
Cardiac Arrhythmias in Muscular Dystrophies Associated with Emerinopathy and Laminopathy: A Cohort Study

 , , , , and
, , , , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Study Design and Population
2.2. Study Protocol
2.3. Outcomes
2.4. Statistical Analysis
3. Results
3.1. Cardiac Arrhythmias at Inclusion
3.2. Prevalence of Atrial Arrhythmias at Follow-Up
3.3. Prevalence of Ventricular Arrhythmias at Follow-Up
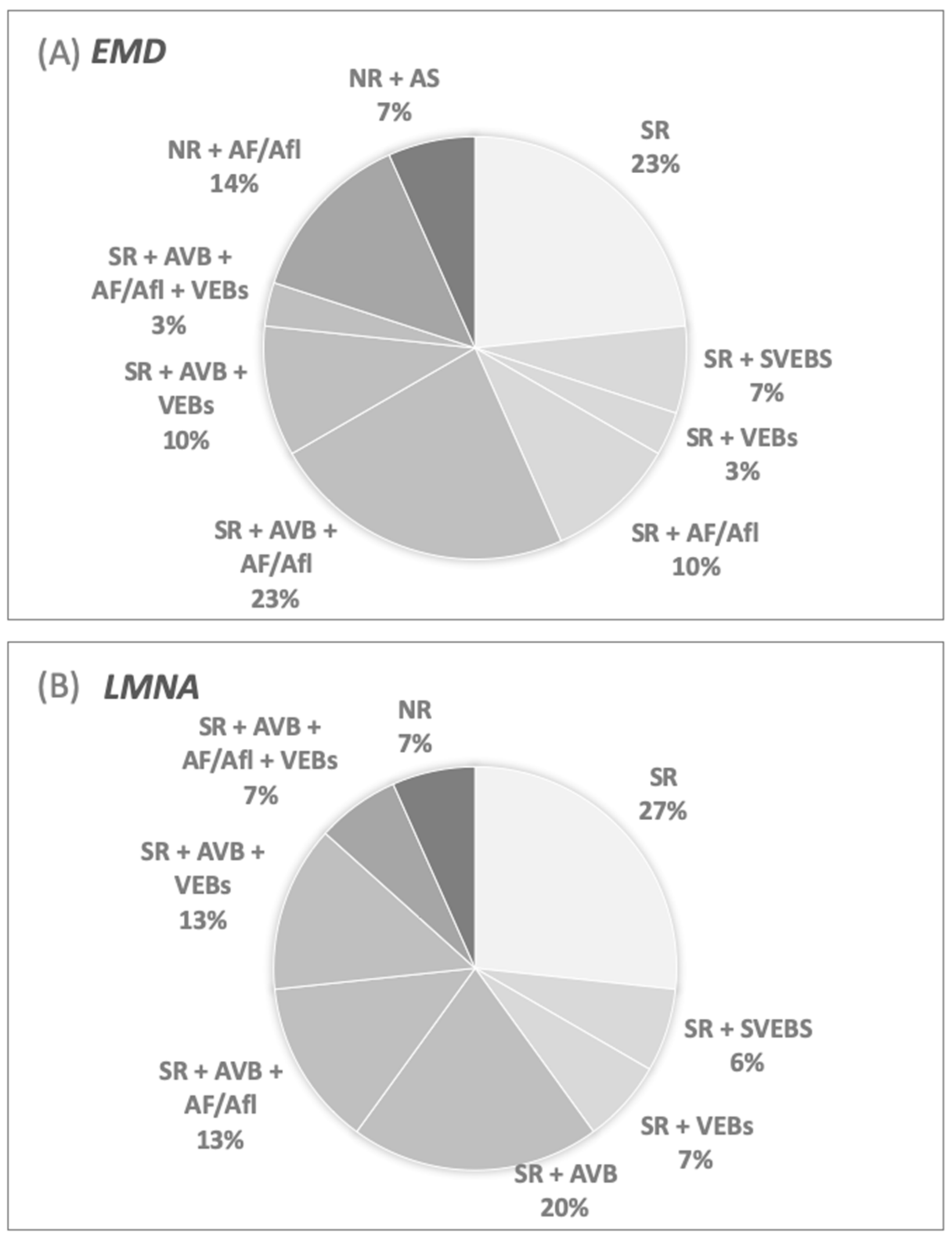
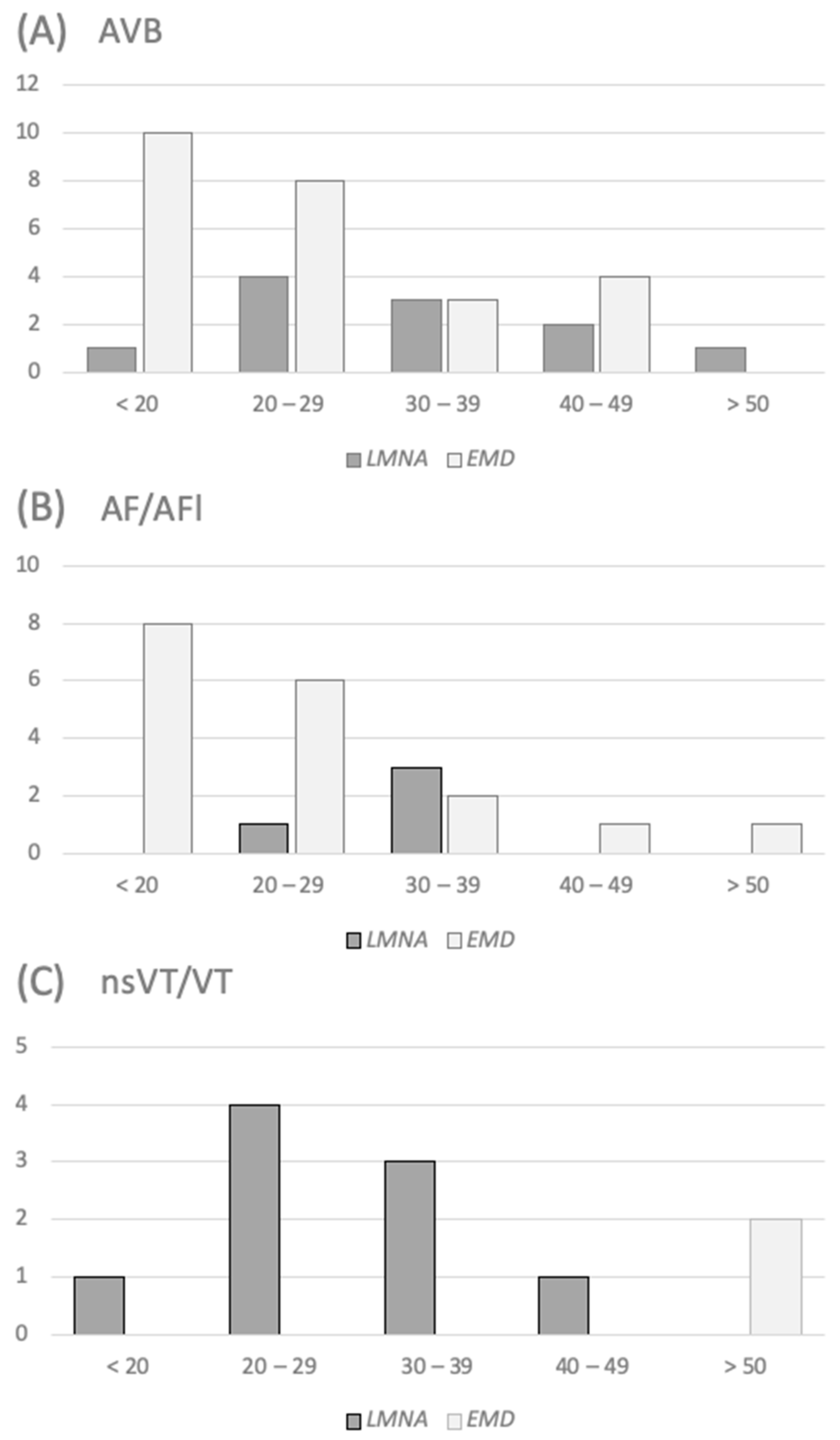
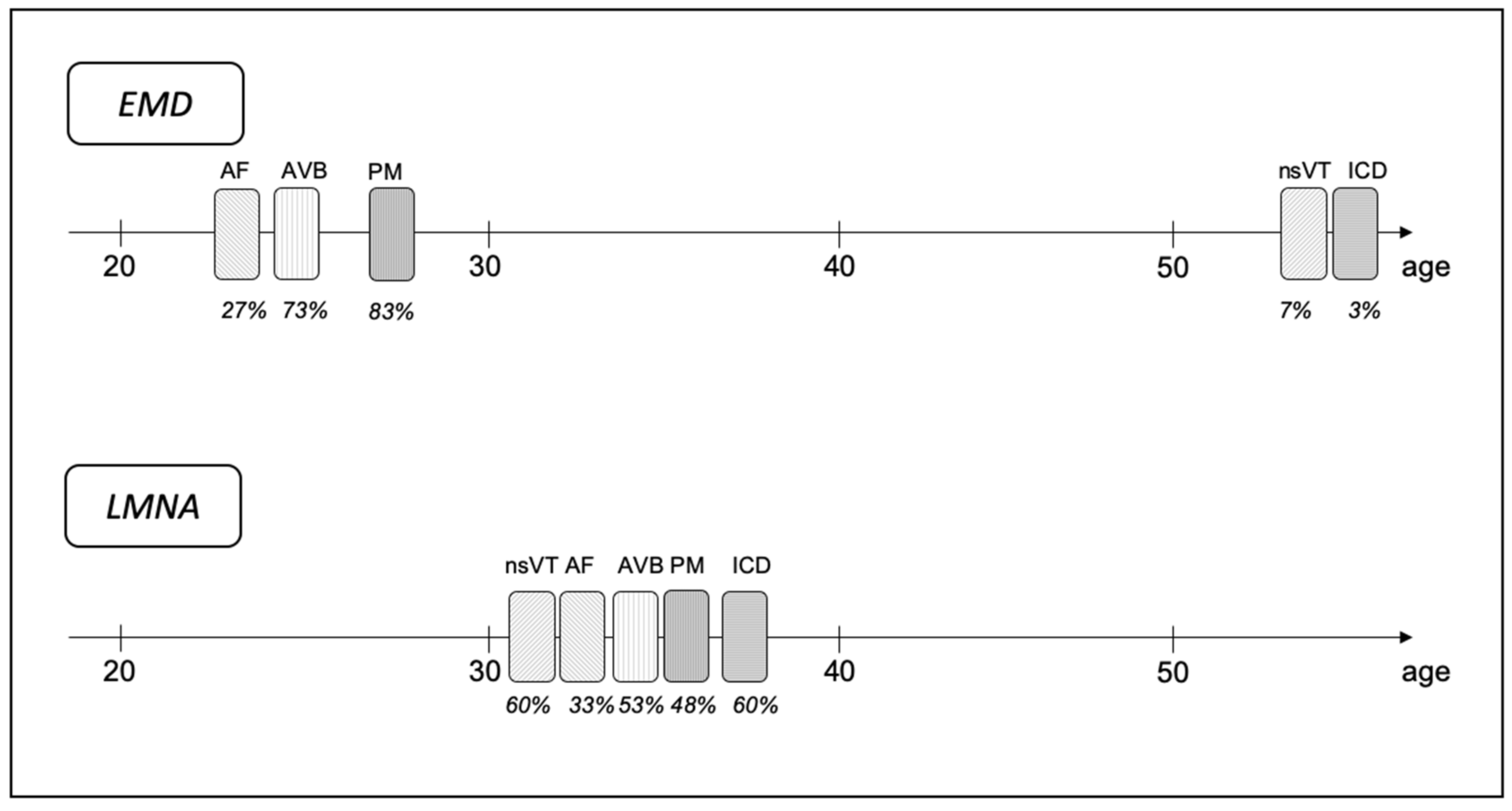
3.4. Timing of Arrhythmia’s Occurrence
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Captur, G.; Arbustini, E.; Bonne, G.; Syrris, P.; Mills, K.; Wahbi, K.; Mohiddin, S.A.; McKenna, S.; Pettit, S.; Ho, C.Y.; et al. Lamin and the heart. Heart 2017, 104, 468–479. [Google Scholar] [CrossRef] [PubMed]
- Birnkrant, D.J.; Bushby, K.; Bann, C.M.; Alman, B.A.; Apkon, S.D.; Blackwell, A.; Case, L.E.; Cripe, L.; Hadjiyannakis, S.; Olson, A.K.; et al. Diagnosis and management of Duchenne muscular dystrophy, part 2: Respiratory, cardiac, bone health, and orthopaedic management. Lancet Neurol. 2018, 17, 347–361. [Google Scholar] [CrossRef] [Green Version]
- Mah, J.K.; Korngut, L.; Fiest, K.M.; Dykeman, J.; Day, L.J.; Pringsheim, T.; Jette, N. A Systematic Review and Meta-analysis on the Epi-demiology of the Muscular Dystrophies. Can. J. Neurol. Sci. 2016, 43, 163–177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Berlo, J.H.; de Voogt, W.G.; van der Kooi, A.J.; van Tintelen, J.P.; Bonne, G.; Yaou, R.B.; Duboc, D.; Rossenbacker, T.; Heidbüchel, H.; de Visser, M.; et al. Meta-analysis of clinical characteristics of 299 carriers of LMNA gene mutations: Do lamin A/C mutations portend a high risk of sudden death? J. Mol. Med. 2005, 83, 79–83. [Google Scholar] [CrossRef]
