
Left Ventricular Noncompaction in Children: The Role of Genetics, Morphology, and Function for Outcome

 , ,
, , {kind=link}
{kind=link}
Abstract
:1. Introduction
2. Phenotype and Definition
3. Pathogenesis and Genetics
3.1. Developmental Aspects
3.2. Molecular Genetics
3.3. Functional Models
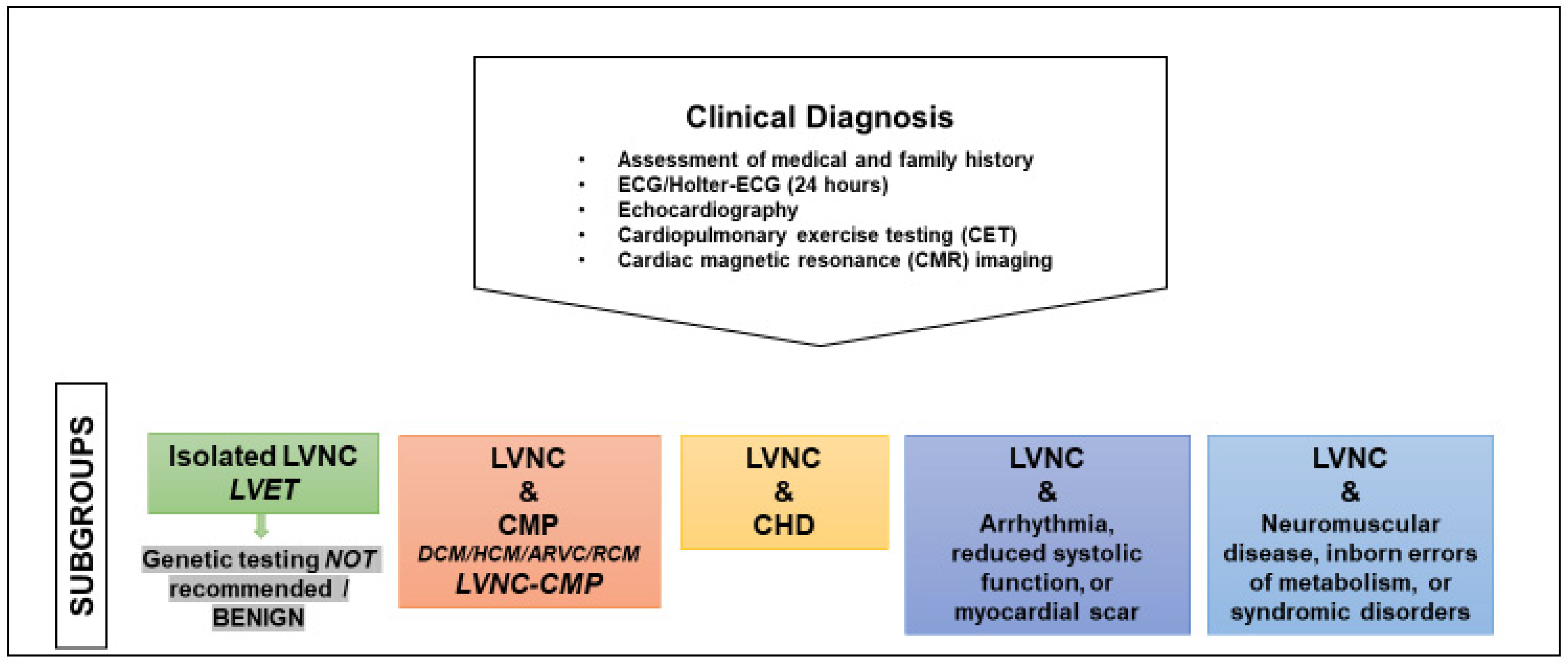
4. Diagnosis
4.1. How to Reach and Confirm a Diagnosis
- -
- Assessment of medical and family history
- -
- Electrocardiography (ECG)
- -
- Echocardiography with functional assessment of LV size and function
- -
- Holter-ECG (24 h)
- -
- Cardiopulmonary exercise testing (CET)
- -
- Cardiac magnetic resonance (CMR) imaging
- -
- Genetic testing
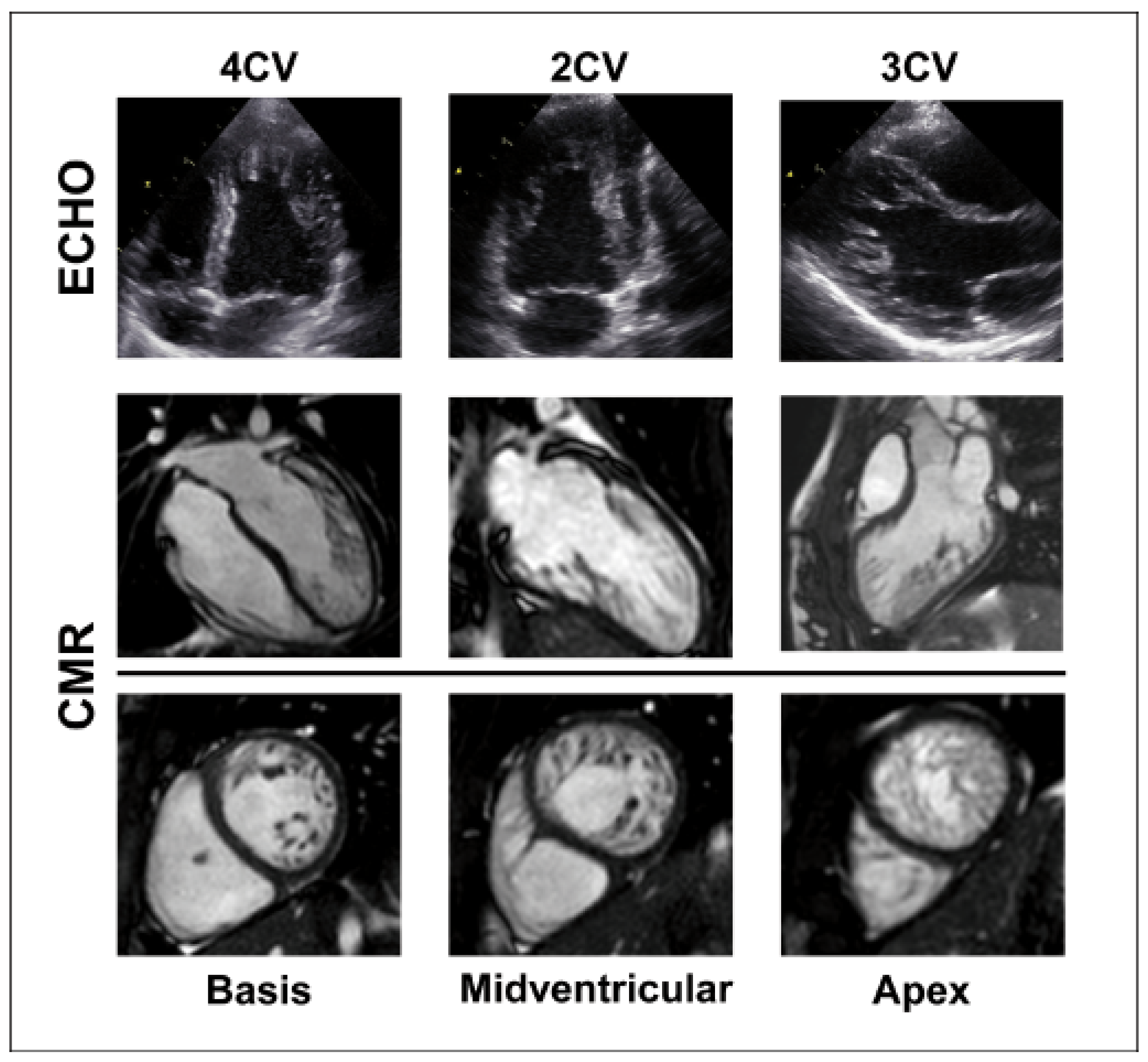
4.2. The Importance of Imaging Modalities
4.3. Indication for Genetic Testing
5. Prognosis and Outcome
5.1. Adult and Paediatric LVNC Outcome
5.2. Genetic Variants and Risk of MACE
5.3. Paediatric LVNC Subtypes
6. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Lee, T.M.; Hsu, D.T.; Kantor, P.; Towbin, J.A.; Ware, S.M.; Colan, S.D.; Chung, W.K.; Jefferies, J.L.; Rossano, J.W.; Castleberry, C.D.; et al. Pediatric Cardiomyopathies. Circ. Res. 2017, 121, 855–873. [Google Scholar] [CrossRef] [PubMed]
- Maron, B.J.; Towbin, J.A.; Thiene, G.; Antzelevitch, C.; Corrado, D.; Arnett, D.; Moss, A.J.; Seidman, C.E.; Young, J.B. Contemporary definitions and classification of the cardiomyopathies: An American Heart Association Scientific Statement from the Council on Clinical Cardiology, Heart Failure and Transplantation Committee; Quality of Care and Outcomes Research and Functional Genomics and Translational Biology Interdisciplinary Working Groups; and Council on Epidemiology and Prevention. Circulation 2006, 113, 1807–1816. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elliott, P.; Andersson, B.; Arbustini, E.; Bilinska, Z.; Cecchi, F.; Charron, P.; Dubourg, O.; Kuhl, U.; Maisch, B.; McKenna, W.J.; et al. Classification of the cardiomyopathies: A position statement from the European Society of Cardiology Working Group on Myocardial and Pericardial Diseases. Eur. Heart J. 2008, 29, 270–276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arbustini, E.; Favalli, V.; Narula, N.; Serio, A.; Grasso, M. Left Ventricular Noncompaction: A Distinct Genetic Cardiomyopathy? J. Am. Coll. Cardiol. 2016, 68, 949–966. [Google Scholar] [CrossRef]
- Anderson, R.H.; Jensen, B.; Mohun, T.J.; Petersen, S.E.; Aung, N.; Zemrak, F.; Planken, R.N.; MacIver, D.H. Key Questions Relating to Left Ventricular Noncompaction Cardiomyopathy: Is the Emperor Still Wearing Any Clothes? Can. J. Cardiol. 2017, 33, 747–757. [Google Scholar] [CrossRef]
- Postma, A.V.; van Engelen, K.; van de Meerakker, J.; Rahman, T.; Probst, S.; Baars, M.J.; Bauer, U.; Pickardt, T.; Sperling, S.R.; Berger, F.; et al. Mutations in the sarcomere gene MYH7 in Ebstein anomaly. Circ. Cardiovasc. Genet. 2011, 4, 43–50. [Google Scholar] [CrossRef] [Green Version]
- Klaassen, S.; Probst, S.; Oechslin, E.; Gerull, B.; Krings, G.; Schuler, P.; Greutmann, M.; Hurlimann, D.; Yegitbasi, M.; Pons, L.; et al. Mutations in sarcomere protein genes in left ventricular noncompaction. Circulation 2008, 117, 2893–2901. [Google Scholar] [CrossRef]
- Towbin, J.A.; Lorts, A.; Jefferies, J.L. Left ventricular non-compaction cardiomyopathy. Lancet 2015, 386, 813–825. [Google Scholar] [CrossRef]
