
Celia’s Encephalopathy (BSCL2-Gene-Related): Current Understanding

 ,
,  ,
,  ,
,  , ,
, , 
Abstract
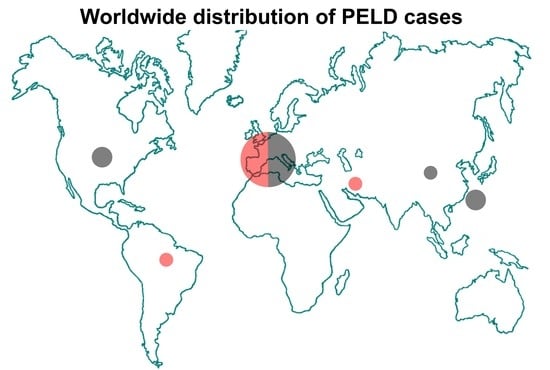
:
1. Introduction
2. BSCL2 Gene
3. Seipin Protein
3.1. Structure and Location
3.2. Role in Adipogenesis
3.3. Role in Central Nervous System
4. Seipin-Associated Diseases: The Seipinopathies
4.1. Upper and/or Lower Motor Neuron Diseases
4.2. Congenital Generalized Lipodystrophy type 2 (CGL2)
4.3. Celia’s Encephalopathy
4.3.1. Clinical Course of the Classic Form
4.3.2. Molecular Bases and Pathogenetic Mechanisms
4.4. Other Variants in BSCL2
4.4.1. Variant c.974dupG
4.4.2. Variant c.1048C>T
4.4.3. Variant c.1076dupC
4.4.4. Variant c.566T>A
5. Treatment
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Guillen-Navarro, E.; Sanchez-Iglesias, S.; Domingo-Jimenez, R.; Victoria, B.; Ruiz-Riquelme, A.; Rabano, A.; Loidi, L.; Beiras, A.; Gonzalez-Mendez, B.; Ramos, A.; et al. A new seipin-associated neurodegenerative syndrome. J. Med Genet. 2013, 50, 401–409. [Google Scholar] [CrossRef]
- Magre, J.; Delepine, M.; Khallouf, E.; Gedde-Dahl, T., Jr.; Van Maldergem, L.; Sobel, E.; Papp, J.; Meier, M.; Megarbane, A.; Bachy, A.; et al. Identification of the gene altered in Berardinelli-Seip congenital lipodystrophy on chromosome 11q13. Nat. Genet. 2001, 28, 365–370. [Google Scholar] [CrossRef]
- Sanchez-Iglesias, S.; Fernandez-Liste, A.; Guillin-Amarelle, C.; Rabano, A.; Rodriguez-Canete, L.; Gonzalez-Mendez, B.; Fernandez-Pombo, A.; Senra, A.; Araujo-Vilar, D. Does Seipin Play a Role in Oxidative Stress Protection and Peroxisome Biogenesis? New Insights from Human Brain Autopsies. Neuroscience 2019, 396, 119–137. [Google Scholar] [CrossRef]
- Lundin, C.; Nordstrom, R.; Wagner, K.; Windpassinger, C.; Andersson, H.; von Heijne, G.; Nilsson, I. Membrane topology of the human seipin protein. FEBS Lett. 2006, 580, 2281–2284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Binns, D.; Lee, S.; Hilton, C.L.; Jiang, Q.X.; Goodman, J.M. Seipin is a discrete homooligomer. Biochemistry 2010, 49, 10747–10755. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, R.; Qian, H.; Lukmantara, I.; Gao, M.; Du, X.; Yan, N.; Yang, H. Human SEIPIN Binds Anionic Phospholipids. Dev. Cell 2018, 47, 248–256.e244. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sui, X.; Arlt, H.; Brock, K.P.; Lai, Z.W.; Di Maio, F.; Marks, D.S.; Liao, M.; Farese, R.V., Jr.; Walther, T.C. Cryo-electron microscopy structure of the lipid droplet-formation protein seipin. J. Cell Biol. 2018, 217, 4080–4091. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bohnert, M. New friends for seipin—Implications of seipin partner proteins in the life cycle of lipid droplets. Semin. Cell Dev. Biol. 2020, 108, 24–32. [Google Scholar] [CrossRef]
- Talukder, M.M.; Sim, M.F.; O’Rahilly, S.; Edwardson, J.M.; Rochford, J.J. Seipin oligomers can interact directly with AGPAT2 and lipin 1, physically scaffolding critical regulators of adipogenesis. Mol. Metab. 2015, 4, 199–209. [Google Scholar] [CrossRef] [Green Version]
- Pagac, M.; Cooper, D.E.; Qi, Y.; Lukmantara, I.E.; Mak, H.Y.; Wu, Z.; Tian, Y.; Liu, Z.; Lei, M.; Du, X.; et al. SEIPIN Regulates Lipid Droplet Expansion and Adipocyte Development by Modulating the Activity of Glycerol-3-phosphate Acyltransferase. Cell Rep. 2016, 17, 1546–1559. [Google Scholar] [CrossRef] [Green Version]
- Salo, V.T.; Hölttä-Vuori, M.; Ikonen, E. Seipin-mediated contacts as gatekeepers of lipid flux at the endoplasmic reticulum–lipid droplet nexus. Contact 2020, 3, 1–16. [Google Scholar] [CrossRef]
- Wei, S.; Soh, S.L.-Y.; Qiu, W.; Yang, W.; Seah, C.J.-Y.; Guo, J.; Ong, W.-Y.; Pang, Z.P.; Han, W. Seipin regulates excitatory synaptic transmission in cortical neurons. J. Neurochem. 2013, 124, 478–489. [Google Scholar] [CrossRef]
- Yang, W.; Thein, S.; Guo, X.; Xu, F.; Venkatesh, B.; Sugii, S.; Radda, G.K.; Han, W. Seipin differentially regulates lipogenesis and adipogenesis through a conserved core sequence and an evolutionarily acquired C-terminus. Biochem. J. 2013, 452, 37–44. [Google Scholar] [CrossRef]
- Choudhary, V.; Schneiter, R. Lipid droplet biogenesis from specialized ER subdomains. Microb. Cell 2020, 7, 218–221. [Google Scholar] [CrossRef] [PubMed]
- Olzmann, J.A.; Carvalho, P. Dynamics and functions of lipid droplets. Nat. Rev. Mol. Cell Biol. 2019, 20, 137–155. [Google Scholar] [CrossRef]
- Cartwright, B.R.; Binns, D.D.; Hilton, C.L.; Han, S.; Gao, Q.; Goodman, J.M. Seipin performs dissectible functions in promoting lipid droplet biogenesis and regulating droplet morphology. Mol. Biol. Cell 2015, 26, 726–739. [Google Scholar] [CrossRef] [PubMed]