- Sanna, T.; Russo, A.D.; Toniolo, D.; Vytopil, M.; Pelargonio, G.; De Martino, G.; Ricci, E.; Silvestri, G.; Giglio, V.; Messano, L.; et al. Cardiac features of Emery-Dreifuss muscular dystrophy caused by lamin A/C gene mutations. Eur. Heart J. 2003, 24, 2227–2236. [Google Scholar] [CrossRef] [Green Version]
- Wang, S.; Peng, D. Cardiac Involvement in Emery-Dreifuss Muscular Dystrophy and Related Management Strategies. Int. Heart J. 2019, 60, 12–18. [Google Scholar] [CrossRef] [Green Version]
- Fishbein, M.C.; Siegel, R.J.; Thompson, C.E.; Hopkins, L.C. Sudden Death of a Carrier of X-Linked Emery-Dreifuss Muscular Dystrophy. Ann. Intern. Med. 1993, 119, 900. [Google Scholar] [CrossRef] [PubMed]
- Tesson, F.; Saj, M.; Uvaize, M.M.; Nicolas, H.; Płoski, R.; Bilińska, Z. Lamin A/C mutations in dilated cardiomyopathy. Cardiol. J. 2014, 21, 331–342. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heller, S.A.; Shih, R.; Kalra, R.; Kang, P.B. Emery-Dreifuss muscular dystrophy. Muscle Nerve 2020, 61, 436–448. [Google Scholar] [CrossRef] [PubMed]
- Bonne, G.; Mercuri, E.; Muchir, A.; Urtizberea, A.; Becane, H.M.; Recan, D.; Merlini, L.; WEhnert, M.; Boor, R.; Reuner, U. Clinical and molecular genetic spectrum of autosomal dominant Emery-Dreifuss muscular dystrophy due to mutations of the lamin A/C gene. Ann. Neurol. 2000, 48, 70–80. [Google Scholar] [CrossRef]
- Emery, A.E.; Dreifuss, F.E. Unusual type of benign x-linked muscular dystrophy. J. Neurol. Neurosurg. Psychiatry 1966, 29, 338–342. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ditaranto, R.; Boriani, G.; Biffi, M.; Lorenzini, M.; Graziosi, M.; Ziacchi, M.; Pasquale, F.; Vitale, G.; Berardini, A.; Rinaldi, R.; et al. Differences in cardiac phenotype and natural history of laminopathies with and without neuromuscular onset. Orphanet J. Rare Dis. 2019, 14, 263. [Google Scholar] [CrossRef]