- Arndt, A.-K.; Schafer, S.; Drenckhahn, J.D.; Sabeh, M.K.; Plovie, E.R.; Caliebe, A.; Klopocki, E.; Musso, G.; Werdich, A.A.; Kalwa, H.; et al. Fine mapping of the 1p36 deletion syndrome identifies mutation of PRDM16 as a cause of cardiomyopathy. Am. J. Hum. Genet. 2013, 93, 67–77. [Google Scholar] [CrossRef] [Green Version]
- Bleyl, S.B.; Mumford, B.R.; Thompson, V.; Carey, J.C.; Pysher, T.J.; Chin, T.K.; Ward, K. Neonatal, lethal noncompaction of the left ventricular myocardium is allelic with Barth syndrome. Am. J. Hum. Genet. 1997, 61, 868–872. [Google Scholar] [CrossRef] [Green Version]
- Gerecke, B.J.; Engberding, R. Noncompaction Cardiomyopathy-History and Current Knowledge for Clinical Practice. J. Clin. Med. 2021, 10, 2457. [Google Scholar] [CrossRef] [PubMed]
- Jenni, R.; Oechslin, E.; Schneider, J.; Attenhofer Jost, C.; Kaufmann, P.A. Echocardiographic and pathoanatomical characteristics of isolated left ventricular non-compaction: A step towards classification as a distinct cardiomyopathy. Heart 2001, 86, 666–671. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gati, S.; Rajani, R.; Carr-White, G.S.; Chambers, J.B. Adult left ventricular noncompaction: Reappraisal of current diagnostic imaging modalities. JACC Cardiovasc. Imaging 2014, 7, 1266–1275. [Google Scholar] [CrossRef] [Green Version]
- Petersen, S.E.; Selvanayagam, J.B.; Wiesmann, F.; Robson, M.D.; Francis, J.M.; Anderson, R.H.; Watkins, H.; Neubauer, S. Left ventricular non-compaction: Insights from cardiovascular magnetic resonance imaging. J. Am. Coll. Cardiol. 2005, 46, 101–105. [Google Scholar] [CrossRef] [Green Version]
- Gebhard, C.; Stahli, B.E.; Greutmann, M.; Biaggi, P.; Jenni, R.; Tanner, F.C. Reduced left ventricular compacta thickness: A novel echocardiographic criterion for non-compaction cardiomyopathy. J. Am. Soc. Echocardiogr. 2012, 25, 1050–1057. [Google Scholar] [CrossRef]
- Stollberger, C.; Gerecke, B.; Engberding, R.; Grabner, B.; Wandaller, C.; Finsterer, J.; Gietzelt, M.; Balzereit, A. Interobserver Agreement of the Echocardiographic Diagnosis of LV Hypertrabeculation/Noncompaction. JACC Cardiovasc. Imaging 2015, 8, 1252–1257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chin, T.K.; Perloff, J.K.; Williams, R.G.; Jue, K.; Mohrmann, R. Isolated noncompaction of left ventricular myocardium. A study of eight cases. Circulation 1990, 82, 507–513. [Google Scholar] [CrossRef] [Green Version]
- Grego-Bessa, J.; Luna-Zurita, L.; del Monte, G.; Bolos, V.; Melgar, P.; Arandilla, A.; Garratt, A.N.; Zang, H.; Mukouyama, Y.S.; Chen, H.; et al. Notch signaling is essential for ventricular chamber development. Dev. Cell 2007, 12, 415–429. [Google Scholar] [CrossRef] [Green Version]
- Misra, C.; Garg, V. Compacting the heart with Notch. Nat. Med. 2013, 19, 133–134. [Google Scholar] [CrossRef]
- Rojanasopondist, P.; Nesheiwat, L.; Piombo, S.; Porter, G.A., Jr.; Ren, M.; Phoon, C.K.L. Genetic Basis of Left Ventricular Noncompaction. Circ. Genom. Precis. Med. 2022, 15, e003517. [Google Scholar] [CrossRef]
- Jensen, B.; van der Wal, A.C.; Moorman, A.F.M.; Christoffels, V.M. Excessive trabeculations in noncompaction do not have the embryonic identity. Int. J. Cardiol. 2017, 227, 325–330. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Faber, J.W.; D’Silva, A.; Christoffels, V.M.; Jensen, B. Lack of morphometric evidence for ventricular compaction in humans. J. Cardiol. 2021, 78, 397–405. [Google Scholar] [CrossRef] [PubMed]
