
Functional and Structural Characterization of Nucleic Acid Ligands That Bind to Activated Coagulation Factor XIII

 ,
,  ,
,  , , ,
, , ,
Abstract
:1. Introduction
2. Experimental Section
2.1. Materials
2.2. Formulations of Used Buffers
2.3. Capillary Electrophoresis-Systematic Evolution of Ligands by Exponential Enrichment (CE-SELEX)
2.4. Next Generation Sequencing (NGS)
2.5. Determination of Dissociation Constants
2.6. Determination of the Impact of Aptamer Binding on Isopeptidase and Transglutaminase Activity of FXIIIa
2.7. Determination of the Capturing Capacity of the Aptamers
2.8. Interference of Aptamers on Fibrinogen Binding of FXIIIa and Alpha2-Antiplasmin (α2AP) Incorporation
2.9. Rotational Thromboelastometry Analysis (ROTEM)
2.10. Molecular Modeling of FXIIIa-Aptamer Binding
2.11. Statistical Analysis
3. Results
3.1. Aptamer Selection and Characterisation
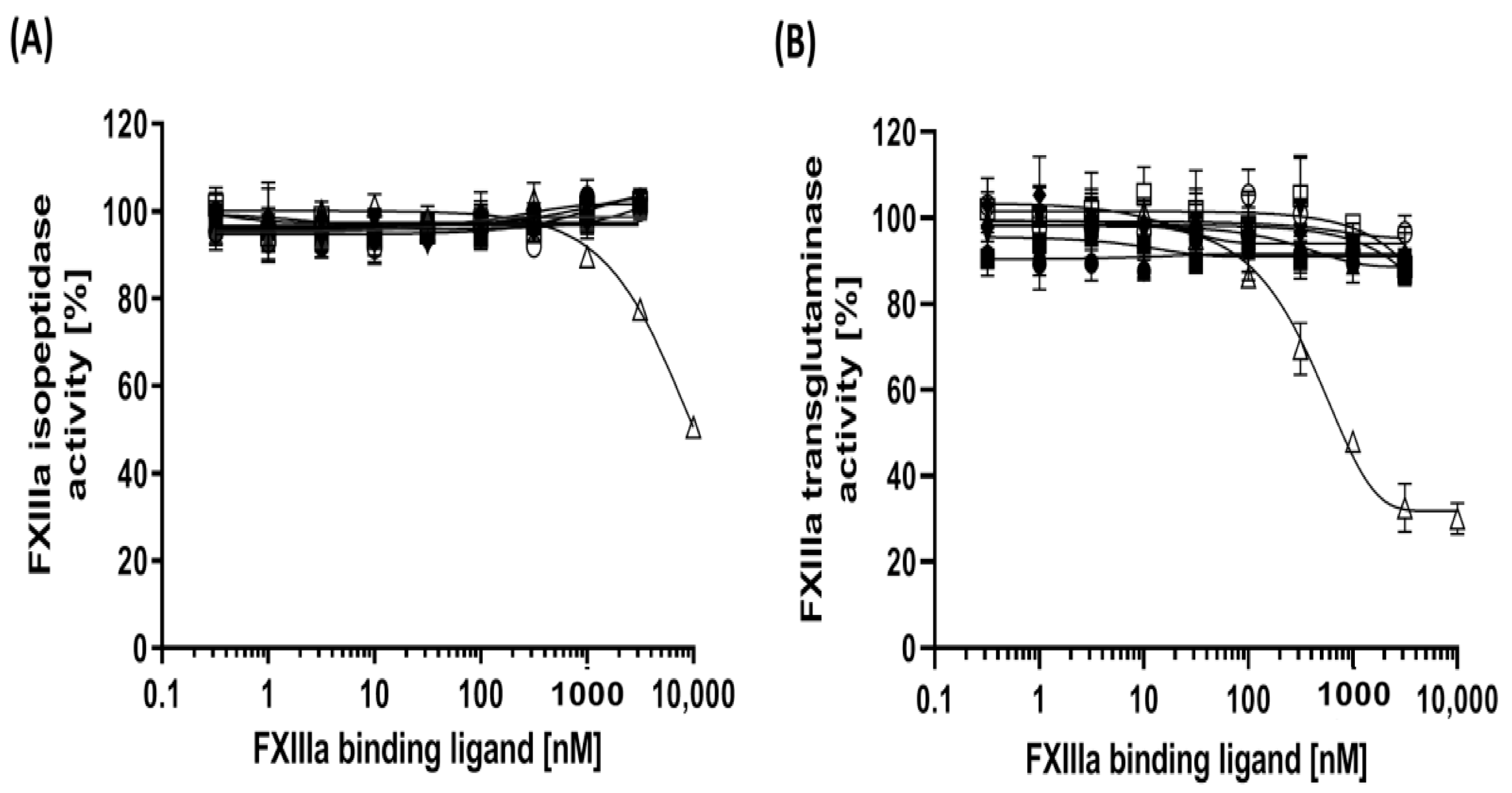
3.2. Impact of Aptamer Binding on Isopeptidase and Transglutaminase Activity of FXIIIa
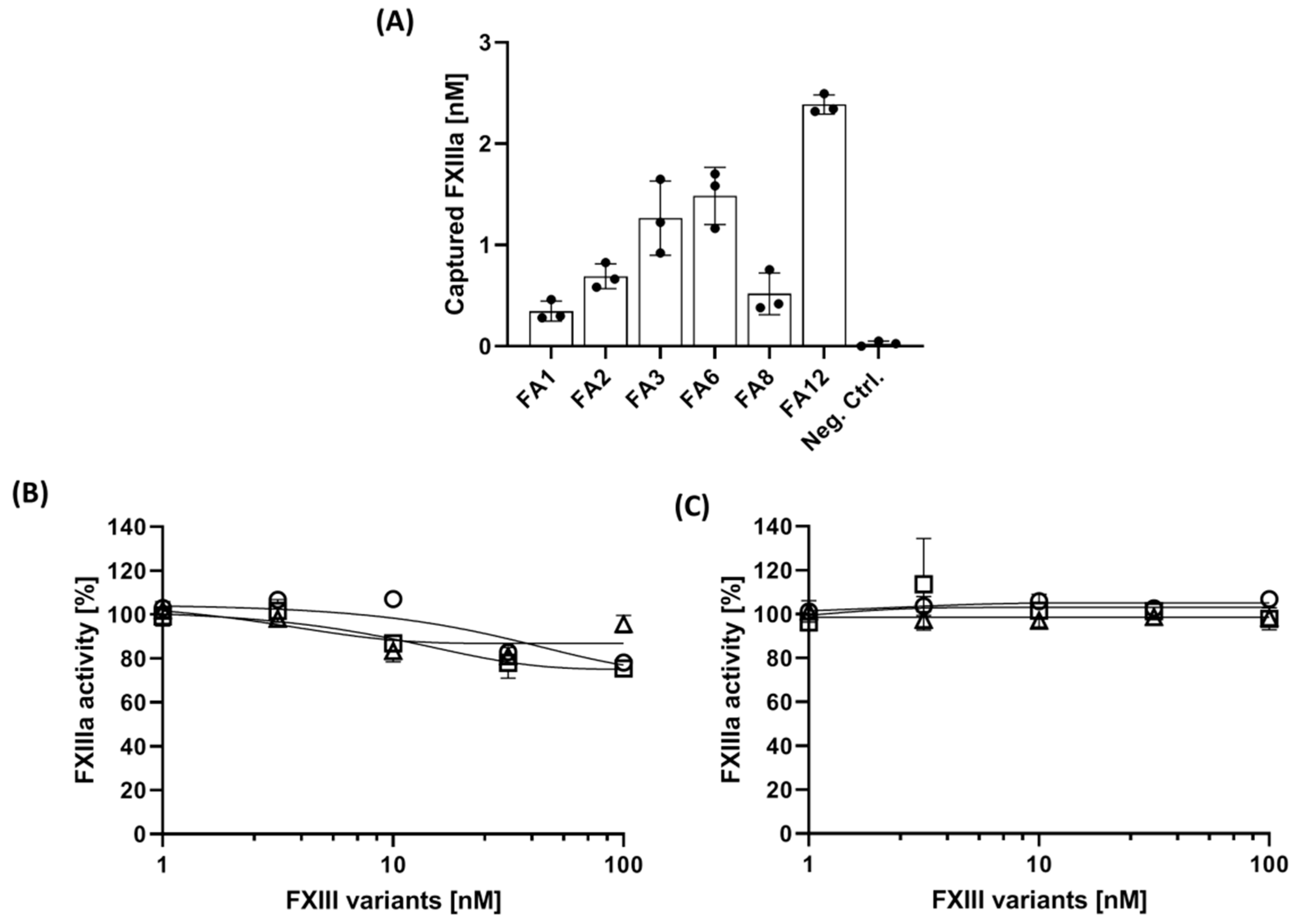
3.3. Determination of Capturing Capacity of Aptamers
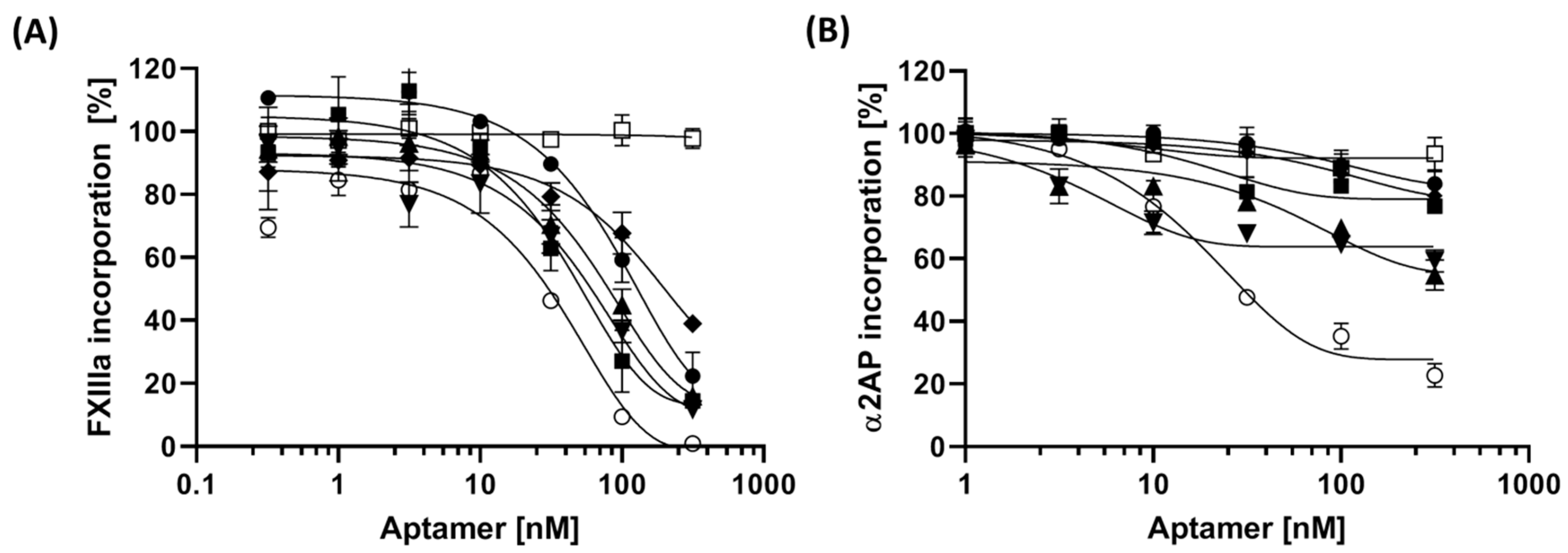
3.4. The Impact of Aptamer on Fibrinogen-Binding of FXIIIa
3.5. Influence of Aptamers on Clot Formation as Assessed by ROTEM-Analysis
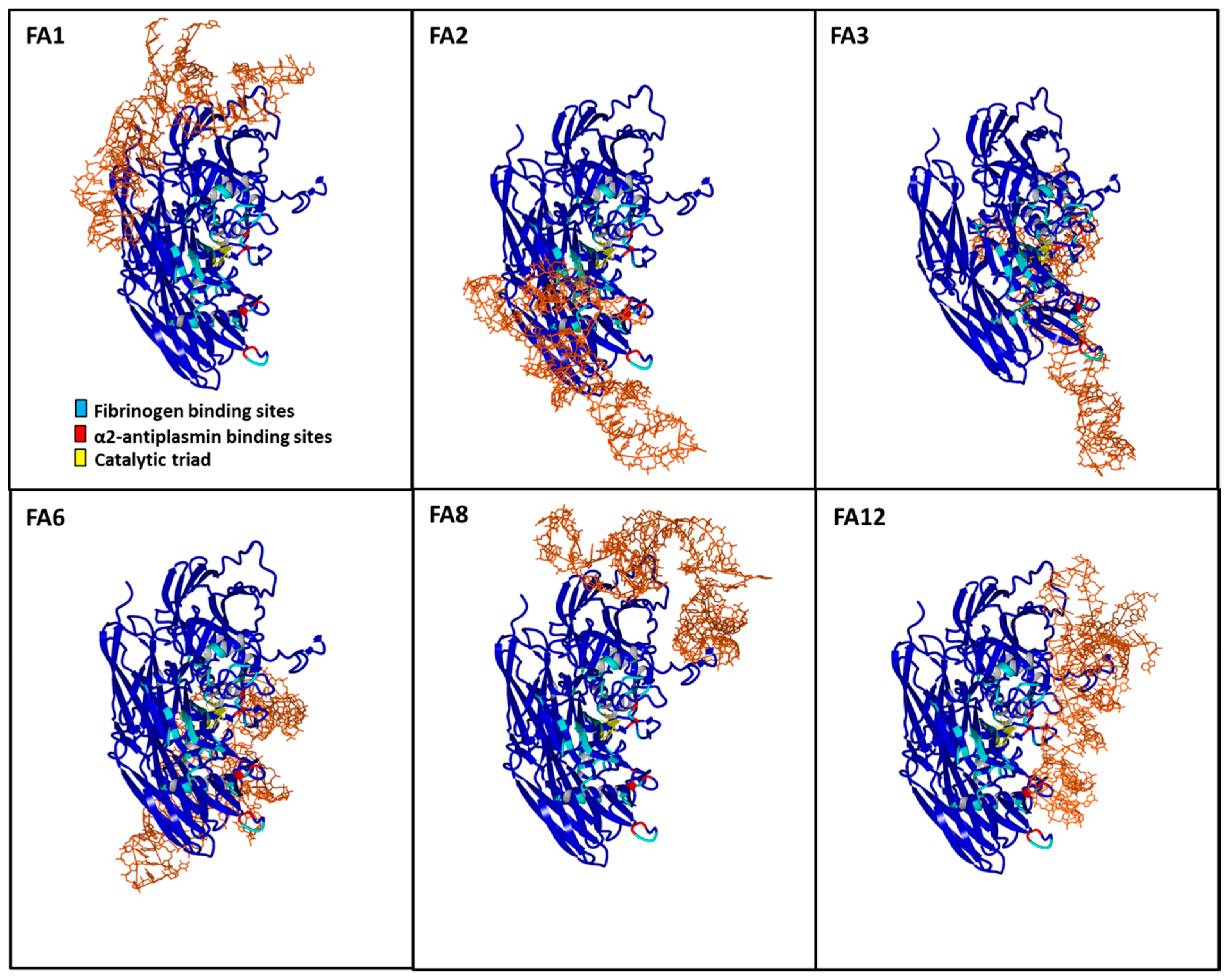
3.6. Aptamer Binding Poses on FXIIIa and Implications for Substrate Access
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Muszbek, L.; Yee, V.C.; Hevessy, Z. Blood coagulation factor XIII: Structure and function. Thromb. Res. 1999, 94, 271–305. [Google Scholar] [CrossRef]
- Lorand, L. Activation of blood coagulation factor XIII. Ann. N. Y. Acad. Sci. 1986, 485, 144–158. [Google Scholar] [CrossRef] [PubMed]
- Greenberg, C.S.; Shuman, M.A. The zymogen forms of blood coagulation factor XIII bind specifically to fibrinogen. J. Biol. Chem. 1982, 257, 6096–6101. [Google Scholar] [CrossRef]