- Peretto, G.; Di Resta, C.; Perversi, J.; Forleo, C.; Maggi, L.; Politano, L.; Barison, A.; Previtali, S.C.; Carboni, N.; Brun, F.; et al. Cardiac and Neuromuscular Features of Patients WithLMNA-Related Cardiomyopathy. Ann. Intern. Med. 2019, 171, 458. [Google Scholar] [CrossRef]
- Priori, S.G.; Blomström-Lundqvist, C.; Mazzanti, A.; Blom, N.; Borggrefe, M.; Camm, J.; Elliott, P.M.; Fitzsimons, D.; Hatala, R.; Hindricks, G.; et al. 2015 ESC Guidelines for the management of patients with ventricular arrhythmias and the prevention of sudden cardiac Death. The Task Force for the Management of Patients with Ventricular Arrhythmias and the Prevention of Sudden Cardiac Death of the European Society of Cardiology (ESC). G. Ital. Cardiol. 2016, 17. [Google Scholar] [CrossRef]
- Hong, J.-S.; Ki, C.-S.; Kim, J.-W.; Suh, Y.-L.; Kim, J.S.; Baek, K.K.; Kim, B.J.; Ahn, K.J.; Kim, D.-K. Cardiac Dysrhythmias, Cardiomyopathy and Muscular Dystrophy in Patients with Emery-Dreifuss Muscular Dystrophy and Limb-Girdle Muscular Dystrophy Type 1B. J. Korean Med. Sci. 2005, 20, 283–290. [Google Scholar] [CrossRef] [PubMed]
- Hasselberg, N.E.; Haland, T.F.; Saberniak, J.; Brekke, P.H.; Berge, K.E.; Leren, T.P.; Edvardsen, T.; Haugaa, K.H. Lamin A/C cardiomyopathy: Young onset, high penetrance, and frequent need for heart transplantation. Eur. Heart J. 2017, 39, 853–860. [Google Scholar] [CrossRef]
- Steckiewicz, R.; Stolarz, P.; Świętoń, E.; Madej-Pilarczyk, A.; Grabowski, M.; Marchel, M.; Pieniak, M.; Filipiak, K.J.; Hausmanowa-Petrusewicz, I.; Opolski, G. Cardiac pacing in 21 patients with Emery-Dreifuss muscular dystrophy: A single-centre study with a 39-year follow-up. Kardiol. Polska 2015, 576–583. [Google Scholar] [CrossRef]