- Riekerk, H.C.E.; Coolen, B.F.; Strijkers, G.J.; van der Wal, A.C.; Petersen, S.E.; Sheppard, M.N.; Oostra, R.J.; Christoffels, V.M.; Jensen, B. Higher spatial resolution improves the interpretation of the extent of ventricular trabeculation. J Anat. 2021, 240, 357–375. [Google Scholar] [CrossRef] [PubMed]
- Meyer, H.V.; Dawes, T.J.W.; Serrani, M.; Bai, W.; Tokarczuk, P.; Cai, J.; de Marvao, A.; Henry, A.; Lumbers, R.T.; Gierten, J.; et al. Genetic and functional insights into the fractal structure of the heart. Nature 2020, 584, 589–594. [Google Scholar] [CrossRef]
- Hershberger, R.E.; Givertz, M.M.; Ho, C.Y.; Judge, D.P.; Kantor, P.F.; McBride, K.L.; Morales, A.; Taylor, M.R.G.; Vatta, M.; Ware, S.M. Genetic Evaluation of Cardiomyopathy-A Heart Failure Society of America Practice Guideline. J. Card. Fail. 2018, 24, 281–302. [Google Scholar] [CrossRef] [Green Version]
- Oechslin, E.; Jenni, R.; Klaassen, S. Left Ventricular Noncompaction Is a Myocardial Phenotype: Cardiomyopathy—Yes or No? Can. J. Cardiol. 2021, 37, 366–369. [Google Scholar] [CrossRef]
- Sasse-Klaassen, S.; Probst, S.; Gerull, B.; Oechslin, E.; Nurnberg, P.; Heuser, A.; Jenni, R.; Hennies, H.C.; Thierfelder, L. Novel gene locus for autosomal dominant left ventricular noncompaction maps to chromosome 11p15. Circulation 2004, 109, 2720–2723. [Google Scholar] [CrossRef] [Green Version]
- Hoedemaekers, Y.M.; Caliskan, K.; Michels, M.; Frohn-Mulder, I.; van der Smagt, J.J.; Phefferkorn, J.E.; Wessels, M.W.; ten Cate, F.J.; Sijbrands, E.J.; Dooijes, D.; et al. The importance of genetic counseling, DNA diagnostics, and cardiologic family screening in left ventricular noncompaction cardiomyopathy. Circ. Cardiovasc. Genet. 2010, 3, 232–239. [Google Scholar] [CrossRef] [Green Version]
- Kuhnisch, J.; Herbst, C.; Al-Wakeel-Marquard, N.; Dartsch, J.; Holtgrewe, M.; Baban, A.; Mearini, G.; Hardt, J.; Kolokotronis, K.; Gerull, B.; et al. Targeted panel sequencing in pediatric primary cardiomyopathy supports a critical role of TNNI3. Clin. Genet. 2019, 96, 549–559. [Google Scholar] [CrossRef] [Green Version]
- Wang, C.; Hata, Y.; Hirono, K.; Takasaki, A.; Ozawa, S.W.; Nakaoka, H.; Saito, K.; Miyao, N.; Okabe, M.; Ibuki, K.; et al. A Wide and Specific Spectrum of Genetic Variants and Genotype-Phenotype Correlations Revealed by Next-Generation Sequencing in Patients with Left Ventricular Noncompaction. J. Am. Heart Assoc. 2017, 6, e006210. [Google Scholar] [CrossRef]
- Kolokotronis, K.; Kuhnisch, J.; Klopocki, E.; Dartsch, J.; Rost, S.; Huculak, C.; Mearini, G.; Stork, S.; Carrier, L.; Klaassen, S.; et al. Biallelic mutation in MYH7 and MYBPC3 leads to severe cardiomyopathy with left ventricular noncompaction phenotype. Hum. Mutat. 2019, 40, 1101–1114. [Google Scholar] [CrossRef] [PubMed]
- Van Waning, J.I.; Moesker, J.; Heijsman, D.; Boersma, E.; Majoor-Krakauer, D. Systematic Review of Genotype-Phenotype Correlations in Noncompaction Cardiomyopathy. J. Am. Heart Assoc. 2019, 8, e012993. [Google Scholar] [CrossRef] [PubMed]
- van Waning, J.I.; Caliskan, K.; Michels, M.; Schinkel, A.F.L.; Hirsch, A.; Dalinghaus, M.; Hoedemaekers, Y.M.; Wessels, M.W.; AS, I.J.; Hofstra, R.M.W.; et al. Cardiac Phenotypes, Genetics, and Risks in Familial Noncompaction Cardiomyopathy. J. Am. Coll. Cardiol. 2019, 73, 1601–1611. [Google Scholar] [CrossRef]