- Szymanski, K.M.; Binns, D.; Bartz, R.; Grishin, N.V.; Li, W.P.; Agarwal, A.K.; Garg, A.; Anderson, R.G.; Goodman, J.M. The lipodystrophy protein seipin is found at endoplasmic reticulum lipid droplet junctions and is important for droplet morphology. Proc. Natl. Acad. Sci. USA 2007, 104, 20890–20895. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fei, W.; Shui, G.; Gaeta, B.; Du, X.; Kuerschner, L.; Li, P.; Brown, A.J.; Wenk, M.R.; Parton, R.G.; Yang, H. Fld1p, a functional homologue of human seipin, regulates the size of lipid droplets in yeast. J. Cell Biol. 2008, 180, 473–482. [Google Scholar] [CrossRef]
- Wang, H.; Becuwe, M.; Housden, B.E.; Chitraju, C.; Porras, A.J.; Graham, M.M.; Liu, X.N.; Thiam, A.R.; Savage, D.B.; Agarwal, A.K.; et al. Seipin is required for converting nascent to mature lipid droplets. eLife 2016, 5, e16582. [Google Scholar] [CrossRef] [Green Version]
- Salo, V.T.; Belevich, I.; Li, S.; Karhinen, L.; Vihinen, H.; Vigouroux, C.; Magre, J.; Thiele, C.; Holtta-Vuori, M.; Jokitalo, E.; et al. Seipin regulates ER-lipid droplet contacts and cargo delivery. Embo J. 2016, 35, 2699–2716. [Google Scholar] [CrossRef]
- Ben M’barek, K.; Ajjaji, D.; Chorlay, A.; Vanni, S.; Foret, L.; Thiam, A.R. ER Membrane Phospholipids and Surface Tension Control Cellular Lipid Droplet Formation. Dev. Cell 2017, 41, 591–604.e597. [Google Scholar] [CrossRef] [Green Version]
- Schuldiner, M.; Bohnert, M. A different kind of love—Lipid droplet contact sites. Biochim. Biophys. Acta. Mol. Cell Biol. Lipids 2017, 1862, 1188–1196. [Google Scholar] [CrossRef]
- Geltinger, F.; Schartel, L.; Wiederstein, M.; Tevini, J.; Aigner, E.; Felder, T.K.; Rinnerthaler, M. Friend or Foe: Lipid Droplets as Organelles for Protein and Lipid Storage in Cellular Stress Response, Aging and Disease. Molecules 2020, 25, 5053. [Google Scholar] [CrossRef]
- Eisenberg-Bord, M.; Mari, M.; Weill, U.; Rosenfeld-Gur, E.; Moldavski, O.; Castro, I.G.; Soni, K.G.; Harpaz, N.; Levine, T.P.; Futerman, A.H.; et al. Identification of seipin-linked factors that act as determinants of a lipid droplet subpopulation. J. Cell Biol. 2018, 217, 269–282. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Castro, I.G.; Eisenberg-Bord, M.; Persiani, E.; Rochford, J.J.; Schuldiner, M.; Bohnert, M. Promethin Is a Conserved Seipin Partner Protein. Cells 2019, 8, 268. [Google Scholar] [CrossRef] [Green Version]
- Chung, J.; Wu, X.; Lambert, T.J.; Lai, Z.W.; Walther, T.C.; Farese, R.V., Jr. LDAF1 and Seipin Form a Lipid Droplet Assembly Complex. Dev. Cell 2019, 51, 551–563.e557. [Google Scholar] [CrossRef]
- Prasanna, X.; Salo, V.T.; Li, S.; Ven, K.; Vihinen, H.; Jokitalo, E.; Vattulainen, I.; Ikonen, E. Seipin traps triacylglycerols to facilitate their nanoscale clustering in the endoplasmic reticulum membrane. PLoS Biol. 2021, 19, e3000998. [Google Scholar] [CrossRef]
- Wang, S.; Idrissi, F.Z.; Hermansson, M.; Grippa, A.; Ejsing, C.S.; Carvalho, P. Seipin and the membrane-shaping protein Pex30 cooperate in organelle budding from the endoplasmic reticulum. Nat. Commun. 2018, 9, 2939. [Google Scholar] [CrossRef] [PubMed]
- Joshi, A.S.; Huang, X.; Choudhary, V.; Levine, T.P.; Hu, J.; Prinz, W.A. A family of membrane-shaping proteins at ER subdomains regulates pre-peroxisomal vesicle biogenesis. J. Cell Biol. 2016, 215, 515–529. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jin, Y.; Tan, Y.; Zhao, P.; Ren, Z. SEIPIN: A Key Factor for Nuclear Lipid Droplet Generation and Lipid Homeostasis. Int. J. Mol. Sci. 2020, 21, 8208. [Google Scholar] [CrossRef]
- Romanauska, A.; Kohler, A. The Inner Nuclear Membrane Is a Metabolically Active Territory that Generates Nuclear Lipid Droplets. Cell 2018, 174, 700–715.e718. [Google Scholar] [CrossRef] [Green Version]
- Soltysik, K.; Ohsaki, Y.; Tatematsu, T.; Cheng, J.; Maeda, A.; Morita, S.Y.; Fujimoto, T. Nuclear lipid droplets form in the inner nuclear membrane in a seipin-independent manner. J. Cell Biol. 2021, 220. [Google Scholar] [CrossRef]
- Du, X.; Yang, H. Seipin regulates the formation of nuclear lipid droplets from a distance. J. Cell Biol. 2021, 220. [Google Scholar] [CrossRef] [PubMed]