- Procyk, R.; Bishop, P.D.; Kudryk, B. Fibrin—Recombinant human factor XIII a-subunit association. Thromb. Res. 1993, 71, 127–138. [Google Scholar] [CrossRef]
- Standeven, K.F.; Carter, A.M.; Grant, P.J.; Weisel, J.W.; Chernysh, I.; Masova, L.; Lord, S.T.; Ariens, R.A.S. Functional analysis of fibrin {gamma}-chain cross-linking by activated factor XIII: Determination of a cross-linking pattern that maximizes clot stiffness. Blood 2007, 110, 902–907. [Google Scholar] [CrossRef]
- Sakata, Y.; Aoki, N. Cross-linking of alpha 2-plasmin inhibitor to fibrin by fibrin-stabilizing factor. J. Clin. Investig. 1980, 65, 290–297. [Google Scholar] [CrossRef]
- Bereczky, Z.; Balogh, E.; Katona, E.; Czuriga, I.; Edes, I.; Muszbek, L. Elevated factor XIII level and the risk of myocardial infarction in women. Haematologica 2007, 92, 287–288. [Google Scholar] [CrossRef] [Green Version]
- Kalaria, V.G.; Zareba, W.; Moss, A.J.; Pancio, G.; Marder, V.J.; Morrissey, J.H.; Weiss, H.J.; Sparks, C.E.; Greenberg, H.; Dwyer, E.; et al. Gender-related differences in thrombogenic factors predicting recurrent cardiac events in patients after acute myocardial infarction. The THROMBO Investigators. Am. J. Cardiol. 2000, 85, 1401–1408. [Google Scholar] [CrossRef]
- Shemirani, A.H.; Szomjak, E.; Csiki, Z.; Katona, E.; Bereczky, Z.; Muszbek, L. Elevated factor XIII level and the risk of peripheral artery disease. Haematologica 2008, 93, 1430–1432. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Agnelli, G.; Buller, H.R.; Cohen, A.; Curto, M.; Gallus, A.S.; Johnson, M.; Masiukiewicz, U.; Pak, R.; Thompson, J.; Raskob, G.E.; et al. Oral apixaban for the treatment of acute venous thromboembolism. N. Engl. J. Med. 2013, 369, 799–808. [Google Scholar] [CrossRef] [Green Version]