- Probst, S.; Oechslin, E.; Schuler, P.; Greutmann, M.; Boye, P.; Knirsch, W.; Berger, F.; Thierfelder, L.; Jenni, R.; Klaassen, S. Sarcomere gene mutations in isolated left ventricular noncompaction cardiomyopathy do not predict clinical phenotype. Circ. Cardiovasc. Genet. 2011, 4, 367–374. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sedaghat-Hamedani, F.; Haas, J.; Zhu, F.; Geier, C.; Kayvanpour, E.; Liss, M.; Lai, A.; Frese, K.; Pribe-Wolferts, R.; Amr, A.; et al. Clinical genetics and outcome of left ventricular non-compaction cardiomyopathy. Eur. Heart J. 2017, 38, 3449–3460. [Google Scholar] [CrossRef] [PubMed]
- Casas, G.; Limeres, J.; Oristrell, G.; Gutierrez-Garcia, L.; Andreini, D.; Borregan, M.; Larranaga-Moreira, J.M.; Lopez-Sainz, A.; Codina-Sola, M.; Teixido-Tura, G.; et al. Clinical Risk Prediction in Patients with Left Ventricular Myocardial Noncompaction. J. Am. Coll. Cardiol. 2021, 78, 643–662. [Google Scholar] [CrossRef]
- Klaassen, S. Ventricular noncompaction: An update. In Braunwald´s Heart Disease, A Textbook of Cardiovascular Medicine, 8th ed.; Libby, P., Bonow, R.O., Mann, D.L., Zipes, D.P., Eds.; Saunders Elsevier: Philadelphia, PA, USA, 2008. [Google Scholar]
- van Waning, J.I.; Caliskan, K.; Hoedemaekers, Y.M.; van Spaendonck-Zwarts, K.Y.; Baas, A.F.; Boekholdt, S.M.; van Melle, J.P.; Teske, A.J.; Asselbergs, F.W.; Backx, A.; et al. Genetics, Clinical Features, and Long-Term Outcome of Noncompaction Cardiomyopathy. J. Am. Coll. Cardiol. 2018, 71, 711–722. [Google Scholar] [CrossRef]
- Schultze-Berndt, A.; Kuhnisch, J.; Herbst, C.; Seidel, F.; Al-Wakeel-Marquard, N.; Dartsch, J.; Theisen, S.; Knirsch, W.; Jenni, R.; Greutmann, M.; et al. Reduced Systolic Function and Not Genetic Variants Determine Outcome in Pediatric and Adult Left Ventricular Noncompaction Cardiomyopathy. Front. Pediatr. 2021, 9, 722926. [Google Scholar] [CrossRef]
- Mazzarotto, F.; Hawley, M.H.; Beltrami, M.; Beekman, L.; de Marvao, A.; McGurk, K.A.; Statton, B.; Boschi, B.; Girolami, F.; Roberts, A.M.; et al. Systematic large-scale assessment of the genetic architecture of left ventricular noncompaction reveals diverse etiologies. Genet. Med. 2021, 23, 856–864. [Google Scholar] [CrossRef]
- Walsh, R.; Thomson, K.L.; Ware, J.S.; Funke, B.H.; Woodley, J.; McGuire, K.J.; Mazzarotto, F.; Blair, E.; Seller, A.; Taylor, J.C.; et al. Reassessment of Mendelian gene pathogenicity using 7,855 cardiomyopathy cases and 60,706 reference samples. Genet. Med. 2017, 19, 192–203. [Google Scholar] [CrossRef] [Green Version]
- Wilsbacher, L.; McNally, E.M. Genetics of Cardiac Developmental Disorders: Cardiomyocyte Proliferation and Growth and Relevance to Heart Failure. Annu Rev Pathol 2016, 11, 395–419. [Google Scholar] [CrossRef] [PubMed]
- Kodo, K.; Ong, S.G.; Jahanbani, F.; Termglinchan, V.; Hirono, K.; InanlooRahatloo, K.; Ebert, A.D.; Shukla, P.; Abilez, O.J.; Churko, J.M.; et al. iPSC-derived cardiomyocytes reveal abnormal TGF-beta signalling in left ventricular non-compaction cardiomyopathy. Nat. Cell Biol. 2016, 18, 1031–1042. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cibi, D.M.; Bi-Lin, K.W.; Shekeran, S.G.; Sandireddy, R.; Tee, N.; Singh, A.; Wu, Y.; Srinivasan, D.K.; Kovalik, J.P.; Ghosh, S.; et al. Prdm16 Deficiency Leads to Age-Dependent Cardiac Hypertrophy, Adverse Remodeling, Mitochondrial Dysfunction, and Heart Failure. Cell Rep. 2020, 33, 108288. [Google Scholar] [CrossRef] [PubMed]
