
A Review of the Molecular Mechanisms Underlying Cardiac Fibrosis and Atrial Fibrillation


Abstract
:1. Introduction
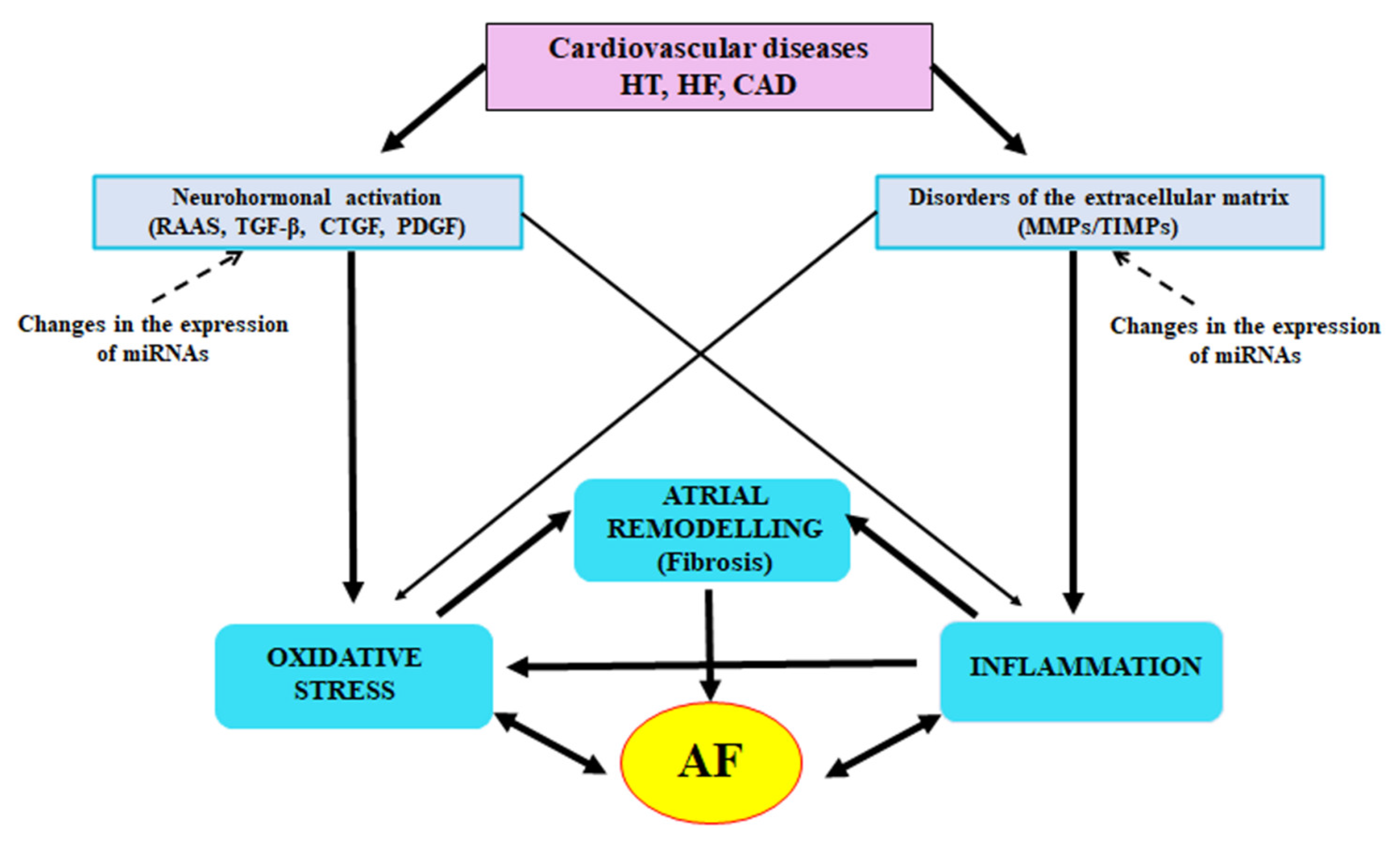
2. The Role of Inflammatory Condition in AF Induction
