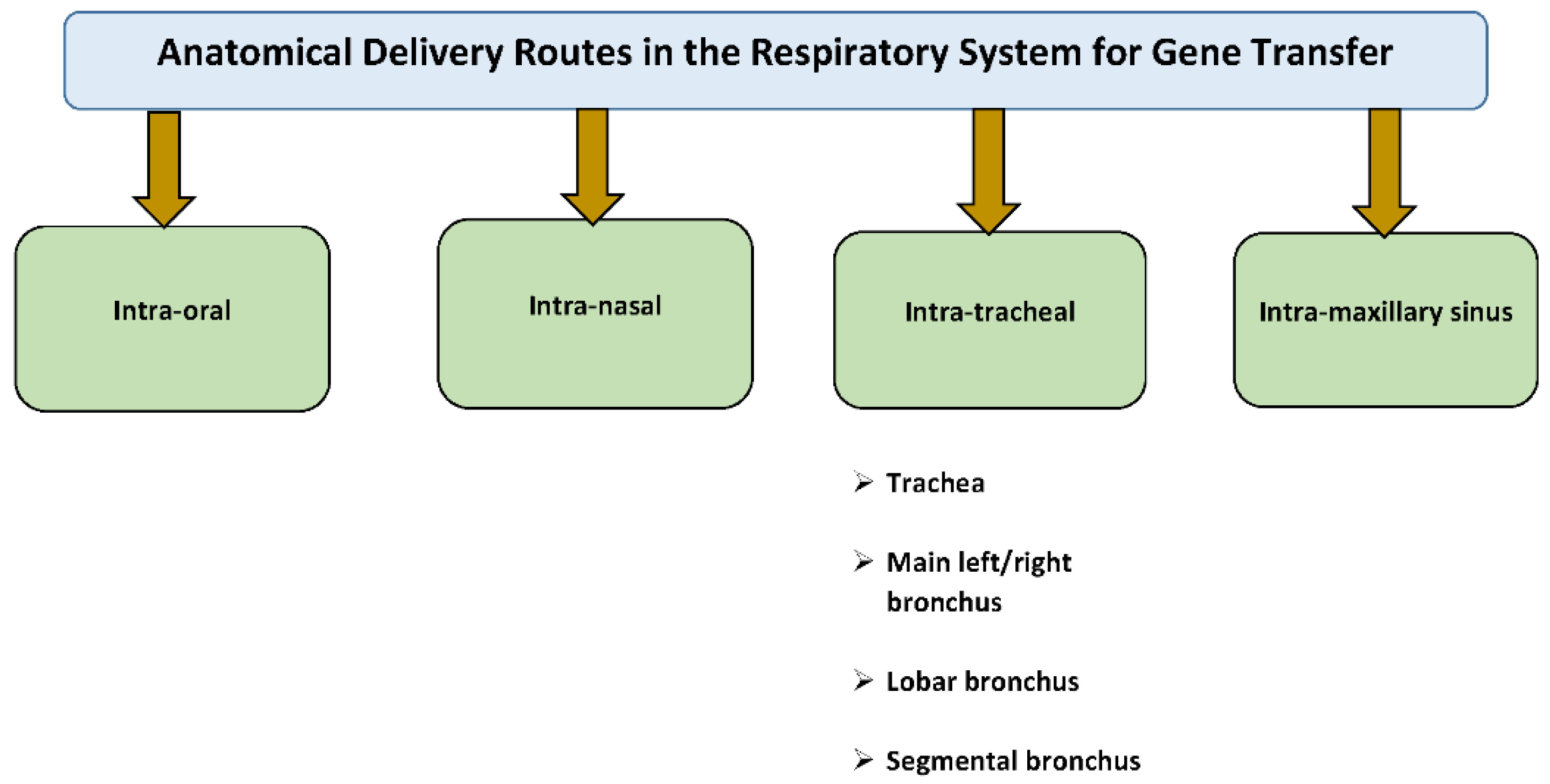
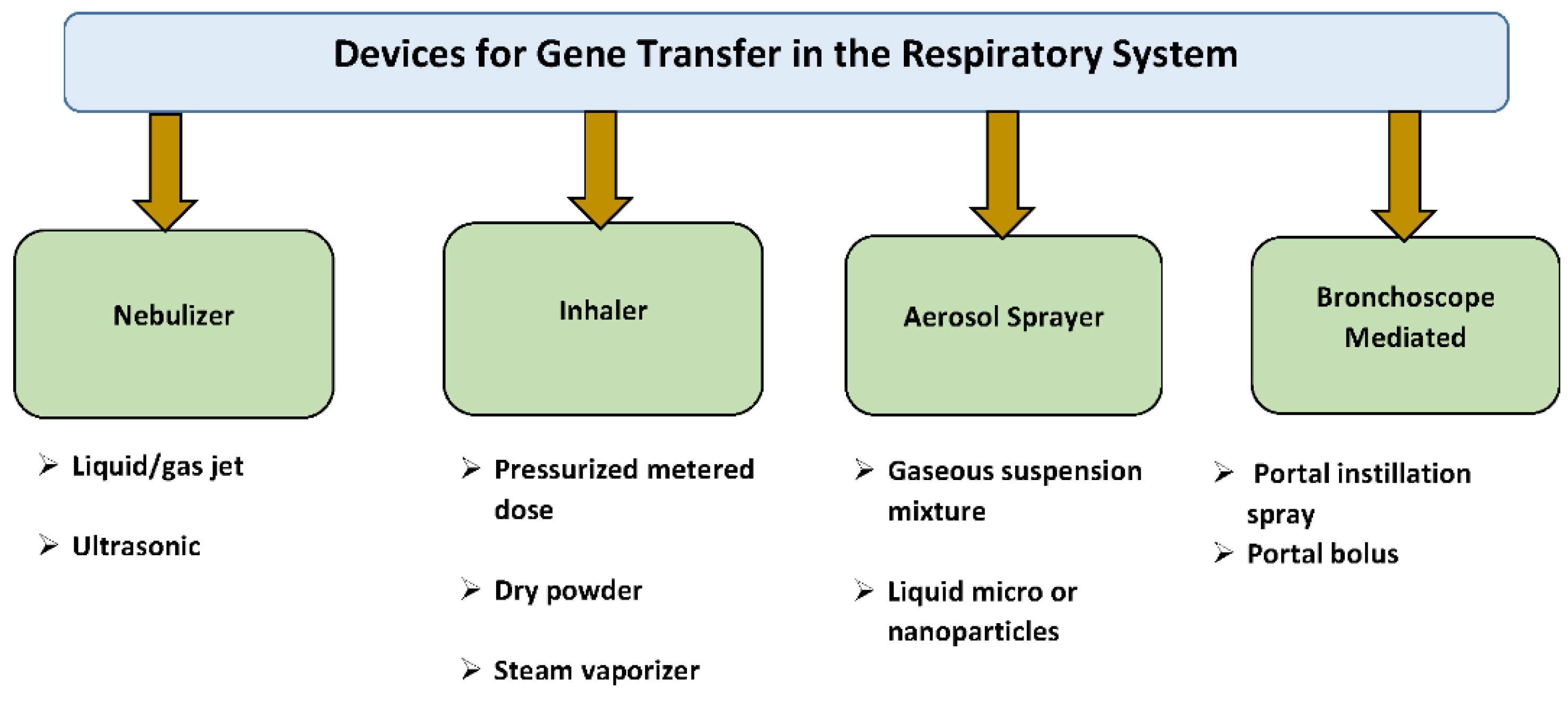
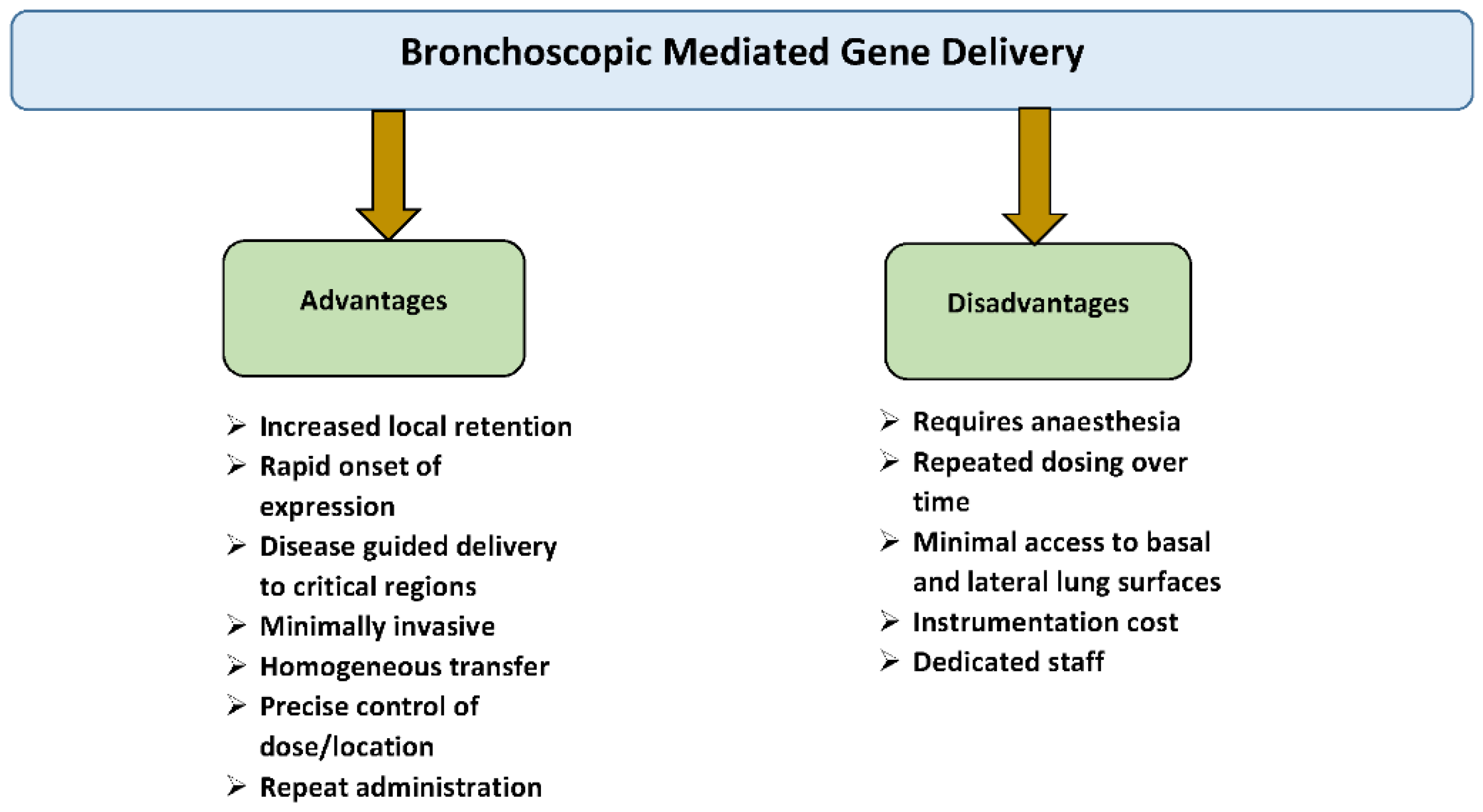
5. Rationale for Intratracheal Gene Delivery to the Lung
6. Intratracheal Delivery with Different Viral Vectors in Small Animals and Clinical Trials
6.1. Adenovirus
6.2. Adeno-Associated Virus
7. Intratracheal Delivery with Different Viral Vectors in Large Animals
7.1. Adenovirus
7.2. Adeno-Associated Virus
8. Re-Administration of AAV Vectors for Lung Gene Transfer
9. Conclusions
Funding
Conflicts of Interest
References
- Katz, M.G.; Fargnoli, A.S.; Williams, R.D.; Bridges, C.R. Gene therapy delivery systems for enhancing viral and nonviral vectors for cardiac diseases: Current concepts and future applications. Hum. Gene Ther. 2013, 24, 914–927. [Google Scholar] [CrossRef] [PubMed]
- Katz, M.G.; Fargnoli, A.S.; Weber, T.; Hajjar, R.J.; Bridges, C.R. Use of Adeno-Associated Virus Vector for Cardiac Gene Delivery in Large-Animal Surgical Models of Heart Failure. Hum. Gene. Ther. Clin. Dev. 2017, 28, 157–164. [Google Scholar] [CrossRef] [PubMed]
- Senís, E.; Fatouros, C.; Große, S.; Wiedtke, E.; Niopek, D.; Mueller, A.-K.; Börner, K.; Grimm, D. CRISPR/Cas9-mediated genome engineering: An adeno-associated viral (AAV) vector toolbox. Biotechnol. J. 2014, 9, 1402–1412. [Google Scholar] [CrossRef] [PubMed]
- Chew, W.L.; Tabebordbar, M.; Cheng, J.K.W.; Mali, P.; Wu, E.Y.; Ng, A.H.M.; Zhu, K.; Wagers, A.J.; Church, G.M. A multifunctional AAV–CRISPR–Cas9 and its host response. Nat. Methods 2016, 13, 868. [Google Scholar] [CrossRef] [PubMed]
- Marangi, M.; Pistritto, G. Innovative Therapeutic Strategies for Cystic Fibrosis: Moving Forward to CRISPR Technique. Front. Pharm. 2018, 9, 396. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Li, J.; Xiao, X. Overcoming adeno-associated virus vector size limitation through viral DNA heterodimerization. Nat. Med. 2000, 6, 599. [Google Scholar] [CrossRef] [PubMed]
- Lee, T.W.R.; Matthews, D.A.; Blair, G.E. Novel molecular approaches to cystic fibrosis gene therapy. Biochem. J. 2005, 387, 1–15. [Google Scholar] [CrossRef]
- Ostedgaard, L.S.; Rokhlina, T.; Karp, P.H.; Lashmit, P.; Afione, S.; Schmidt, M.; Zabner, J.; Stinski, M.F.; Chiorini, J.A.; Welsh, M.J. A shortened adeno-associated virus expression cassette for CFTR gene transfer to cystic fibrosis airway epithelia. Proc. Natl. Acad. Sci. USA 2005, 102, 2952–2957. [Google Scholar] [CrossRef] [Green Version]
- Franks, T.J.; Colby, T.V.; Travis, W.D.; Tuder, R.M.; Reynolds, H.Y.; Brody, A.R.; Cardoso, W.V.; Crystal, R.G.; Drake, C.J.; Engelhardt, J.; et al. Resident Cellular Components of the Human Lung. Proc. Am. Thorac. Soc. 2008, 5, 763–766. [Google Scholar] [CrossRef]
- Knight, D.A.; Holgate, S.T. The airway epithelium: Structural and functional properties in health and disease. Respirology 2003, 8, 432–446. [Google Scholar] [CrossRef] [Green Version]