- Nam, J.M.; Lim, J.E.; Ha, T.W.; Oh, B.; Kang, J.O. Cardiac-specific inactivation of Prdm16 effects cardiac conduction abnormalities and cardiomyopathy-associated phenotypes. Am. J. Physiol. Heart Circ. Physiol. 2020, 318, H764–H777. [Google Scholar] [CrossRef]
- Tian, X.; Li, Y.; He, L.; Zhang, H.; Huang, X.; Liu, Q.; Pu, W.; Zhang, L.; Li, Y.; Zhao, H.; et al. Identification of a hybrid myocardial zone in the mammalian heart after birth. Nat. Commun. 2017, 8, 87. [Google Scholar] [CrossRef] [PubMed]
- Rhee, S.; Chung, J.I.; King, D.A.; D’Amato, G.; Paik, D.T.; Duan, A.; Chang, A.; Nagelberg, D.; Sharma, B.; Jeong, Y.; et al. Endothelial deletion of Ino80 disrupts coronary angiogenesis and causes congenital heart disease. Nat. Commun. 2018, 9, 368. [Google Scholar] [CrossRef] [Green Version]
- Wu, T.; Liang, Z.; Zhang, Z.; Liu, C.; Zhang, L.; Gu, Y.; Peterson, K.L.; Evans, S.M.; Fu, X.D.; Chen, J. PRDM16 Is a Compact Myocardium-Enriched Transcription Factor Required to Maintain Compact Myocardial Cardiomyocyte Identity in Left Ventricle. Circulation 2022, 145, 586–602. [Google Scholar] [CrossRef]
- Gifford, C.A.; Ranade, S.S.; Samarakoon, R.; Salunga, H.T.; de Soysa, T.Y.; Huang, Y.; Zhou, P.; Elfenbein, A.; Wyman, S.K.; Bui, Y.K.; et al. Oligogenic inheritance of a human heart disease involving a genetic modifier. Science 2019, 364, 865–870. [Google Scholar] [CrossRef]
- Miszalski-Jamka, K.; Jefferies, J.L.; Mazur, W.; Glowacki, J.; Hu, J.; Lazar, M.; Gibbs, R.A.; Liczko, J.; Klys, J.; Venner, E.; et al. Novel Genetic Triggers and Genotype-Phenotype Correlations in Patients with Left Ventricular Noncompaction. Circ. Cardiovasc. Genet. 2017, 10, e001763. [Google Scholar] [CrossRef] [Green Version]
- Hershberger, R.E.; Morales, A.; Cowan, J. Is Left Ventricular Noncompaction a Trait, Phenotype, or Disease? The Evidence Points to Phenotype. Circ. Cardiovasc. Genet. 2017, 10, e001968. [Google Scholar] [CrossRef]
- Oechslin, E.; Klaassen, S. Left Ventricular Noncompaction: Phenotype in an Integrated Model of Cardiomyopathy? J. Am. Coll. Cardiol. 2019, 73, 1612–1615. [Google Scholar] [CrossRef] [PubMed]
- Weir-McCall, J.R.; Yeap, P.M.; Papagiorcopulo, C.; Fitzgerald, K.; Gandy, S.J.; Lambert, M.; Belch, J.J.; Cavin, I.; Littleford, R.; Macfarlane, J.A.; et al. Left Ventricular Noncompaction: Anatomical Phenotype or Distinct Cardiomyopathy? J. Am. Coll. Cardiol. 2016, 68, 2157–2165. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andreini, D.; Pontone, G.; Bogaert, J.; Roghi, A.; Barison, A.; Schwitter, J.; Mushtaq, S.; Vovas, G.; Sormani, P.; Aquaro, G.D.; et al. Long-Term Prognostic Value of Cardiac Magnetic Resonance in Left Ventricle Noncompaction: A Prospective Multicenter Study. J. Am. Coll. Cardiol. 2016, 68, 2166–2181. [Google Scholar] [CrossRef] [PubMed]
- Zemrak, F.; Ahlman, M.A.; Captur, G.; Mohiddin, S.A.; Kawel-Boehm, N.; Prince, M.R.; Moon, J.C.; Hundley, W.G.; Lima, J.A.; Bluemke, D.A.; et al. The relationship of left ventricular trabeculation to ventricular function and structure over a 9.5-year follow-up: The MESA study. J. Am. Coll. Cardiol. 2014, 64, 1971–1980. [Google Scholar] [CrossRef] [Green Version]