- Buckley, A.E.; Dean, J.; Mahy, I.R. Cardiac involvement in Emery Dreifuss muscular dystrophy: A case series. Heart 1999, 82, 105–108. [Google Scholar] [CrossRef]
- Steckiewicz, R.; Stolarz, P.; Kosior, D.A.; Marchel, M.; Pieniak, M.; Świętoń, E.; Piotrowska-Kownacka, E.; Grabowski, M. Atrial mechanical and electrical dysfunction in patient with Emery-Dreifuss muscular dystrophy reason of change in electrotherapeutical approach: Frequent result of rare disease. Kardiol. Pol. 2013, 71, 406–409. [Google Scholar] [CrossRef] [PubMed]
- Bialer, M.G.; McDaniel, N.L.; E Kelly, T. Progression of cardiac disease in Emery-Dreifuss muscular dystrophy. Clin. Cardiol. 1991, 14, 411–416. [Google Scholar] [CrossRef]
- Boriani, G.; Gallina, M.; Merlini, L.; Bonne, G.; Toniolo, D.; Amati, S.; Biffi, M.; Martignani, C.; Frabetti, L.; Bonvicini, M.; et al. Clinical relevance of atrial fibrillation/flutter, stroke, pacemaker implant, and heart failure in Emery-Dreifuss muscular dystrophy: A long-term longitudinal study. Stroke 2003, 34, 901–908. [Google Scholar] [CrossRef] [Green Version]
- Sakata, K.; Shimizu, M.; Ino, H.; Yamaguchi, M.; Terai, H.; Fujino, N.; Hayashi, K.; Kaneda, T.; Inoue, M.; Oda, Y.; et al. High Incidence of Sudden Cardiac Death With Conduction Disturbances and Atrial Cardiomyopathy Caused by a Nonsense Mutation in the STA Gene. Circulation 2005, 111, 3352–3358. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Madej-Pilarczyk, A.; Marchel, M.; Fidziańska, A.; Opolski, G.; Hausmanowa-Petrusewicz, I. Low symptomatic malignant cardiac arrhythmia in a patient with lamin-related congenital muscular dystrophy. Kardiol. Polska 2015, 73, 942. [Google Scholar] [CrossRef]