- Barkauskas, C.E.; Cronce, M.J.; Rackley, C.R.; Bowie, E.J.; Keene, D.R.; Stripp, B.R.; Randell, S.H.; Noble, P.W.; Hogan, B.L.M. Type 2 alveolar cells are stem cells in adult lung. J. Clin. Investig. 2013, 123, 3025–3036. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rock, J.R.; Onaitis, M.W.; Rawlins, E.L.; Lu, Y.; Clark, C.P.; Xue, Y.; Randell, S.H.; Hogan, B.L.M. Basal cells as stem cells of the mouse trachea and human airway epithelium. Proc. Natl. Acad. Sci. USA 2009, 106, 12771–12775. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Groneberg, D.A.; Witt, C.; Wagner, U.; Chung, K.F.; Fischer, A. Fundamentals of pulmonary drug delivery. Respir. Med. 2003, 97, 382–387. [Google Scholar] [CrossRef] [Green Version]
- Scheuch, G. Novel approaches to enhance pulmonary delivery of proteins and peptides. J. Physiol. Pharm. 2007, 58, 615–625. [Google Scholar]
- Siekmeier, R.; Scheuch, G. Systemic treatment by inhalation of macromolecules - principles, problems, and examples. J. Physiol. Pharm. 2008, 59, 53–79. [Google Scholar]
- Gessler, T.; Schmehl, T.; Olschewski, H.; Grimminger, F.; Seeger, W. Aerosolized vasodilators in pulmonary hypertension. J. Aerosol Med. Depos. Clear. Eff. Lung 2002, 15, 117–122. [Google Scholar] [CrossRef] [PubMed]
- Gessler, T.; Seeger, W.; Schmehl, T. Inhaled Prostanoids in the Therapy of Pulmonary Hypertension. J. Aerosol Med. Pulm. Drug Deliv. 2008, 21, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Reynolds, A.M.; Xia, W.; Holmes, M.D.; Hodge, S.J.; Danilov, S.; Curiel, D.T.; Morrell, N.W.; Reynolds, P.N. Bone morphogenetic protein type 2 receptor gene therapy attenuates hypoxic pulmonary hypertension. Am. J. Physiol. Lung Cell Mol. Physiol. 2007, 292, L1182–L1192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bonfield, T.L.; Caplan, A.I. Adult Mesenchymal Stem Cells: An Innovative Therapeutic for Lung Diseases. Discov. Med. 2010, 9, 337–345. [Google Scholar] [PubMed]
- Zuckerman, J.B.; Robinson, C.B.; M, K.S.; Shell, R.; Sferra, T.J.; Chirmule, N.; Magosin, S.A.; Propert, K.J.; Brown-Parr, E.C.; Hughes, J.V.; et al. A Phase I Study of Adenovirus-Mediated Transfer of the Human Cystic Fibrosis Transmembrane Conductance Regulator Gene to a Lung Segment of Individuals with Cystic Fibrosis. Hum. Gene Ther. 1999, 10, 2973–2985. [Google Scholar] [CrossRef]
- Harvey, B.G.; Leopold, P.L.; Hackett, N.R.; Grasso, T.M.; Williams, P.M.; Tucker, A.L.; Kaner, R.J.; Ferris, B.; Gonda, I.; Sweeney, T.D.; et al. Airway epithelial CFTR mRNA expression in cystic fibrosis patients after repetitive administration of a recombinant adenovirus. J. Clin. Investig. 1999, 104, 1245–1255. [Google Scholar] [CrossRef] [Green Version]