- Ruiz-Riquelme, A.; Sanchez-Iglesias, S.; Rabano, A.; Guillen-Navarro, E.; Domingo-Jimenez, R.; Ramos, A.; Rosa, I.; Senra, A.; Nilsson, P.; Garcia, A.; et al. Larger aggregates of mutant seipin in Celia’s Encephalopathy, a new protein misfolding neurodegenerative disease. Neurobiol. Dis. 2015, 83, 44–53. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Yechoor, V.K.; Chang, B.H.; Li, M.V.; March, K.L.; Chan, L. The human lipodystrophy gene product Berardinelli-Seip congenital lipodystrophy 2/seipin plays a key role in adipocyte differentiation. Endocrinology 2009, 150, 4552–4561. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Payne, V.A.; Grimsey, N.; Tuthill, A.; Virtue, S.; Gray, S.L.; Dalla Nora, E.; Semple, R.K.; O’Rahilly, S.; Rochford, J.J. The human lipodystrophy gene BSCL2/seipin may be essential for normal adipocyte differentiation. Diabetes 2008, 57, 2055–2060. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, W.; Thein, S.; Wang, X.; Bi, X.; Ericksen, R.E.; Xu, F.; Han, W. BSCL2/seipin regulates adipogenesis through actin cytoskeleton remodelling. Hum. Mol. Genet. 2014, 23, 502–513. [Google Scholar] [CrossRef]
- Bi, J.; Wang, W.; Liu, Z.; Huang, X.; Jiang, Q.; Liu, G.; Wang, Y.; Huang, X. Seipin promotes adipose tissue fat storage through the ER Ca(2)(+)-ATPase SERCA. Cell Metab. 2014, 19, 861–871. [Google Scholar] [CrossRef] [Green Version]
- Islinger, M.; Voelkl, A.; Fahimi, H.D.; Schrader, M. The peroxisome: An update on mysteries 2.0. Histochem. Cell Biol. 2018, 150, 443–471. [Google Scholar] [CrossRef] [Green Version]
- Berger, J.; Dorninger, F.; Forss-Petter, S.; Kunze, M. Peroxisomes in brain development and function. Biochim. Et Biophys. Acta 2016, 1863, 934–955. [Google Scholar] [CrossRef] [Green Version]
- Hamilton, J.A.; Hillard, C.J.; Spector, A.A.; Watkins, P.A. Brain uptake and utilization of fatty acids, lipids and lipoproteins: Application to neurological disorders. J. Mol. Neurosci. Mn 2007, 33, 2–11. [Google Scholar] [CrossRef]
- Zhang, J.; Liu, Q. Cholesterol metabolism and homeostasis in the brain. Protein Cell 2015, 6, 254–264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Puchkov, D.; Haucke, V. Greasing the synaptic vesicle cycle by membrane lipids. Trends Cell Biol. 2013, 23, 493–503. [Google Scholar] [CrossRef]
- Bruce, K.D.; Zsombok, A.; Eckel, R.H. Lipid Processing in the Brain: A Key Regulator of Systemic Metabolism. Front. Endocrinol. 2017, 8, 60. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Montesinos, J.; Guardia-Laguarta, C.; Area-Gomez, E. The fat brain. Curr. Opin. Clin. Nutr. Metab. Care 2020, 23, 68–75. [Google Scholar] [CrossRef]
- Hamilton, L.K.; Dufresne, M.; Joppe, S.E.; Petryszyn, S.; Aumont, A.; Calon, F.; Barnabe-Heider, F.; Furtos, A.; Parent, M.; Chaurand, P.; et al. Aberrant Lipid Metabolism in the Forebrain Niche Suppresses Adult Neural Stem Cell Proliferation in an Animal Model of Alzheimer’s Disease. Cell Stem Cell 2015, 17, 397–411. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cole, N.B.; Murphy, D.D.; Grider, T.; Rueter, S.; Brasaemle, D.; Nussbaum, R.L. Lipid droplet binding and oligomerization properties of the Parkinson’s disease protein alpha-synuclein. J. Biol. Chem. 2002, 277, 6344–6352. [Google Scholar] [CrossRef] [Green Version]
- Farmer, B.C.; Walsh, A.E.; Kluemper, J.C.; Johnson, L.A. Lipid Droplets in Neurodegenerative Disorders. Front. Neurosci. 2020, 14, 742. [Google Scholar] [CrossRef]
- Marschallinger, J.; Iram, T.; Zardeneta, M.; Lee, S.E.; Lehallier, B.; Haney, M.S.; Pluvinage, J.V.; Mathur, V.; Hahn, O.; Morgens, D.W.; et al. Lipid-droplet-accumulating microglia represent a dysfunctional and proinflammatory state in the aging brain. Nat. Neurosci. 2020, 23, 194–208. [Google Scholar] [CrossRef]
- Doetsch, F.; Garcia-Verdugo, J.M.; Alvarez-Buylla, A. Cellular composition and three-dimensional organization of the subventricular germinal zone in the adult mammalian brain. J. Neurosci. Off. J. Soc. Neurosci. 1997, 17, 5046–5061. [Google Scholar] [CrossRef]
- Brawer, J.R.; Walsh, R.J. Response of tanycytes to aging in the median eminence of the rat. Am. J. Anat. 1982, 163, 247–256. [Google Scholar] [CrossRef]
- Liu, L.; MacKenzie, K.R.; Putluri, N.; Maletic-Savatic, M.; Bellen, H.J. The Glia-Neuron Lactate Shuttle and Elevated ROS Promote Lipid Synthesis in Neurons and Lipid Droplet Accumulation in Glia via APOE/D. Cell Metab. 2017, 26, 719–737.e716. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ioannou, M.S.; Jackson, J.; Sheu, S.H.; Chang, C.L.; Weigel, A.V.; Liu, H.; Pasolli, H.A.; Xu, C.S.; Pang, S.; Matthies, D.; et al. Neuron-Astrocyte Metabolic Coupling Protects against Activity-Induced Fatty Acid Toxicity. Cell 2019, 177, 1522–1535.e1514. [Google Scholar] [CrossRef] [PubMed]
- Bozza, P.T.; Viola, J.P. Lipid droplets in inflammation and cancer. Prostaglandins Leukot. Essent. Fat. Acids 2010, 82, 243–250. [Google Scholar] [CrossRef]