3. Neurohormonal Mechanisms
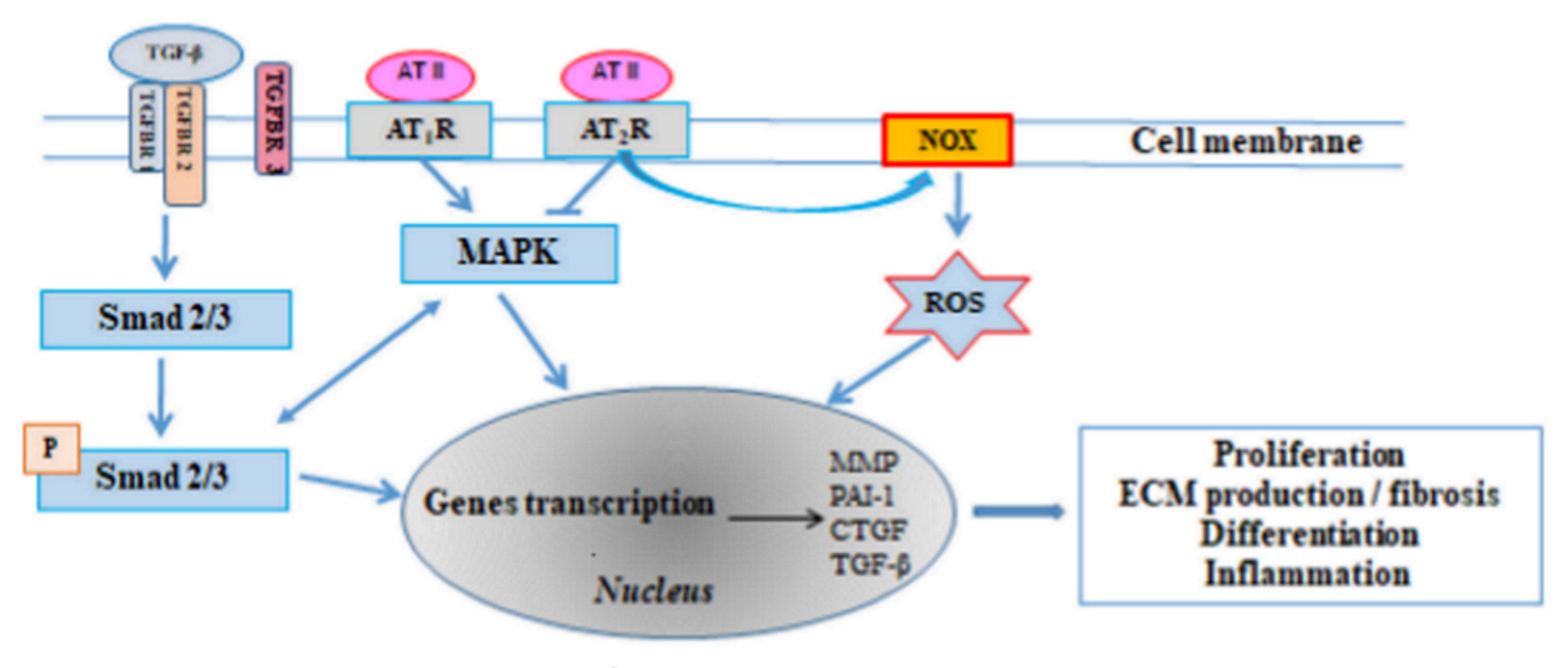
3.1. The Renin-Angiotensin-Aldosterone System (RAAS)
3.2. Transforming Growth Factor β (TGF-β)
3.3. Platelet-Derived Growth Factor (PDGF)