- Pasotti, M.; Klersy, C.; Pilotto, A.; Marziliano, N.; Rapezzi, C.; Serio, A.; Mannarino, S.; Gambarin, F.; Favalli, V.; Grasso, M.; et al. Long-Term Outcome and Risk Stratification in Dilated Cardiolaminopathies. J. Am. Coll. Cardiol. 2008, 52, 1250–1260. [Google Scholar] [CrossRef] [Green Version]
- Wahbi, K.; Ben Yaou, R.; Gandjbakhch, E.; Anselme, F.; Gossios, T.; Lakdawala, N.K.; Stalens, C.; Sacher, F.; Babuty, D.; Trochu, J.-N.; et al. Development and Validation of a New Risk Prediction Score for Life-Threatening Ventricular Tachyarrhythmias in Laminopathies. Circulation 2019, 140, 293–302. [Google Scholar] [CrossRef] [PubMed]
- Van Rijsingen, I.A.; Arbustini, E.; Elliott, P.M.; Mogensen, J.; Hermans-van Ast, J.F.; van der Kooi, A.J.; van Tintele, J.P.; van den Berg, M.P.; Pilotto, A.; Pasotti, M.; et al. Risk factors for malignant ventricular arrhythmias in lamin a/c mutation carriers a European cohort study. J. Am. Coll. Cardiol. 2012, 59, 493–500. [Google Scholar] [CrossRef] [PubMed]
- Carboni, N.; Mura, M.; Mercuri, E.; Marrosu, G.; Manzi, R.C.; Cocco, E.; Nissardi, V.; Isola, F.; Mateddu, A.; Solla, E.; et al. Cardiac and muscle imaging findings in a family with X-linked Emery–Dreifuss muscular dystrophy. Neuromuscul. Disord. 2012, 22, 152–158. [Google Scholar] [CrossRef]
- Nigro, G.; Russo, V.; Ventriglia, V.M.; Della Cioppa, N.; Palladino, A.; Nigro, V.; Calabrò, R.; Nigro, G.; Politano, L. Early onset of cardiomyopathy and primary prevention of sudden death in X-linked Emery–Dreifuss muscular dystrophy. Neuromuscul. Disord. 2010, 20, 174–177. [Google Scholar] [CrossRef]
- Madej-Pilarczyk, A.; Marchel, M.; Ochman, K.; Cegielska, J.; Steckiewicz, R. Low-symptomatic skeletal muscle disease in patients with a cardiac disease—Diagnostic approach in skeletal muscle laminopathies. Neurol. Neurochir. Polska 2018, 52, 174–180. [Google Scholar] [CrossRef]