- Bailey, A.P.; Koster, G.; Guillermier, C.; Hirst, E.M.; MacRae, J.I.; Lechene, C.P.; Postle, A.D.; Gould, A.P. Antioxidant Role for Lipid Droplets in a Stem Cell Niche of Drosophila. Cell 2015, 163, 340–353. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garfield, A.S.; Chan, W.S.; Dennis, R.J.; Ito, D.; Heisler, L.K.; Rochford, J.J. Neuroanatomical characterisation of the expression of the lipodystrophy and motor-neuropathy gene Bscl2 in adult mouse brain. PLoS ONE 2012, 7, e45790. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, X.; Xie, B.; Qi, Y.; Du, X.; Wang, S.; Zhang, Y.; Paxinos, G.; Yang, H.; Liang, H. The expression of SEIPIN in the mouse central nervous system. Brain Struct. Funct. 2016, 221, 4111–4127. [Google Scholar] [CrossRef]
- Poisson, A.; Chatron, N.; Labalme, A.; Till, M.; Broussolle, E.; Sanlaville, D.; Demily, C.; Lesca, G. Regressive Autism Spectrum Disorder Expands the Phenotype of BSCL2/Seipin-Associated Neurodegeneration. Biol. Psychiatry 2019, 85, e17–e19. [Google Scholar] [CrossRef]
- Opri, R.; Fabrizi, G.M.; Cantalupo, G.; Ferrarini, M.; Simonati, A.; Dalla Bernardina, B.; Darra, F. Progressive Myoclonus Epilepsy in Congenital Generalized Lipodystrophy type 2: Report of 3 cases and literature review. Seizure 2016, 42, 1–6. [Google Scholar] [CrossRef]
- Sanchez-Iglesias, S.; Crocker, M.; O’Callaghan, M.; Darling, A.; Garcia-Cazorla, A.; Domingo-Jimenez, R.; Castro, A.; Fernandez-Pombo, A.; Ruibal, A.; Aguiar, P.; et al. Celia’s encephalopathy and c.974dupG in BSCL2 gene: A hidden change in a known variant. Neurogenetics 2019, 20, 73–82. [Google Scholar] [CrossRef]
- Ferranti, S.; Lo Rizzo, C.; Renieri, A.; Galluzzi, P.; Grosso, S. Focus on progressive myoclonic epilepsy in Berardinelli-Seip syndrome. Neurol. Sci. Off. J. Ital. Neurol. Soc. Ital. Soc. Clin. Neurophysiol. 2020, 41, 3345–3348. [Google Scholar] [CrossRef]
- Wei, S.; Soh, S.L.; Xia, J.; Ong, W.Y.; Pang, Z.P.; Han, W. Motor neuropathy-associated mutation impairs Seipin functions in neurotransmission. J. Neurochem. 2014, 129, 328–338. [Google Scholar] [CrossRef] [PubMed]
- Ebihara, C.; Ebihara, K.; Aizawa-Abe, M.; Mashimo, T.; Tomita, T.; Zhao, M.; Gumbilai, V.; Kusakabe, T.; Yamamoto, Y.; Aotani, D.; et al. Seipin is necessary for normal brain development and spermatogenesis in addition to adipogenesis. Hum. Mol. Genet. 2015, 24, 4238–4249. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, G.; Zhou, L.; Zhu, Y.; Wang, C.; Sha, S.; Xian, X.; Ji, Y.; Liu, G.; Chen, L. Seipin knockout in mice impairs stem cell proliferation and progenitor cell differentiation in the adult hippocampal dentate gyrus via reduced levels of PPARgamma. Dis. Models Mech. 2015, 8, 1615–1624. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, L.; Chen, T.; Li, G.; Wu, C.; Wang, C.; Li, L.; Sha, S.; Chen, L.; Liu, G.; Chen, L. Activation of PPARgamma Ameliorates Spatial Cognitive Deficits through Restoring Expression of AMPA Receptors in Seipin Knock-Out Mice. J. Neurosci. Off. J. Soc. Neurosci. 2016, 36, 1242–1253. [Google Scholar] [CrossRef] [PubMed]
- Ito, D.; Suzuki, N. Seipinopathy: A novel endoplasmic reticulum stress-associated disease. Brain A J. Neurol. 2009, 132, 8–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lattanzi, G.; Maggi, L.; Araujo-Vilar, D. Laminopathies. Nucleus 2018, 9, 543–544. [Google Scholar] [CrossRef] [Green Version]
- Windpassinger, C.; Auer-Grumbach, M.; Irobi, J.; Patel, H.; Petek, E.; Horl, G.; Malli, R.; Reed, J.A.; Dierick, I.; Verpoorten, N.; et al. Heterozygous missense mutations in BSCL2 are associated with distal hereditary motor neuropathy and Silver syndrome. Nat. Genet. 2004, 36, 271–276. [Google Scholar] [CrossRef]
- Fernandez-Marmiesse, A.; Sanchez-Iglesias, S.; Darling, A.; O’Callaghan, M.M.; Tonda, R.; Jou, C.; Araujo-Vilar, D. A de novo heterozygous missense BSCL2 variant in 2 siblings with intractable developmental and epileptic encephalopathy. Seizure 2019, 71, 161–165. [Google Scholar] [CrossRef]
- Irobi, J.; Van den Bergh, P.; Merlini, L.; Verellen, C.; Van Maldergem, L.; Dierick, I.; Verpoorten, N.; Jordanova, A.; Windpassinger, C.; De Vriendt, E.; et al. The phenotype of motor neuropathies associated with BSCL2 mutations is broader than Silver syndrome and distal HMN type V. Brain A J. Neurol. 2004, 127, 2124–2130. [Google Scholar] [CrossRef]
- Choi, B.O.; Park, M.H.; Chung, K.W.; Woo, H.M.; Koo, H.; Chung, H.K.; Choi, K.G.; Park, K.D.; Lee, H.J.; Hyun, Y.S.; et al. Clinical and histopathological study of Charcot-Marie-Tooth neuropathy with a novel S90W mutation in BSCL2. Neurogenetics 2013, 14, 35–42. [Google Scholar] [CrossRef]
- Hsiao, C.T.; Tsai, P.C.; Lin, C.C.; Liu, Y.T.; Huang, Y.H.; Liao, Y.C.; Huang, H.W.; Lin, K.P.; Soong, B.W.; Lee, Y.C. Clinical and Molecular Characterization of BSCL2 Mutations in a Taiwanese Cohort with Hereditary Neuropathy. PLoS ONE 2016, 11, e0147677. [Google Scholar] [CrossRef]