- Elliott, P.R.; Pei, X.Y.; Dafforn, T.R.; Lomas, D.A. Topography of a 2.0 A structure of alpha1-antitrypsin reveals targets for rational drug design to prevent conformational disease. Protein Sci. A Publ. Protein Soc. 2000, 9, 1274–1281. [Google Scholar] [CrossRef] [PubMed]
- Serban, K.A.; Petrache, I. Alpha-1 Antitrypsin and Lung Cell Apoptosis. Ann. Am. Thorac. Soc. 2016, 13 (Suppl. 2), S146–S149. [Google Scholar] [CrossRef]
- Nogee, L.M. Alterations in SP-B and SP-C Expression in Neonatal Lung Disease. Annu. Rev. Physiol. 2004, 66, 601–623. [Google Scholar] [CrossRef] [PubMed]
- Aneja, M.K.; Rudolph, C. Gene therapy of surfactant protein B deficiency. Curr. Opin. Mol. Ther. 2006, 8, 432–438. [Google Scholar] [PubMed]
- Pickles, R.J.; Fahrner, J.A.; Petrella, J.M.; Boucher, R.C.; Bergelson, J.M. Retargeting the coxsackievirus and adenovirus receptor to the apical surface of polarized epithelial cells reveals the glycocalyx as a barrier to adenovirus-mediated gene transfer. J. Virol. 2000, 74, 6050–6057. [Google Scholar] [CrossRef] [PubMed]
- Knowles, M.R.; Boucher, R.C. Mucus clearance as a primary innate defense mechanism for mammalian airways. J. Clin. Investig. 2002, 109, 571–577. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, N.; Duncan, G.A.; Hanes, J.; Suk, J.S. Barriers to inhaled gene therapy of obstructive lung diseases: A review. J. Control. Release 2016, 240, 465–488. [Google Scholar] [CrossRef] [Green Version]
- Sinn, P.L.; Shah, A.J.; Donovan, M.D.; Paul, B. McCray, J. Viscoelastic Gel Formulations Enhance Airway Epithelial Gene Transfer with Viral Vectors. Am. J. Respir. Cell Mol. Biol. 2005, 32, 404–410. [Google Scholar] [CrossRef]
- Goerke, J. Pulmonary surfactant: Functions and molecular composition. Biochim. Biophys. Acta Mol. Basis Dis. 1998, 1408, 79–89. [Google Scholar] [CrossRef]
- Duncan, J.E.; Whitsett, J.A.; Horowitz, A.D. Pulmonary Surfactant Inhibits Cationic Liposome-Mediated Gene Delivery to Respiratory Epithelial Cells In Vitro. Hum. Gene Ther. 1997, 8, 431–438. [Google Scholar] [CrossRef] [PubMed]
- Sibille, Y.; Reynolds, H.Y. Macrophages and Polymorphonuclear Neutrophils in Lung Defense and Injury. Am. Rev. Respir. Dis. 1990, 141, 471–501. [Google Scholar] [CrossRef] [PubMed]
- McCray, P.B., Jr.; Wang, G.; Kline, J.N.; Zabner, J.; Chada, S.; Jolly, D.J.; Chang, S.M.W.; Davidson, B.L. Alveolar Macrophages Inhibit Retrovirus-Mediated Gene Transfer to Airway Epithelia. Hum. Gene Ther. 1997, 8, 1087–1093. [Google Scholar] [CrossRef] [PubMed]
- Zsengellér, Z.; Otake, K.; Hossain, S.A.; Berclaz, P.Y.; Trapnell, B.C. Internalization of adenovirus by alveolar macrophages initiates early proinflammatory signaling during acute respiratory tract infection. J. Virol. 2000, 74, 9655–9667. [Google Scholar] [CrossRef]
- Pickles, R.J.; McCarty, D.; Matsui, H.; Hart, P.J.; Randell, S.H.; Boucher, R.C. Limited entry of adenovirus vectors into well-differentiated airway epithelium is responsible for inefficient gene transfer. J. Virol. 1998, 72, 6014–6023. [Google Scholar] [PubMed]