- Gati, S.; Papadakis, M.; Papamichael, N.D.; Zaidi, A.; Sheikh, N.; Reed, M.; Sharma, R.; Thilaganathan, B.; Sharma, S. Reversible de novo left ventricular trabeculations in pregnant women: Implications for the diagnosis of left ventricular noncompaction in low-risk populations. Circulation 2014, 130, 475–483. [Google Scholar] [CrossRef]
- Gati, S.; Chandra, N.; Bennett, R.L.; Reed, M.; Kervio, G.; Panoulas, V.F.; Ghani, S.; Sheikh, N.; Zaidi, A.; Wilson, M.; et al. Increased left ventricular trabeculation in highly trained athletes: Do we need more stringent criteria for the diagnosis of left ventricular non-compaction in athletes? Heart 2013, 99, 401–408. [Google Scholar] [CrossRef]
- Kohli, S.K.; Pantazis, A.A.; Shah, J.S.; Adeyemi, B.; Jackson, G.; McKenna, W.J.; Sharma, S.; Elliott, P.M. Diagnosis of left-ventricular non-compaction in patients with left-ventricular systolic dysfunction: Time for a reappraisal of diagnostic criteria? Eur. Heart J. 2008, 29, 89–95. [Google Scholar] [CrossRef] [Green Version]
- Al-Wakeel-Marquard, N.; Degener, F.; Herbst, C.; Kuhnisch, J.; Dartsch, J.; Schmitt, B.; Kuehne, T.; Messroghli, D.; Berger, F.; Klaassen, S. RIKADA Study Reveals Risk Factors in Pediatric Primary Cardiomyopathy. J. Am. Heart Assoc. 2019, 8, e012531. [Google Scholar] [CrossRef] [Green Version]
- Negri, F.; De Luca, A.; Fabris, E.; Korcova, R.; Cernetti, C.; Grigoratos, C.; Aquaro, G.D.; Nucifora, G.; Camici, P.G.; Sinagra, G. Left ventricular noncompaction, morphological, and clinical features for an integrated diagnosis. Heart Fail. Rev. 2019, 24, 315–323. [Google Scholar] [CrossRef]
- Nucifora, G.; Aquaro, G.D.; Pingitore, A.; Masci, P.G.; Lombardi, M. Myocardial fibrosis in isolated left ventricular non-compaction and its relation to disease severity. Eur. J. Heart Fail. 2011, 13, 170–176. [Google Scholar] [CrossRef] [Green Version]
- Zhou, H.; Lin, X.; Fang, L.; Zhao, X.; Ding, H.; Chen, W.; Xu, R.; Bai, X.; Wang, Y.; Fang, Q. Characterization of Compacted Myocardial Abnormalities by Cardiac Magnetic Resonance with Native T1 Mapping in Left Ventricular Non-Compaction Patients- A Comparison with Late Gadolinium Enhancement. Circ. J. 2016, 80, 1210–1216. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Al-Wakeel-Marquard, N.; Seidel, F.; Herbst, C.; Kuhnisch, J.; Kuehne, T.; Berger, F.; Klaassen, S.; Messroghli, D.R. Diffuse myocardial fibrosis by T1 mapping is associated with heart failure in pediatric primary dilated cardiomyopathy. Int. J. Cardiol. 2021, 333, 219–225. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Yang, F.; Wan, K.; Mui, D.; Han, Y.; Chen, Y. Left ventricular midwall fibrosis as a predictor of sudden cardiac death in non-ischaemic dilated cardiomyopathy: A meta-analysis. ESC Heart Fail. 2020, 7, 2184–2192. [Google Scholar] [CrossRef] [PubMed]
- Al-Wakeel-Marquard, N.; Seidel, F.; Kuhnisch, J.; Kuehne, T.; Berger, F.; Messroghli, D.R.; Klaassen, S. Midwall Fibrosis and Cardiac Mechanics: Rigid Body Rotation Is a Novel Marker of Disease Severity in Pediatric Primary Dilated Cardiomyopathy. Front. Cardiovasc. Med. 2021, 8, 810005. [Google Scholar] [CrossRef] [PubMed]
- Anwer, S.; Heiniger, P.S.; Rogler, S.; Erhart, L.; Cassani, D.; Kuzo, N.; Rebellius, L.; Schoenenberger-Berzins, R.; Schmid, D.; Nussbaum, S.; et al. Left ventricular mechanics and cardiovascular outcomes in non-compaction phenotype. Int. J. Cardiol. 2021, 336, 73–80. [Google Scholar] [CrossRef]