- Auer-Grumbach, M.; Loscher, W.N.; Wagner, K.; Petek, E.; Korner, E.; Offenbacher, H.; Hartung, H.P. Phenotypic and genotypic heterogeneity in hereditary motor neuronopathy type V: A clinical, electrophysiological and genetic study. Brain A J. Neurol. 2000, 123, 1612–1623. [Google Scholar] [CrossRef] [Green Version]
- Ito, D.; Suzuki, N. Molecular pathogenesis of seipin/BSCL2-related motor neuron diseases. Ann. Neurol. 2007, 61, 237–250. [Google Scholar] [CrossRef]
- Araujo-Vilar, D.; Santini, F. Diagnosis and treatment of lipodystrophy: A step-by-step approach. J. Endocrinol. Investig. 2019, 42, 61–73. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patni, N.; Garg, A. Congenital generalized lipodystrophies--new insights into metabolic dysfunction. Nat. Rev. Endocrinol. 2015, 11, 522–534. [Google Scholar] [CrossRef]
- Agarwal, A.K.; Simha, V.; Oral, E.A.; Moran, S.A.; Gorden, P.; O’Rahilly, S.; Zaidi, Z.; Gurakan, F.; Arslanian, S.A.; Klar, A.; et al. Phenotypic and genetic heterogeneity in congenital generalized lipodystrophy. J. Clin. Endocrinol. Metab. 2003, 88, 4840–4847. [Google Scholar] [CrossRef] [PubMed]
- Brown, R.J.; Oral, E.A.; Cochran, E.; Araujo-Vilar, D.; Savage, D.B.; Long, A.; Fine, G.; Salinardi, T.; Gorden, P. Long-term effectiveness and safety of metreleptin in the treatment of patients with generalized lipodystrophy. Endocrine 2018, 60, 479–489. [Google Scholar] [CrossRef] [PubMed]
- Akinci, B.; Oral, E.A.; Neidert, A.; Rus, D.; Cheng, W.Y.; Thompson-Leduc, P.; Cheung, H.C.; Bradt, P.; Foss de Freitas, M.C.; Montenegro, R.M.; et al. Comorbidities and Survival in Patients With Lipodystrophy: An International Chart Review Study. J. Clin. Endocrinol. Metab. 2019, 104, 5120–5135. [Google Scholar] [CrossRef] [PubMed]
- Lima, J.G.; Nobrega, L.H.C.; Lima, N.N.; Dos Santos, M.C.F.; Silva, P.H.D.; Baracho, M.F.P.; Lima, D.N.; de Melo Campos, J.T.A.; Ferreira, L.C.; Freire Neto, F.P.; et al. Causes of death in patients with Berardinelli-Seip congenital generalized lipodystrophy. PLoS ONE 2018, 13, e0199052. [Google Scholar] [CrossRef] [Green Version]
- Alaei, M.R.; Talebi, S.; Ghofrani, M.; Taghizadeh, M.; Keramatipour, M. Whole Exome Sequencing Reveals a BSCL2 Mutation Causing Progressive Encephalopathy with Lipodystrophy (PELD) in an Iranian Pediatric Patient. Iran. Biomed. J. 2016, 20, 295–301. [Google Scholar] [CrossRef]
- Havel, L.S.; Wang, C.E.; Wade, B.; Huang, B.; Li, S.; Li, X.J. Preferential accumulation of N-terminal mutant huntingtin in the nuclei of striatal neurons is regulated by phosphorylation. Hum. Mol. Genet. 2011, 20, 1424–1437. [Google Scholar] [CrossRef] [PubMed]
- Aguzzi, A.; De Cecco, E. Shifts and drifts in prion science. Science 2020, 370, 32–34. [Google Scholar] [CrossRef]
- Wu, Y.R.; Hung, S.I.; Chang, Y.C.; Chen, S.T.; Lin, Y.L.; Chung, W.H. Complementary mutations in seipin gene in a patient with Berardinelli-Seip congenital lipodystrophy and dystonia: Phenotype variability suggests multiple roles of seipin gene. J. Neurol. Neurosurg. Psychiatry 2009, 80, 1180–1181. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.H.; Chen, T.H.; Hsiao, H.P.; Huang, C.T.; Wang, C.C.; Shiau, Y.H.; Chao, M.C. A Taiwanese boy with congenital generalized lipodystrophy caused by homozygous Ile262fs mutation in the BSCL2 gene. Kaohsiung J. Med Sci. 2010, 26, 615–620. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Chen, X.; Luo, F.; Jiang, L.; Xu, J.; Chen, S. Medical management of a child with congenital generalized lipodystrophy accompanied with progressive myoclonic epilepsy: A case report. Medicine 2019, 98, e18121. [Google Scholar] [CrossRef]
- Sanchez-Iglesias, S.; Fernandez-Pombo, A.; Araujo-Vilar, D. Focus on progressive myoclonic epilepsy in Berardinelli-Seip syndrome. Neurol. Sci. Off. J. Ital. Neurol. Soc. Ital. Soc. Clin. Neurophysiol. 2020. [Google Scholar] [CrossRef]
- Nagy, E.; Maquat, L.E. A rule for termination-codon position within intron-containing genes: When nonsense affects RNA abundance. Trends Biochem. Sci. 1998, 23, 198–199. [Google Scholar] [CrossRef]
- Serino, D.; Davico, C.; Specchio, N.; Marras, C.E.; Fioretto, F. Berardinelli-Seip syndrome and progressive myoclonus epilepsy. Epileptic Disord. Int. Epilepsy J. Videotape 2019, 21, 117–121. [Google Scholar] [CrossRef]
- Pedicelli, S.; de Palma, L.; Pelosini, C.; Cappa, M. Metreleptin for the treatment of progressive encephalopathy with/without lipodystrophy (PELD) in a child with progressive myoclonic epilepsy: A case report. Ital. J. Pediatr. 2020, 46, 158. [Google Scholar] [CrossRef]