- Büller, H.R.; Décousus, H.; Grosso, M.A.; Mercuri, M.; Middeldorp, S.; Prins, M.H.; Raskob, G.E.; Schellong, S.M.; Schwocho, L.; Segers, A.; et al. Edoxaban versus warfarin for the treatment of symptomatic venous thromboembolism. N. Engl. J. Med. 2013, 369, 1406–1415. [Google Scholar] [CrossRef] [Green Version]
- van Es, N.; Coppens, M.; Schulman, S.; Middeldorp, S.; Büller, H.R. Direct oral anticoagulants compared with vitamin K antagonists for acute venous thromboembolism: Evidence from phase 3 trials. Blood 2014, 124, 1968–1975. [Google Scholar] [CrossRef]
- Steiner, T.; Weitz, J.I.; Veltkamp, R. Anticoagulant-Associated Intracranial Hemorrhage in the Era of Reversal Agents. Stroke 2017, 48, 1432–1437. [Google Scholar] [CrossRef]
- Catani, M.V.; Bernassola, F.; Rossi, A.; Melino, G. Inhibition of clotting factor XIII activity by nitric oxide. Biochem. Biophys. Res. Commun. 1998, 249, 275–278. [Google Scholar] [CrossRef]
- Reinhardt, G. alpha-Halogenmethyl carbonyl compounds as very potent inhibitors of factor XIIIa in vitro. Ann. N. Y. Acad. Sci. 1981, 370, 836–842. [Google Scholar] [CrossRef]
- Lukacova, D.; Matsueda, G.R.; Haber, E.; Reed, G.L. Inhibition of factor XIII activation by an anti-peptide monoclonal antibody. Biochemistry 1991, 30, 10164–10170. [Google Scholar] [CrossRef]
- Reed, G.L.; Houng, A.K. The contribution of activated factor XIII to fibrinolytic resistance in experimental pulmonary embolism. Circulation 1999, 99, 299–304. [Google Scholar] [CrossRef] [Green Version]
- Schmitz, T.; Bauml, C.A.; Imhof, D. Inhibitors of blood coagulation factor XIIIa. Anal. Biochem. 2020, 113708. [Google Scholar] [CrossRef] [PubMed]
- Lorand, L.; Rule, N.G.; Ong, H.H.; Furlanetto, R.; Jacobsen, A.; Downey, J.; Oner, N.; Bruner-Lorand, J. Amine specificity in transpeptidation. Inhibition of fibrin cross-linking. Biochemistry 1968, 7, 1214–1223. [Google Scholar] [CrossRef]