- Cai, J.; Bryant, J.A.; Le, T.T.; Su, B.; de Marvao, A.; O’Regan, D.P.; Cook, S.A.; Chin, C.W. Fractal analysis of left ventricular trabeculations is associated with impaired myocardial deformation in healthy Chinese. J. Cardiovasc. Magn. Reson. 2017, 19, 102. [Google Scholar] [CrossRef] [Green Version]
- Nucifora, G.; Sree Raman, K.; Muser, D.; Shah, R.; Perry, R.; Awang Ramli, K.A.; Selvanayagam, J.B. Cardiac magnetic resonance evaluation of left ventricular functional, morphological, and structural features in children and adolescents vs. young adults with isolated left ventricular non-compaction. Int. J. Cardiol. 2017, 246, 68–73. [Google Scholar] [CrossRef]
- Sarnecki, J.; Paszkowska, A.; Petryka-Mazurkiewicz, J.; Kubik, A.; Feber, J.; Jurkiewicz, E.; Ziolkowska, L. Left and Right Ventricular Morphology, Function and Myocardial Deformation in Children with Left Ventricular Non-Compaction Cardiomyopathy: A Case-Control Cardiovascular Magnetic Resonance Study. J. Clin. Med. 2022, 11, 1104. [Google Scholar] [CrossRef]
- Guigui, S.A.; Horvath, S.A.; Arenas, I.A.; Mihos, C.G. Cardiac geometry, function and mechanics in left ventricular non-compaction cardiomyopathy with preserved ejection fraction. J. Echocardiogr. 2022, 1–7. [Google Scholar] [CrossRef]
- Sabatino, J.; Di Salvo, G.; Krupickova, S.; Fraisse, A.; Prota, C.; Bucciarelli, V.; Josen, M.; Paredes, J.; Sirico, D.; Voges, I.; et al. Left Ventricular Twist Mechanics to Identify Left Ventricular Noncompaction in Childhood. Circ. Cardiovasc. Imaging 2019, 12, e007805. [Google Scholar] [CrossRef] [Green Version]
- Hershberger, R.E.; Givertz, M.M.; Ho, C.Y.; Judge, D.P.; Kantor, P.F.; McBride, K.L.; Morales, A.; Taylor, M.R.G.; Vatta, M.; Ware, S.M.; et al. Genetic evaluation of cardiomyopathy: A clinical practice resource of the American College of Medical Genetics and Genomics (ACMG). Genet. Med. 2018, 20, 899–909. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Musunuru, K.; Hershberger, R.E.; Day, S.M.; Klinedinst, N.J.; Landstrom, A.P.; Parikh, V.N.; Prakash, S.; Semsarian, C.; Sturm, A.C. Genetic Testing for Inherited Cardiovascular Diseases: A Scientific Statement from the American Heart Association. Circ. Genom. Precis. Med. 2020, 13, e000067. [Google Scholar] [CrossRef]
- Ross, S.B.; Jones, K.; Blanch, B.; Puranik, R.; McGeechan, K.; Barratt, A.; Semsarian, C. A systematic review and meta-analysis of the prevalence of left ventricular non-compaction in adults. Eur. Heart J. 2020, 41, 1428–1436. [Google Scholar] [CrossRef]
- Oechslin, E.N.; Attenhofer Jost, C.H.; Rojas, J.R.; Kaufmann, P.A.; Jenni, R. Long-term follow-up of 34 adults with isolated left ventricular noncompaction: A distinct cardiomyopathy with poor prognosis. J. Am. Coll. Cardiol. 2000, 36, 493–500. [Google Scholar] [CrossRef] [Green Version]
- Vaidya, V.R.; Lyle, M.; Miranda, W.R.; Farwati, M.; Isath, A.; Patlolla, S.H.; Hodge, D.O.; Asirvatham, S.J.; Kapa, S.; Deshmukh, A.J.; et al. Long-Term Survival of Patients with Left Ventricular Noncompaction. J. Am. Heart Assoc. 2021, 10, e015563. [Google Scholar] [CrossRef]
- Aung, N.; Doimo, S.; Ricci, F.; Sanghvi, M.M.; Pedrosa, C.; Woodbridge, S.P.; Al-Balah, A.; Zemrak, F.; Khanji, M.Y.; Munroe, P.B.; et al. Prognostic Significance of Left Ventricular Noncompaction: Systematic Review and Meta-Analysis of Observational Studies. Circ. Cardiovasc. Imaging 2020, 13, e009712. [Google Scholar] [CrossRef]