- Coyne, C.B.; Kelly, M.M.; Boucher, R.C.; Johnson, L.G. Enhanced Epithelial Gene Transfer by Modulation of Tight Junctions with Sodium Caprate. Am. J. Respir. Cell Mol. Biol. 2000, 23, 602–609. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walters, R.W.; Grunst, T.; Bergelson, J.M.; Finberg, R.W.; Welsh, M.J.; Zabner, J. Basolateral Localization of Fiber Receptors Limits Adenovirus Infection from the Apical Surface of Airway Epithelia. J. Biol. Chem. 1999, 274, 10219–10226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Henning, A.; Hein, S.; Schneider, M.; Bur, M.; Lehr, C.M. Pulmonary Drug Delivery: Medicines for Inhalation. Handb. Exp. Pharmacol. 2010, 171–192. [Google Scholar] [CrossRef]
- Laube, B.L. Aerosolized Medications for Gene and Peptide Therapy. Respir. Care 2015, 60, 806–824. [Google Scholar] [CrossRef] [Green Version]
- Chono, S.; Tanino, T.; Seki, T.; Morimoto, K. Influence of particle size on drug delivery to rat alveolar macrophages following pulmonary administration of ciprofloxacin incorporated into liposomes. J. Drug Target. 2006, 14, 557–566. [Google Scholar] [CrossRef]
- Zhou, J.; Wu, Y.; Henderson, F.; McCoy, D.M.; Salome, R.G.; McGowan, S.E.; Mallampalli, R.K. Adenoviral gene transfer of a mutant surfactant enzyme ameliorates pseudomonas-induced lung injury. Gene Ther. 2006, 13, 974. [Google Scholar] [CrossRef] [PubMed]
- Dong, J.-Y.; Wang, D.; Ginkel, F.W.v.; Pascual, D.W.; Frizzell, R.A. Systematic Analysis of Repeated Gene Delivery into Animal Lungs with a Recombinant Adenovirus Vector. Hum. Gene Ther. 1996, 7, 319–331. [Google Scholar] [CrossRef] [PubMed]
- Joseph, P.M.; O’Sullivan, B.P.; Lapey, A.; Dorkin, H.; Oren, J.; Balfour, R.; Perricone, M.A.; Rosenberg, M.; Wadsworth, S.C.; Smith, A.E.; et al. Aerosol and Lobar Administration of a Recombinant Adenovirus to Individuals with Cystic Fibrosis. I. Methods, Safety, and Clinical Implications. Hum. Gene Ther. 2001, 12, 1369–1382. [Google Scholar] [CrossRef]
- Vieillard-Baron, A.; Frisdal, E.; Raffestin, B.; H Baker, A.; Eddahibi, S.; Adnot, S.; d’Ortho, M.-P. Inhibition of Matrix Metalloproteinases by Lung TIMP-1 Gene Transfer Limits Monocrotaline-Induced Pulmonary Vascular Remodeling in Rats. Hum. Gene Ther. 2003, 14, 861–869. [Google Scholar] [CrossRef] [PubMed]
- McMurtry, M.S.; Moudgil, R.; Hashimoto, K.; Bonnet, S.; Michelakis, E.D.; Archer, S.L. Overexpression of human bone morphogenetic protein receptor 2 does not ameliorate monocrotaline pulmonary arterial hypertension. Am. J. Physiol. Lung Cell Mol. Physiol. 2007, 292, L872–L878. [Google Scholar] [CrossRef] [PubMed]