- Muller, J.; Isermann, B.; Ducker, C.; Salehi, M.; Meyer, M.; Friedrich, M.; Madhusudhan, T.; Oldenburg, J.; Mayer, G.; Potzsch, B. An exosite-specific ssDNA aptamer inhibits the anticoagulant functions of activated protein C and enhances inhibition by protein C inhibitor. Chem. Biol. 2009, 16, 442–451. [Google Scholar] [CrossRef] [Green Version]
- Muller, J.; Wulffen, B.; Potzsch, B.; Mayer, G. Multidomain targeting generates a high-affinity thrombin-inhibiting bivalent aptamer. Chembiochem 2007, 8, 2223–2226. [Google Scholar] [CrossRef]
- Hamedani, N.S.; Müller, J.; Tolle, F.; Rühl, H.; Pezeshkpoor, B.; Liphardt, K.; Oldenburg, J.; Mayer, G.; Pötzsch, B. Selective Modulation of the Protease Activated Protein C Using Exosite Inhibiting Aptamers. Nucleic Acid Ther. 2020. [Google Scholar] [CrossRef]
- Muller, J.; Freitag, D.; Mayer, G.; Potzsch, B. Anticoagulant characteristics of HD1-22, a bivalent aptamer that specifically inhibits thrombin and prothrombinase. J. Thromb. Haemost. 2008, 6, 2105–2112. [Google Scholar] [CrossRef]
- Rusconi, C.P.; Scardino, E.; Layzer, J.; Pitoc, G.A.; Ortel, T.L.; Monroe, D.; Sullenger, B.A. RNA aptamers as reversible antagonists of coagulation factor IXa. Nature 2002, 419, 90–94. [Google Scholar] [CrossRef]
- Hamedani, N.S.; Ruhl, H.; Zimmermann, J.J.; Heiseler, T.; Oldenburg, J.; Mayer, G.; Potzsch, B.; Muller, J. In Vitro Evaluation of Aptamer-Based Reversible Inhibition of Anticoagulant Activated Protein C as a Novel Supportive Hemostatic Approach. Nucleic Acid Ther. 2016, 26, 355–362. [Google Scholar] [CrossRef]
- Hamedani, N.S.; Muller, J. Capillary Electrophoresis for the Selection of DNA Aptamers Recognizing Activated Protein C. Methods Mol. Biol. 2016, 1380, 61–75. [Google Scholar] [CrossRef]
- Hamedani, N.S.; Blumke, F.; Tolle, F.; Rohrbach, F.; Ruhl, H.; Oldenburg, J.; Mayer, G.; Potzsch, B.; Muller, J. Capture and Release (CaR): A simplified procedure for one-tube isolation and concentration of single-stranded DNA during SELEX. Chem. Commun. 2015, 51, 1135–1138. [Google Scholar] [CrossRef] [PubMed]