- Hug, N.; Longman, D.; Caceres, J.F. Mechanism and regulation of the nonsense-mediated decay pathway. Nucleic Acids Res. 2016, 44, 1483–1495. [Google Scholar] [CrossRef] [Green Version]
- Nickless, A.; Bailis, J.M.; You, Z. Control of gene expression through the nonsense-mediated RNA decay pathway. Cell Biosci. 2017, 7, 26. [Google Scholar] [CrossRef]
- Brown, R.J.; Araujo-Vilar, D.; Cheung, P.T.; Dunger, D.; Garg, A.; Jack, M.; Mungai, L.; Oral, E.A.; Patni, N.; Rother, K.I.; et al. The Diagnosis and Management of Lipodystrophy Syndromes: A Multi-Society Practice Guideline. J. Clin. Endocrinol. Metab. 2016, 101, 4500–4511. [Google Scholar] [CrossRef]
- Araujo-Vilar, D.; Domingo-Jimenez, R.; Ruibal, A.; Aguiar, P.; Ibanez-Mico, S.; Garrido-Pumar, M.; Martinez-Olmos, M.A.; Lopez-Soler, C.; Guillin-Amarelle, C.; Gonzalez-Rodriguez, M.; et al. Association of metreleptin treatment and dietary intervention with neurological outcomes in Celia’s encephalopathy. Eur. J. Hum. Genet. EJHG 2018, 26, 396–406. [Google Scholar] [CrossRef] [Green Version]
- Aotani, D.; Ebihara, K.; Sawamoto, N.; Kusakabe, T.; Aizawa-Abe, M.; Kataoka, S.; Sakai, T.; Iogawa, H.; Ebihara, C.; Fujikura, J.; et al. Functional magnetic resonance imaging analysis of food-related brain activity in patients with lipodystrophy undergoing leptin replacement therapy. J. Clin. Endocrinol. Metab. 2012, 97, 3663–3671. [Google Scholar] [CrossRef] [Green Version]
- Paz-Filho, G.J. The Effects of Leptin Replacement on Neural Plasticity. Neural Plast. 2016, 2016, 8528934. [Google Scholar] [CrossRef] [Green Version]
- Farr, S.A.; Banks, W.A.; Morley, J.E. Effects of leptin on memory processing. Peptides 2006, 27, 1420–1425. [Google Scholar] [CrossRef]
- Fewlass, D.C.; Noboa, K.; Pi-Sunyer, F.X.; Johnston, J.M.; Yan, S.D.; Tezapsidis, N. Obesity-related leptin regulates Alzheimer’s Abeta. Faseb J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2004, 18, 1870–1878. [Google Scholar] [CrossRef]
- O’Malley, D.; MacDonald, N.; Mizielinska, S.; Connolly, C.N.; Irving, A.J.; Harvey, J. Leptin promotes rapid dynamic changes in hippocampal dendritic morphology. Mol. Cell. Neurosci. 2007, 35, 559–572. [Google Scholar] [CrossRef] [PubMed]
- Udagawa, J.; Nimura, M.; Otani, H. Leptin affects oligodendroglial development in the mouse embryonic cerebral cortex. Neuro Endocrinol. Lett. 2006, 27, 177–182. [Google Scholar] [PubMed]
- Hadley, K.B.; Ryan, A.S.; Nelson, E.B.; Salem, N. An assessment of dietary docosahexaenoic acid requirements for brain accretion and turnover during early childhood. World Rev. Nutr. Diet. 2009, 99, 97–104. [Google Scholar] [CrossRef]
- Innis, S.M. Dietary omega 3 fatty acids and the developing brain. Brain Res. 2008, 1237, 35–43. [Google Scholar] [CrossRef] [PubMed]
- Calderon, F.; Kim, H.Y. Docosahexaenoic acid promotes neurite growth in hippocampal neurons. J. Neurochem. 2004, 90, 979–988. [Google Scholar] [CrossRef] [PubMed]
- Moriguchi, T.; Harauma, A.; Salem, N., Jr. Plasticity of mouse brain docosahexaenoic acid: Modulation by diet and age. Lipids 2013, 48, 343–355. [Google Scholar] [CrossRef]
- Lei, E.; Vacy, K.; Boon, W.C. Fatty acids and their therapeutic potential in neurological disorders. Neurochem. Int. 2016, 95, 75–84. [Google Scholar] [CrossRef]
- Sanchez-Iglesias, S.; Unruh-Pinheiro, A.; Guillin-Amarelle, C.; Gonzalez-Mendez, B.; Ruiz-Riquelme, A.; Rodriguez-Canete, B.L.; Rodriguez-Garcia, S.; Guillen-Navarro, E.; Domingo-Jimenez, R.; Araujo-Vilar, D. Skipped BSCL2 Transcript in Celia’s Encephalopathy (PELD): New Insights on Fatty Acids Involvement, Senescence and Adipogenesis. PLoS ONE 2016, 11, e0158874. [Google Scholar] [CrossRef] [PubMed]
- Vargas, C.R.; Wajner, M.; Sirtori, L.R.; Goulart, L.; Chiochetta, M.; Coelho, D.; Latini, A.; Llesuy, S.; Bello-Klein, A.; Giugliani, R.; et al. Evidence that oxidative stress is increased in patients with X-linked adrenoleukodystrophy. Biochim. Et Biophys. Acta 2004, 1688, 26–32. [Google Scholar] [CrossRef] [Green Version]
- Muller, C.C.; Nguyen, T.H.; Ahlemeyer, B.; Meshram, M.; Santrampurwala, N.; Cao, S.; Sharp, P.; Fietz, P.B.; Baumgart-Vogt, E.; Crane, D.I. PEX13 deficiency in mouse brain as a model of Zellweger syndrome: Abnormal cerebellum formation, reactive gliosis and oxidative stress. Dis. Models Mech. 2011, 4, 104–119. [Google Scholar] [CrossRef] [Green Version]
- Kiaei, M.; Kipiani, K.; Chen, J.; Calingasan, N.Y.; Beal, M.F. Peroxisome proliferator-activated receptor-gamma agonist extends survival in transgenic mouse model of amyotrophic lateral sclerosis. Exp. Neurol. 2005, 191, 331–336. [Google Scholar] [CrossRef]