- Farkas, L.; Farkas, D.; Ask, K.; Möller, A.; Gauldie, J.; Margetts, P.; Inman, M.; Kolb, M. VEGF ameliorates pulmonary hypertension through inhibition of endothelial apoptosis in experimental lung fibrosis in rats. J. Clin. Investig. 2009, 119, 1298–1311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pozeg, Z.I.; Michelakis, E.D.; McMurtry, M.S.; Thébaud, B.; Wu, X.-C.; Dyck, J.R.B.; Hashimoto, K.; Wang, S.; Moudgil, R.; Harry, G.; et al. In Vivo Gene Transfer of the O2-Sensitive Potassium Channel Kv1.5 Reduces Pulmonary Hypertension and Restores Hypoxic Pulmonary Vasoconstriction in Chronically Hypoxic Rats. Circulation 2003, 107, 2037–2044. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Luo, M.; Guo, C.; Yan, Z.; Wang, Y.; Lei-Butters, D.C.M.; Engelhardt, J.F. Analysis of adeno-associated virus progenitor cell transduction in mouse lung. Mol. Ther. J. Am. Soc. Gene Ther. 2009, 17, 285–293. [Google Scholar] [CrossRef] [PubMed]
- Limberis, M.P.; Wilson, J.M. Adeno-associated virus serotype 9 vectors transduce murine alveolar and nasal epithelia and can be readministered. Proc. Natl. Acad. Sci. USA 2006, 103, 12993–12998. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fleurence, E.; Riviere, C.; Lacaze-Masmonteil, T.; Franco-Motoya, M.-L.; Waszak, P.; Bourbon, J.; Danos, O.; Douar, A.-M.; Delacourt, P.C. Comparative Efficacy of Intratracheal Adeno-Associated Virus Administration to Newborn Rats. Hum. Gene Ther. 2005, 16, 1298–1306. [Google Scholar] [CrossRef]
- Carlon, M.; Toelen, J.; Van der Perren, A.; Vandenberghe, L.H.; Reumers, V.; Sbragia, L.; Gijsbers, R.; Baekelandt, V.; Himmelreich, U.; Wilson, J.M.; et al. Efficient gene transfer into the mouse lung by fetal intratracheal injection of rAAV2/6.2. Mol. Ther. 2010, 18, 2130–2138. [Google Scholar] [CrossRef] [PubMed]
- Dragana, V.; Rik, G.; Ana, Q.-J.; James, D.; Chris, V.d.H.; Anke, V.d.P.; Adrian, L.; Veerle, B.; Zeger, D.; Sylvia, C.M. Noninvasive Imaging Reveals Stable Transgene Expression in Mouse Airways After Delivery of a Nonintegrating Recombinant Adeno-Associated Viral Vector. Hum. Gene Ther. 2016, 27, 60–71. [Google Scholar] [CrossRef]
- Rogers, C.S.; Hao, Y.; Rokhlina, T.; Samuel, M.; Stoltz, D.A.; Li, Y.; Petroff, E.; Vermeer, D.W.; Kabel, A.C.; Yan, Z.; et al. Production of CFTR-null and CFTR-ΔF508 heterozygous pigs by adeno-associated virus–mediated gene targeting and somatic cell nuclear transfer. J. Clin. Investig. 2008, 118, 1571–1577. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rogers, C.S.; Abraham, W.M.; Brogden, K.A.; Engelhardt, J.F.; Fisher, J.T.; Paul, B.; McCray, J.; McLennan, G.; Meyerholz, D.K.; Namati, E.; et al. The porcine lung as a potential model for cystic fibrosis. Am. J. Physiol. Lung Cell Mol. Physiol. 2008, 295, L240–L263. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Luo, M.; Zhang, L.; Ding, W.; Yan, Z.; Engelhardt, J.F. Bioelectric properties of chloride channels in human, pig, ferret, and mouse airway epithelia. Am. J. Respir. Cell Mol. Biol. 2007, 36, 313–323. [Google Scholar] [CrossRef]