- Tolle, F.; Mayer, G. Preparation of SELEX Samples for Next-Generation Sequencing. Methods Mol. Biol. 2016, 1380, 77–84. [Google Scholar] [CrossRef]
- Jeddi, I.; Saiz, L. Three-dimensional modeling of single stranded DNA hairpins for aptamer-based biosensors. Sci. Rep. 2017, 7, 1178. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Wang, J.; Huang, Y.; Xiao, Y. 3dRNA v2.0: An Updated Web Server for RNA 3D Structure Prediction. Int. J. Mol. Sci. 2019, 20, 4116. [Google Scholar] [CrossRef] [Green Version]
- Zuker, M. Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res. 2003, 31, 3406–3415. [Google Scholar] [CrossRef] [PubMed]
- Land, H.; Humble, M.S. YASARA: A Tool to Obtain Structural Guidance in Biocatalytic Investigations. Methods Mol. Biol. 2018, 1685, 43–67. [Google Scholar] [CrossRef]
- Yan, Y.; Tao, H.; He, J.; Huang, S.-Y. The HDOCK server for integrated protein-protein docking. Nat. Protoc. 2020, 15, 1829–1852. [Google Scholar] [CrossRef] [PubMed]
- Stieler, M.; Weber, J.; Hils, M.; Kolb, P.; Heine, A.; Büchold, C.; Pasternack, R.; Klebe, G. Structure of active coagulation factor XIII triggered by calcium binding: Basis for the design of next-generation anticoagulants. Angew. Chem. Int. Ed Engl. 2013, 52, 11930–11934. [Google Scholar] [CrossRef]
- Singh, S.; Nazabal, A.; Kaniyappan, S.; Pellequer, J.-L.; Wolberg, A.S.; Imhof, D.; Oldenburg, J.; Biswas, A. The Plasma Factor XIII Heterotetrameric Complex Structure: Unexpected Unequal Pairing within a Symmetric Complex. Biomolecules 2019, 9, 765. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thomas, A.; Biswas, A.; Dodt, J.; Philippou, H.; Hethershaw, E.; Ensikat, H.J.; Ivaskevicius, V.; Oldenburg, J. Coagulation Factor XIIIA Subunit Missense Mutations Affect Structure and Function at the Various Steps of Factor XIII Action. Hum. Mutat. 2016, 37, 1030–1041. [Google Scholar] [CrossRef]