- Schutz, B.; Reimann, J.; Dumitrescu-Ozimek, L.; Kappes-Horn, K.; Landreth, G.E.; Schurmann, B.; Zimmer, A.; Heneka, M.T. The oral antidiabetic pioglitazone protects from neurodegeneration and amyotrophic lateral sclerosis-like symptoms in superoxide dismutase-G93A transgenic mice. J. Neurosci. Off. J. Soc. Neurosci. 2005, 25, 7805–7812. [Google Scholar] [CrossRef] [Green Version]
- Esmaeili, M.A.; Yadav, S.; Gupta, R.K.; Waggoner, G.R.; Deloach, A.; Calingasan, N.Y.; Beal, M.F.; Kiaei, M. Preferential PPAR-alpha activation reduces neuroinflammation, and blocks neurodegeneration in vivo. Hum. Mol. Genet. 2016, 25, 317–327. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dumont, M.; Stack, C.; Elipenahli, C.; Jainuddin, S.; Gerges, M.; Starkova, N.; Calingasan, N.Y.; Yang, L.; Tampellini, D.; Starkov, A.A.; et al. Bezafibrate administration improves behavioral deficits and tau pathology in P301S mice. Hum. Mol. Genet. 2012, 21, 5091–5105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johri, A.; Calingasan, N.Y.; Hennessey, T.M.; Sharma, A.; Yang, L.; Wille, E.; Chandra, A.; Beal, M.F. Pharmacologic activation of mitochondrial biogenesis exerts widespread beneficial effects in a transgenic mouse model of Huntington’s disease. Hum. Mol. Genet. 2012, 21, 1124–1137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, J.; Racke, M.K.; Drew, P.D. Peroxisome proliferator-activated receptor-alpha agonist fenofibrate regulates IL-12 family cytokine expression in the CNS: Relevance to multiple sclerosis. J. Neurochem. 2007, 103, 1801–1810. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kreisler, A.; Gele, P.; Wiart, J.F.; Lhermitte, M.; Destee, A.; Bordet, R. Lipid-lowering drugs in the MPTP mouse model of Parkinson’s disease: Fenofibrate has a neuroprotective effect, whereas bezafibrate and HMG-CoA reductase inhibitors do not. Brain Res. 2007, 1135, 77–84. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
| Case | BSCL2 Variant | BSCL2 Reported Variant | Genotype | Sex | Reported Age | GL | Encephalopathy | Seizures | Other Neurological Symptoms | Other Non-Neurological Symptoms | Treatment | Deceased (age, years) | Ref. |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 985C>T | 985C>T | Homozygote | F | D | N | Y | Generalized tonic-clonic | Poor motor coordination, ataxic gait, generalized fine tremor, dystonia, sleep disturbances, severe spasticity, tetraparesis, pyramidal and extrapyramidal signs, loss of motor skills, social, language and cognitive development | Hypertriglyceridemia, hepatic steatosis | Antiepileptic | Y (8) | [1] |
| 2 | 985C>T;507_511del | 985C>T;507_511del | Compound heterozygote | F | 11 | Y | Y | Progressive myoclonic epilepsy | Gait ataxia, intellectual disability, dystonia, difficulty swallowing, loss of language | Hypertriglyceridemia, hepatic steatosis | Anti-epileptic, metreleptin, n-3 FA, fenofibrate, pioglitazone | N | [1] |
| 3 | 985C>T;538G>T | 985C>T;538G>T | Compound heterozygote | M | D | Y | Y | Progressive myoclonic epilepsy | Cognitive impairment, pyramidal signs, language loss, dystonia, difficulty swallowing | N.R. | Antiepileptic | Y (8) | [1] |
| 4 | 985C>T;507_511del | 985C>T;507_511del | Compound heterozygote | M | D | Y | Y | Progressive myoclonic epilepsy | Cognitive impairment, ataxic gait | N.R. | Antiepileptic | Y (7) | [1] |
| 5 | 985C>T;507_511del | 985C>T;507_511del | Compound heterozygote | M | D | Y | Y | Progressive myoclonic epilepsy | Cognitive impairment, ataxic gait | Hypertriglyceridemia, hepatic steatosis | Antiepileptic | Y (7) | [1] |
| 6 | 985C>T | 985C>T | Homozygote | F | D | N | Y | Progressive myoclonic epilepsy | Cognitive impairment, irritability, dysphagia, sleep disorder and pyramidal signs, ataxic gait | N.R. | Antiepileptic | Y (6) | [1] |
| 7 | 985C>T | 985C>T | Homozygote | M | D | Y | Y | Myoclonic | Autism, repetitive and stereotypic hand movements, repetitive upward staring, ataxia, generalized hypertonia, severe global developmental delay | Inguinal hernia, generalized hypertrichosis | Antiepileptic | Y (8) | [81] |
| 8 | 985C>T | Not published | Homozygote | M | 5 | Y | Y | Myoclonic | Gait ataxia, intellectual disability | Hypertriglyceridemia, hepatic steatosis | Antiepileptic | N | (not published) |
| 9 | 985C>T;1004A>C | 985C>T;1004A>C | Compound heterozygote | F | D | N | Y | N | Regressive autism spectrum disorder, atypical Parkinsonism, loss of communication and language skills, dystonic hypertonia, and extrapyramidal and pyramidal features, camptocormia, dysphagia marked frontal lobe syndrome | N.R. | None | Y (28) | [58] |
| 10 | 974dupG | 974dupG | Homozygote | F | D | Y | Y | Myoclonic | Language delay, myoclonus, dystonia, seizures, gait ataxia, abnormal behavior | Hypertriglyceridemia, hepatic steatosis | Antiepileptic | Y (9) | [60] |