- Aguero, J.; Ishikawa, K.; Hadri, L.; Santos-Gallego, C.; Fish, K.; Hammoudi, N.; Chaanine, A.; Torquato, S.; Naim, C.; Ibanez, B.; et al. Characterization of right ventricular remodeling and failure in a chronic pulmonary hypertension model. Am. J. Physiol. Heart Circ. Physiol. 2014, 307, H1204–H1215. [Google Scholar] [CrossRef] [Green Version]
- Pereda, D.; García-Lunar, I.; Sierra, F.; Sánchez-Quintana, D.; Santiago, E.; Ballesteros, C.; Encalada, J.F.; Sánchez-González, J.; Fuster, V.; Ibáñez, B.; et al. Magnetic Resonance Characterization of Cardiac Adaptation and Myocardial Fibrosis in Pulmonary Hypertension Secondary to Systemic-To-Pulmonary Shunt. Circ. Cardiovasc. Imaging 2016, 9, e004566. [Google Scholar] [CrossRef]
- Aguero, J.; Ishikawa, K.; Fish, K.M.; Hammoudi, N.; Hadri, L.; Garcia-Alvarez, A.; Ibanez, B.; Fuster, V.; Hajjar, R.J.; Leopold, J.A. Combination Proximal Pulmonary Artery Coiling and Distal Embolization Induces Chronic Elevations in Pulmonary Artery Pressure in Swine. PLoS ONE 2015, 10, e0124526. [Google Scholar] [CrossRef]
- Tashiro, J.; Rubio, G.A.; Limper, A.H.; Williams, K.; Elliot, S.J.; Ninou, I.; Aidinis, V.; Tzouvelekis, A.; Glassberg, M.K. Exploring Animal Models That Resemble Idiopathic Pulmonary Fibrosis. Front Med. (Lausanne) 2017, 4, 118. [Google Scholar] [CrossRef]
- Organ, L.; Bacci, B.; Koumoundouros, E.; Barcham, G.; Kimpton, W.; Nowell, C.J.; Samuel, C.; Snibson, K. A novel segmental challenge model for bleomycin-induced pulmonary fibrosis in sheep. Exp. Lung Res. 2015, 41, 115–134. [Google Scholar] [CrossRef]
- Crystal, R.G. Adenovirus: The first effective in vivo gene delivery vector. Hum. Gene Ther. 2014, 25, 3–11. [Google Scholar] [CrossRef] [PubMed]
- Rosenfeld, M.A.; Yoshimura, K.; Trapnell, B.C.; Yoneyama, K.; Rosenthal, E.R.; Dalemans, W.; Fukayama, M.; Bargon, J.; Stier, L.E.; Stratford-Perricaudet, L.; et al. In vivo transfer of the human cystic fibrosis transmembrane conductance regulator gene to the airway epithelium. Cell 1992, 68, 143–155. [Google Scholar] [CrossRef]
- Bout, A.; Perricaudet, M.; Baskin, G.; Imler, J.-L.; Scholte, B.J.; Pavirani, A.; Valerio, D. Lung Gene Therapy: In Vivo Adenovirus-Mediated Gene Transfer to Rhesus Monkey Airway Epithelium. Hum. Gene Ther. 1994, 5, 3–10. [Google Scholar] [CrossRef] [PubMed]
- Flotte, T.R.; Afione, S.A.; Conrad, C.; McGrath, S.A.; Solow, R.; Oka, H.; Zeitlin, P.L.; Guggino, W.B.; Carter, B.J. Stable in vivo expression of the cystic fibrosis transmembrane conductance regulator with an adeno-associated virus vector. Proc. Natl. Acad. Sci. USA 1993, 90, 10613–10617. [Google Scholar] [CrossRef] [PubMed]