- Durda, M.A.; Wolberg, A.S.; Kerlin, B.A. State of the art in factor XIII laboratory assessment. Transfus. Apher. Sci. 2018, 57, 700–704. [Google Scholar] [CrossRef] [PubMed]
- Mosesson, M.W.; Siebenlist, K.R.; Hernandez, I.; Lee, K.N.; Christiansen, V.J.; McKee, P.A. Evidence that alpha2-antiplasmin becomes covalently ligated to plasma fibrinogen in the circulation: A new role for plasma factor XIII in fibrinolysis regulation. J. Thromb. Haemost. 2008, 6, 1565–1570. [Google Scholar] [CrossRef] [PubMed]
- Kohler, H.P. Interaction between FXIII and fibrinogen. Blood 2013, 121, 1931–1932. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, K.A.; Pease, R.J.; Avery, C.A.; Brown, J.M.; Adamson, P.J.; Cooke, E.J.; Neergaard-Petersen, S.; Cordell, P.A.; Ariëns, R.A.S.; Fishwick, C.W.G.; et al. The activation peptide cleft exposed by thrombin cleavage of FXIII-A(2) contains a recognition site for the fibrinogen α chain. Blood 2013, 121, 2117–2126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Byrnes, J.R.; Wilson, C.; Boutelle, A.M.; Brandner, C.B.; Flick, M.J.; Philippou, H.; Wolberg, A.S. The interaction between fibrinogen and zymogen FXIII-A2B2 is mediated by fibrinogen residues γ390-396 and the FXIII-B subunits. Blood 2016, 128, 1969–1978. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mouapi, K.N.; Wagner, L.J.; Stephens, C.A.; Hindi, M.M.; Wilkey, D.W.; Merchant, M.L.; Maurer, M.C. Evaluating the Effects of Fibrinogen αC Mutations on the Ability of Factor XIII to Crosslink the Reactive αC Glutamines (Q237, Q328, Q366). Thromb. Haemost. 2019, 119, 1048–1057. [Google Scholar] [CrossRef]
- Gupta, S.; Biswas, A.; Akhter, M.S.; Krettler, C.; Reinhart, C.; Dodt, J.; Reuter, A.; Philippou, H.; Ivaskevicius, V.; Oldenburg, J. Revisiting the mechanism of coagulation factor XIII activation and regulation from a structure/functional perspective. Sci. Rep. 2016, 6, 30105. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.; Dodt, J.; Volkers, P.; Hethershaw, E.; Philippou, H.; Ivaskevicius, V.; Imhof, D.; Oldenburg, J.; Biswas, A. Structure functional insights into calcium binding during the activation of coagulation factor XIII A. Sci. Rep. 2019, 9, 11324. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Aptamer | Oligonucleotide Sequence (5′–3′) | Binding Affinity (KD, nM) | ||
|---|---|---|---|---|
| FXIIIa | FXIII-A | FXIII | ||
| FA1 | AAGCAGTGGTAAGTAGGTTGACCATCCGCCGATTAGGCAATCACGTGGCTTAGGCACCGTATCTCTTCGAGCAATCCACAC * | 50.4 | >1000 | >500 |
| FA2 | AAGCAGTGGTAAGTAGGTTGAGCTTTCATCACCCGTCGGTTAGACTAAGTCTCCAGCATCCTCTCTTCGAGCAATCCACAC | 79.1 | >1000 | >1000 |
| FA3 | AAGCAGTGGTAAGTAGGTTGACCATCGTAGTTCGTACACCAGTCCATTCTAGGACTCACAATCTCTTCGAGCAATCCACAC | 92.9 | >1000 | >1000 |
| FA6 | AAGCAGTGGTAAGTAGGTTGACGCTGCGGGCATCACGTAAGTTAATATATCACTCGGCTATTCTCTTCGAGCAATCCACAC | 93.5 | >1000 | >1000 |
| FA8 | AAGCAGTGGTAAGTAGGTTGATCATCATGTTATTTTTGTTCCATCACGCCAGTTCTGGTTCTCTCTTCGAGCAATCCACAC | 79.1 | >1000 | >1000 |
| FA12 | AAGCAGTGGTAAGTAGGTTGATCATCCTCTTTAAGTCATCACTTTAGTTTCTCCATCTACATCTCTTCGAGCAATCCACAC | 51.8 | >1000 | >500 |
| Aptamer | FXIIIa Isopeptidase Activity (Max. Inhibition) 1 | FXIIIa Transglutaminase Activity (Max. Inhibition) 1 | IC50 (nM) of Inhibition of FXIIIa Incorporation to Fibrinogen | IC50 (nM) of Inhibition of α2AP Incorporation to Fibrinogen |
|---|---|---|---|---|
| FA1 | <1% | 9.34% | 89.51 (53.3–194) 2 | 86.39 (35–266) |
| FA2 | <1% | 12.87% | 39.27 (24.65–63.1) | 17.52 (9–42.7) |
| FA3 | <1% | 11.1% | 66.15 (43.05–109.5) | 60.32 (24.3–164) |
| FA6 | <1% | 12.47% | 57 (34.6–98.6) | 4.28 (2.9–6.5) |
| FA8 | <1% | 8.3% | 136.3 (60.6–n.d.) | 88.9 (36–159) |
| FA12 | <1% | 3.38% | 37.95 (22.6–64.6) | 17.4 (13.2–23.3) |
| Neg. Ctrl. | <1% | 11.35% | Not affected | Not affected |
| CT (s) | CFT (s) | MCF (mm) | A10 (mm) | A20 (mm) | MCE | |
|---|---|---|---|---|---|---|