- Kalin, K.; Oreziak, A.; Franaszczyk, M.; Bilinska, Z.T.; Ploski, R.; Bilinska, M. Genetic muscle disorder mimicking atrial arrhythmias with conduction defects requiring pacemaker implantation. Pol. Arch. Intern. Med. 2019, 129, 627–629. [Google Scholar] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| EMD Group n = 30 | LMNA Group n = 15 | p-Value | |
|---|---|---|---|
| Age (years) | 21.0 (15.25–30.0) | 26.0 (18.0–33.0) | NS |
| Female (%) | 20 | 73 | <0.001 |
| BMI (kg/m2) | 21.5 (19.4–25.2) | 20.2 (17.3–25.1) | NS |
| Sporadic/familial | 13/17 | 9/6 | NS |
| LVEDV (mL) | 119 (90–169) | 103 (86–125) | NS |
| LAV (mL) | 56.4 (47.8–73.3) | 50.5 (38.5–63) | 0.08 |
| LVEF (%) | 52 (48–58) | 54 (48–58) | NS |
| NTpro-BNP (pg/mL) | 70 (44–102) | 109 (54–347) | NS |
| NYHA I-II (%) | 0 | 20 | 0.08 |
| NYHA III-IV | 0 | 0 | NS |
| Total Group (n = 45) | EMD Group (n = 30) | LMNA Group (n = 15) | p-Value | |
|---|---|---|---|---|
| SR, % (n) | 84.4 (38) | 80 (24) | 93 (14) | 0.40 |
| NR, % (n) | 15.6 (7) | 20 (6) | 6.7 (1) | 0.40 |
| AT, % (n) | 24.4 (11) | 16.7 (5) | 40 (6) | 0.14 |
| AF, % (n) | 28.9 (13) | 33.3 (10) | 20 (3) | 0.49 |
| AFl, % (n) | 17.8 (8) | 23.3 (7) | 6.7 (1) | 0.24 |
| AS, % (n) | 6.7 (3) | 10 (3) | 0 (0) | 0.54 |
| AVB 1st degree, % (n) | 11.1 (5) | 10 (3) | 13.3 (2) | 1.00 |
| AVB 2nd degree, % (n) | 24.4 (11) | 20 (6) | 33.3 (5) | 0.46 |
| AVB 3rd degree, % (n) | 22.2 (10) | 26.7 (8) | 13.3 (2) | 0.46 |
| SVEBs, % (n) | 37.8 (17) | 36.7 (11) | 40 (6) | 1.00 |
| VEBs, % (n) | 13.3 (6) | 6.7 (2) | 26.7 (4) | 0.16 |
| nsVT, % (n) | 0 (0) | 0 (0) | 0 (0) | - |
| VT, % (n) | 0 (0) | 0 (0) | 0 (0) | - |
| Total Group (n = 45) | EMD (n = 30) | LMNA (n = 15) | p-Value | |
|---|---|---|---|---|