- Conrad, C.K.; Allen, S.S.; Afione, S.A.; Reynolds, T.C.; Beck, S.E.; Fee-Maki, M.; Barrazza-Ortiz, X.; Adams, R.F.B.A.; Askin, F.B.; Carter, B.J.; et al. Safety of single-dose administration of an adeno-associated virus (AAV)-CFTR vector in the primate lung. Gene Ther. 1996, 3, 658–668. [Google Scholar] [PubMed]
- Wagner, J.A.; Reynolds, T.; Moran, M.L.; Moss, R.B.; Wine, J.J.; Flotte, T.R.; Gardner, P. Efficient and persistent gene transfer of AAV-CFTR in maxillary sinus. Lancet 1998, 351, 1702–1703. [Google Scholar] [CrossRef]
- Wagner, J.A.; Nepomuceno, I.B.; Messner, A.H.; Moran, M.L.; Batson, E.P.; Dimiceli, S.; Brown, B.W.; Desch, J.K.; Norbash, A.M.; Conrad, C.K.; et al. A Phase II, Double-Blind, Randomized, Placebo-Controlled Clinical Trial of tgAAVCF Using Maxillary Sinus Delivery in Patients with Cystic Fibrosis with Antrostomies. Hum. Gene. Ther. 2002, 13, 1349–1359. [Google Scholar] [CrossRef]
- McClain, L.E.; Davey, M.G.; Zoltick, P.W.; Limberis, M.P.; Flake, A.W.; Peranteau, W.H. Vector serotype screening for use in ovine perinatal lung gene therapy. J. Pediatr. Surg. 2016, 51, 879–884. [Google Scholar] [CrossRef] [Green Version]
- Fischer, A.C.; Smith, C.I.; Cebotaru, L.; Zhang, X.; Askin, F.B.; Wright, J.; Guggino, S.E.; Adams, R.J.; Flotte, T.; Guggino, W.B. Expression of a Truncated Cystic Fibrosis Transmembrane Conductance Regulator with an AAV5-pseudotyped Vector in Primates. Mol. Ther. 2007, 15, 756–763. [Google Scholar] [CrossRef]
- Steines, B.; Dickey, D.D.; Bergen, J.; Excoffon, K.J.D.A.; Weinstein, J.R.; Li, X.; Yan, Z.; Abou Alaiwa, M.H.; Shah, V.S.; Bouzek, D.C.; et al. CFTR gene transfer with AAV improves early cystic fibrosis pig phenotypes. JCI Insight 2016, 1, e88728. [Google Scholar] [CrossRef]
- Liu, X.; Luo, M.; Trygg, C.; Yan, Z.; Lei-Butters, D.C.M.; Smith, C.I.; Fischer, A.C.; Munson, K.; Guggino, W.B.; Bunnell, B.A.; et al. Biological Differences in rAAV Transduction of Airway Epithelia in Humans and in Old World Non-human Primates. Mol. Ther. 2007, 15, 2114–2123. [Google Scholar] [CrossRef] [PubMed]
- Hadri, L.; Kratlian, R.G.; Benard, L.; Maron, B.A.; Dorfmüller, P.; Ladage, D.; Guignabert, C.; Ishikawa, K.; Aguero, J.; Ibanez, B.; et al. Therapeutic efficacy of AAV1.SERCA2a in monocrotaline-induced pulmonary arterial hypertension. Circulation 2013, 128, 512–523. [Google Scholar] [CrossRef] [PubMed]
- Aguero, J.; Ishikawa, K.; Hadri, L.; Santos-Gallego, C.G.; Fish, K.M.; Kohlbrenner, E.; Hammoudi, N.; Kho, C.; Lee, A.; Ibáñez, B.; et al. Intratracheal Gene Delivery of SERCA2a Ameliorates Chronic Post-Capillary Pulmonary Hypertension: A Large Animal Model. J. Am. Coll. Cardiol. 2016, 67, 2032–2046. [Google Scholar] [CrossRef] [PubMed]
- Halbert, C.L.; Rutledge, E.A.; Allen, J.M.; Russell, D.W.; Miller, A.D. Repeat Transduction in the Mouse Lung by Using Adeno-Associated Virus Vectors with Different Serotypes. J. Virol. 2000, 74, 1524–1532. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wallace, P.M.; Rodgers, J.N.; Leytze, G.M.; Johnson, J.S.; Linsley, P.S. Induction and reversal of long-lived specific unresponsiveness to a T-dependent antigen following CTLA4Ig treatment. J. Immunol. 1995, 154, 5885–5895. [Google Scholar] [PubMed]