| 11 | 974dupG;1015C>T | 974dupG;1015C>T | Compound heterozygote | F | 2 | Y | N | N | Language delay | Hypertriglyceridemia, hepatic steatosis, hyperinsulinemia | Metreleptin, n-3 FA | N | [60] |
| 12 | 974dupG;1020_1021delAA | 782_783dupG; 828_829delAA | Compound heterozygote | M | D | Y | Y | Absence seizures; eyelid myoclonus | Pyramidal signs, loss of language, dystonic tetraplegia | Hypertriglyceridemia, hypertransaminasemia, hepatic steatosis, hypertrophic cardiomyopathy | Antiepileptic | Y (9) | [59] |
| 13 | 974dupG | 782_783dupG | Homozygote | F | D | Y | Y | Absence seizures, slight eyelid myoclonus, myoclonic-atonic seizures | Cognitive decline, pyramidal signs, tetraparesis, intellectual disability | Hypertriglyceridemia, muscle hypertrophy, hepatic steatosis, cardiomegaly, acanthosis nigricans | Antiepileptic | Y (7) | [59] |
| 14 | 974dupG | 782_783dupG | Homozygote | F | D | Y | Y | Absence seizures, slight eyelid myoclonus, myoclonic-atonic seizures | Cognitive decline, pyramidal signs, tetraparesis, intellectual disability | Hypertriglyceridemia | Antiepileptic | Y (11) | [59] |
| 15 | 974dupG | 782dupG | Homozygote | M | 17 | Y | Y | Progressive myoclonic epilepsy | Generalized hypertonia | None | Antiepileptic | N | [86] |
| 16 | 974dupG; 757G>T | 783insG;565G>T | Compound heterozygote | M | 28 | Y | N | N | Gait disturbance, torticollis, abnormal posturing of the fingers, axial dystonia abnormal thigh abduction and foot dystonia, mild intellectual disability | Muscular hypertrophy, acromegaloid features, acanthosis nigricans, hypertriglyceridemia, hepatic steatosis | — | N | [84] |
| 17 | 974dupG | 783insG | Homozygote | M | 1.8 | Y | N | N | None | Prominent musculature, eruptive xanthomas, hepatic steatosis, hypertriglyceridemia, hyperinsulinemia. | Fenofibrate | N | [85] |
| 18 | 974dupG | 1126insG | Heterozygote | F | 9 | Y | N.R. | N.R. | Intellectual disability | Diabetes mellitus | N.R. | N.R. | [77] |
| 19 | 1048C>T | 1048C>T | Homozygote | F | 15 | Y | Y | Progressive myoclonic epilepsy | Dyskinetic, distonic, and choreoathetotic movements, psychomotor regression, language impairment, bulbar signs, loss of gait | Coarse facial features, synophrys, bulbous nasal tip, large ear pinnae, wide mouth, long fingers and toes, hypertrichosis | Antiepileptic | N | [61] |
| 20 | 1048C>T | 1048C>T | Homozygote | F | 18 | Y | Y | Progressive myoclonic epilepsy | Dyskinetic, distonic, and choreoathetotic movements, psychomotor regression, language impairment, bulbar signs, loss of gait, aphasia | Coarse facial features, synophrys, bulbous nasal tip, large ear pinnae, wide mouth, long fingers and toes, hypertrichosis, abnormal glucose tolerance | Antiepileptic | Y (18) | [61] |
| 21 | 1076dupC | 1076dupC | Homozygote | M | 6 | Y | Y | Progressive myoclonic epilepsy | Language impairment, severe hyperactivity, drop attacks, ataxic gate, psychomotor delay, ataxic gait | Hypertriglyceridemia, hypertransaminasemia, hepatic steatosis | Antiepileptic | N | [89] |
| 22 | 566T>A | 566T>A | Heterozygote | M | 10 | N | Y | Generalized tonic-clonic | Delays in social skills, eyelid myoclonus, absence, atonic seizures, moderate intellectual disability, autism spectrum disorder, ataxic gait | None | Antiepileptic | N | [69] |
| 23 | 566T>A | 566T>A | Heterozygote | F | 0.9 | N | Y | Asymmetric tonic seizures | Mild psychomotor delay | None | Antiepileptic | Y (0.9) | [69] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sánchez-Iglesias, S.; Fernández-Pombo, A.; Cobelo-Gómez, S.; Hermida-Ameijeiras, Á.; Alarcón-Martínez, H.; Domingo-Jiménez, R.; Ruíz Riquelme, A.I.; Requena, J.R.; Araújo-Vilar, D. Celia’s Encephalopathy (BSCL2-Gene-Related): Current Understanding. J. Clin. Med. 2021, 10, 1435. https://0-doi-org.brum.beds.ac.uk/10.3390/jcm10071435
Sánchez-Iglesias S, Fernández-Pombo A, Cobelo-Gómez S, Hermida-Ameijeiras Á, Alarcón-Martínez H, Domingo-Jiménez R, Ruíz Riquelme AI, Requena JR, Araújo-Vilar D. Celia’s Encephalopathy (BSCL2-Gene-Related): Current Understanding. Journal of Clinical Medicine. 2021; 10(7):1435. https://0-doi-org.brum.beds.ac.uk/10.3390/jcm10071435
Chicago/Turabian StyleSánchez-Iglesias, Sofía, Antía Fernández-Pombo, Silvia Cobelo-Gómez, Álvaro Hermida-Ameijeiras, Helena Alarcón-Martínez, Rosario Domingo-Jiménez, Alejandro Iván Ruíz Riquelme, Jesús R. Requena, and David Araújo-Vilar. 2021. "Celia’s Encephalopathy (BSCL2-Gene-Related): Current Understanding" Journal of Clinical Medicine 10, no. 7: 1435. https://0-doi-org.brum.beds.ac.uk/10.3390/jcm10071435