| SVEBs, % (n) | 48.9 (22) | 46.7 (14) | 53.3 (8) | 0.76 |
| AT only, % (n) | 17.8 (8) | 10 (3) | 33.3 (5) | 0.10 |
| AF/Afl only, % (n) | 28.9 (13) | 26.7 (8) | 33.3 (5) | 0.73 |
| AS only, % (n) | 31.1 (14) | 46.7 (14) | 0 (0) | 0.001 |
| No AT/AF/AFl/AS, % (n) | 22.2 (10) | 16.7 (5) | 33.3 (5) | 0.26 |
| VEBs, % (n) | 40 (18) | 30 (9) | 60 (9) | 0.11 |
| VEBs couplets, % (n) | 22.2 (10) | 13.3 (4) | 40 (6) | 0.06 |
| nsVT, % (n) | 22.2 (10) | 3.3 (1) | 60 (9) | <0.001 |
| VT, % (n) | 8.9 (4) | 6.7 (2) | 13.3 (2) | 0.59 |
| nsVT/VT, % (n) | 24.4 (11) | 6.7 (2) | 60 (9) | <0.001 |
| PM implantation, % (n) | 66.7 (30) | 76.7 (23) | 46.7 (7) | 0.09 |
| ICD implantation, % (n) | 22,2 (10) | 3,3 (1) | 60 (9) | <0.001 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Marchel, M.; Madej-Pilarczyk, A.; Tymińska, A.; Steckiewicz, R.; Ostrowska, E.; Wysińska, J.; Russo, V.; Grabowski, M.; Opolski, G. Cardiac Arrhythmias in Muscular Dystrophies Associated with Emerinopathy and Laminopathy: A Cohort Study. J. Clin. Med. 2021, 10, 732. https://0-doi-org.brum.beds.ac.uk/10.3390/jcm10040732
Marchel M, Madej-Pilarczyk A, Tymińska A, Steckiewicz R, Ostrowska E, Wysińska J, Russo V, Grabowski M, Opolski G. Cardiac Arrhythmias in Muscular Dystrophies Associated with Emerinopathy and Laminopathy: A Cohort Study. Journal of Clinical Medicine. 2021; 10(4):732. https://0-doi-org.brum.beds.ac.uk/10.3390/jcm10040732
Chicago/Turabian StyleMarchel, Michał, Agnieszka Madej-Pilarczyk, Agata Tymińska, Roman Steckiewicz, Ewa Ostrowska, Julia Wysińska, Vincenzo Russo, Marcin Grabowski, and Grzegorz Opolski. 2021. "Cardiac Arrhythmias in Muscular Dystrophies Associated with Emerinopathy and Laminopathy: A Cohort Study" Journal of Clinical Medicine 10, no. 4: 732. https://0-doi-org.brum.beds.ac.uk/10.3390/jcm10040732