- Brescia, S.T.; Rossano, J.W.; Pignatelli, R.; Jefferies, J.L.; Price, J.F.; Decker, J.A.; Denfield, S.W.; Dreyer, W.J.; Smith, O.; Towbin, J.A.; et al. Mortality and sudden death in pediatric left ventricular noncompaction in a tertiary referral center. Circulation 2013, 127, 2202–2208. [Google Scholar] [CrossRef] [Green Version]
- Jefferies, J.L.; Wilkinson, J.D.; Sleeper, L.A.; Colan, S.D.; Lu, M.; Pahl, E.; Kantor, P.F.; Everitt, M.D.; Webber, S.A.; Kaufman, B.D.; et al. Cardiomyopathy Phenotypes and Outcomes for Children with Left Ventricular Myocardial Noncompaction: Results from the Pediatric Cardiomyopathy Registry. J. Card. Fail. 2015, 21, 877–884. [Google Scholar] [CrossRef] [Green Version]
- Miller, E.M.; Hinton, R.B.; Czosek, R.; Lorts, A.; Parrott, A.; Shikany, A.R.; Ittenbach, R.F.; Ware, S.M. Genetic Testing in Pediatric Left Ventricular Noncompaction. Circ. Cardiovasc. Genet. 2017, 10, e001735. [Google Scholar] [CrossRef] [Green Version]
- Ross, S.B.; Singer, E.S.; Driscoll, E.; Nowak, N.; Yeates, L.; Puranik, R.; Sy, R.W.; Rajagopalan, S.; Barratt, A.; Ingles, J.; et al. Genetic architecture of left ventricular noncompaction in adults. Hum. Genome Var. 2020, 7, 33. [Google Scholar] [CrossRef]
- Li, S.; Zhang, C.; Liu, N.; Bai, H.; Hou, C.; Pu, J. Clinical implications of sarcomere and nonsarcomere gene variants in patients with left ventricular noncompaction cardiomyopathy. Mol. Genet. Genom. Med. 2019, 7, e874. [Google Scholar] [CrossRef] [Green Version]
- Li, S.; Zhang, C.; Liu, N.; Bai, H.; Hou, C.; Song, L.; Pu, J. Titin-truncating variants are associated with heart failure events in patients with left ventricular non-compaction cardiomyopathy. Clin. Cardiol. 2019, 42, 530–535. [Google Scholar] [CrossRef]
- Hirono, K.; Hata, Y.; Miyao, N.; Okabe, M.; Takarada, S.; Nakaoka, H.; Ibuki, K.; Ozawa, S.; Origasa, H.; Nishida, N.; et al. Increased Burden of Ion Channel Gene Variants Is Related to Distinct Phenotypes in Pediatric Patients with Left Ventricular Noncompaction. Circ. Genom. Precis. Med. 2020, 13, e002940. [Google Scholar] [CrossRef]
- Saleeb, S.F.; Margossian, R.; Spencer, C.T.; Alexander, M.E.; Smoot, L.B.; Dorfman, A.L.; Bergersen, L.; Gauvreau, K.; Marx, G.R.; Colan, S.D. Reproducibility of echocardiographic diagnosis of left ventricular noncompaction. J. Am. Soc. Echocardiogr. 2012, 25, 194–202. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Klaassen, S.; Kühnisch, J.; Schultze-Berndt, A.; Seidel, F. Left Ventricular Noncompaction in Children: The Role of Genetics, Morphology, and Function for Outcome. J. Cardiovasc. Dev. Dis. 2022, 9, 206. https://0-doi-org.brum.beds.ac.uk/10.3390/jcdd9070206
Klaassen S, Kühnisch J, Schultze-Berndt A, Seidel F. Left Ventricular Noncompaction in Children: The Role of Genetics, Morphology, and Function for Outcome. Journal of Cardiovascular Development and Disease. 2022; 9(7):206. https://0-doi-org.brum.beds.ac.uk/10.3390/jcdd9070206
Chicago/Turabian StyleKlaassen, Sabine, Jirko Kühnisch, Alina Schultze-Berndt, and Franziska Seidel. 2022. "Left Ventricular Noncompaction in Children: The Role of Genetics, Morphology, and Function for Outcome" Journal of Cardiovascular Development and Disease 9, no. 7: 206. https://0-doi-org.brum.beds.ac.uk/10.3390/jcdd9070206