- Sansom, D.M. CD28, CTLA-4 and their ligands: Who does what and to whom? Immunology 2000, 101, 169–177. [Google Scholar] [CrossRef] [PubMed]
- Foy, T.M.; Aruffo, A.; Bajorath, J.; Buhlmann, J.E.; Noelle, R.J. Immune Regulation by CD40 and its Ligand GP39. Annu. Rev. Immunol. 1996, 14, 591–617. [Google Scholar] [CrossRef]
- Auricchio, A.; O’Connor, E.; Weiner, D.; Gao, G.-P.; Hildinger, M.; Wang, L.; Calcedo, R.; Wilson, J.M. Noninvasive gene transfer to the lung for systemic delivery of therapeutic proteins. J. Clin. Investig. 2002, 110, 499–504. [Google Scholar] [CrossRef] [Green Version]
- Sumner-Jones, S.G.; Gill, D.R.; Hyde, S.C. Lack of Repeat Transduction by Recombinant Adeno-Associated Virus Type 5/5 Vectors in the Mouse Airway. J. Virol. 2007, 81, 12360–12367. [Google Scholar] [CrossRef] [Green Version]
- Moss, R.B.; Milla, C.; Colombo, J.; Accurso, F.; Zeitlin, P.L.; Clancy, J.P.; Spencer, L.T.; Pilewski, J.; Waltz, D.A.; Dorkin, H.L.; et al. Repeated Aerosolized AAV-CFTR for Treatment of Cystic Fibrosis: A Randomized Placebo-Controlled Phase 2B Trial. Hum. Gene Ther. 2007, 18, 726–732. [Google Scholar] [CrossRef]
- Kobinger, G.P.; Weiner, D.J.; Yu, Q.-C.; Wilson, J.M. Filovirus-pseudotyped lentiviral vector can efficiently and stably transduce airway epithelia in vivo. Nat. Biotechnol. 2001, 19, 225. [Google Scholar] [CrossRef] [PubMed]
- Stocker, A.G.; Kremer, K.L.; Koldej, R.; Miller, D.S.; Anson, D.S.; Parsons, D.W. Single-dose lentiviral gene transfer for lifetime airway gene expression. J. Gene Med. 2009, 11, 861–867. [Google Scholar] [CrossRef] [PubMed]




© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Katz, M.G.; Fargnoli, A.S.; Gubara, S.M.; Fish, K.; Weber, T.; Bridges, C.R.; Hajjar, R.J.; Ishikawa, K. Targeted Gene Delivery through the Respiratory System: Rationale for Intratracheal Gene Transfer. J. Cardiovasc. Dev. Dis. 2019, 6, 8. https://0-doi-org.brum.beds.ac.uk/10.3390/jcdd6010008
Katz MG, Fargnoli AS, Gubara SM, Fish K, Weber T, Bridges CR, Hajjar RJ, Ishikawa K. Targeted Gene Delivery through the Respiratory System: Rationale for Intratracheal Gene Transfer. Journal of Cardiovascular Development and Disease. 2019; 6(1):8. https://0-doi-org.brum.beds.ac.uk/10.3390/jcdd6010008
Chicago/Turabian StyleKatz, Michael G., Anthony S. Fargnoli, Sarah M. Gubara, Kenneth Fish, Thomas Weber, Charles R. Bridges, Roger J. Hajjar, and Kiyotake Ishikawa. 2019. "Targeted Gene Delivery through the Respiratory System: Rationale for Intratracheal Gene Transfer" Journal of Cardiovascular Development and Disease 6, no. 1: 8. https://0-doi-org.brum.beds.ac.uk/10.3390/jcdd6010008