| Pool plasma | 58.33 ± 6.03 | 716.33 ± 184 | 22 ± 1 | 19.67 ± 0.58 | 21.67 ± 0.58 | 28.22 ± 1.64 |
| FA1 | 48 ± 4.58 | >2700 | 17.67 ± 2.08 | 16.33 ± 1.53 | 17.33 ± 1.53 | 21.51 ± 3.11 |
| FA2 | 55.67 ± 5.51 | >2700 | 12 ± 1 **** | 11.33 ± 0.58 **** | 12 ± 1 **** | 13.65 ± 1.29 **** |
| FA3 | 53.67 ± 1.15 | >2700 | 19.33 ± 0.58 | 17 ± 1 | 19 ± 1 | 23.97 ± 0.89 |
| FA6 | 55.50 ± 2.12 | >2700 | 19.5 ± 0.51 | 17.5 ± 0.71 | 19.5 ± 0.71 | 24.23 ± 1.09 |
| FA8 | 60.33 ± 5.03 | >2700 | 17.33 ± 0.58 | 15.67 ± 0.58 * | 17 ± 0 * | 20.97 ± 0.85 |
| FA12 | 62.33 ± 4.51 | >2700 | 16 ± 1 ** | 14.33 ± 0.58 *** | 15.67 ± 1.15 ** | 19.06 ± 1.42 ** |
| Tridegin | 60.67 ± 2.1 | 2396 ± 74 | 19.67 ± 1.5 | 15.67 ± 0.58 *** | 17.67 ± 0.58 *** | 24.5 ± 2.35 |
| Neg. Ctrl. | 54.3 ± 4.04 | 918.5 ± 105 | 20 ± 1.73 | 18.3 ± 1.15 | 19.66 ± 1.53 | 25.03 ± 2.67 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hamedani, N.S.; Biswas, A.; Rudan, O.; Tönges, R.; Meyring, C.; Tolle, F.; Mayer, G.; Oldenburg, J.; Müller, J.; Pötzsch, B. Functional and Structural Characterization of Nucleic Acid Ligands That Bind to Activated Coagulation Factor XIII. J. Clin. Med. 2021, 10, 677. https://0-doi-org.brum.beds.ac.uk/10.3390/jcm10040677
Hamedani NS, Biswas A, Rudan O, Tönges R, Meyring C, Tolle F, Mayer G, Oldenburg J, Müller J, Pötzsch B. Functional and Structural Characterization of Nucleic Acid Ligands That Bind to Activated Coagulation Factor XIII. Journal of Clinical Medicine. 2021; 10(4):677. https://0-doi-org.brum.beds.ac.uk/10.3390/jcm10040677
Chicago/Turabian StyleHamedani, Nasim Shahidi, Arijit Biswas, Oliver Rudan, Rosa Tönges, Carlotta Meyring, Fabian Tolle, Günter Mayer, Johannes Oldenburg, Jens Müller, and Bernd Pötzsch. 2021. "Functional and Structural Characterization of Nucleic Acid Ligands That Bind to Activated Coagulation Factor XIII" Journal of Clinical Medicine 10, no. 4: 677. https://0-doi-org.brum.beds.ac.uk/10.3390/jcm10040677