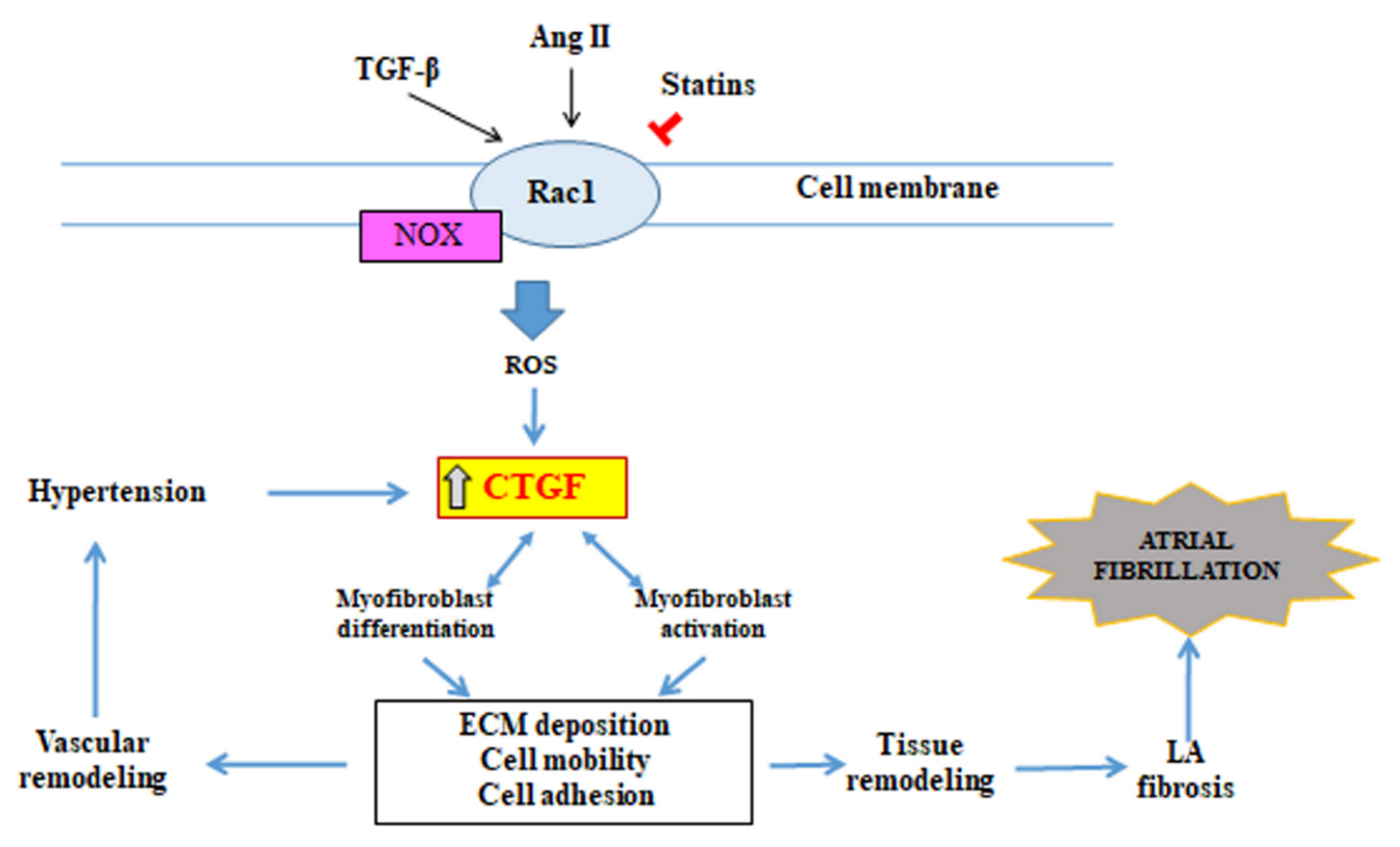
3.4. Connective Tissue Growth Factor (CTGF)
4. Participation of Extracellular Matrix, Matrix Metalloproteinases and Their Inhibitors in Fibrotic Process
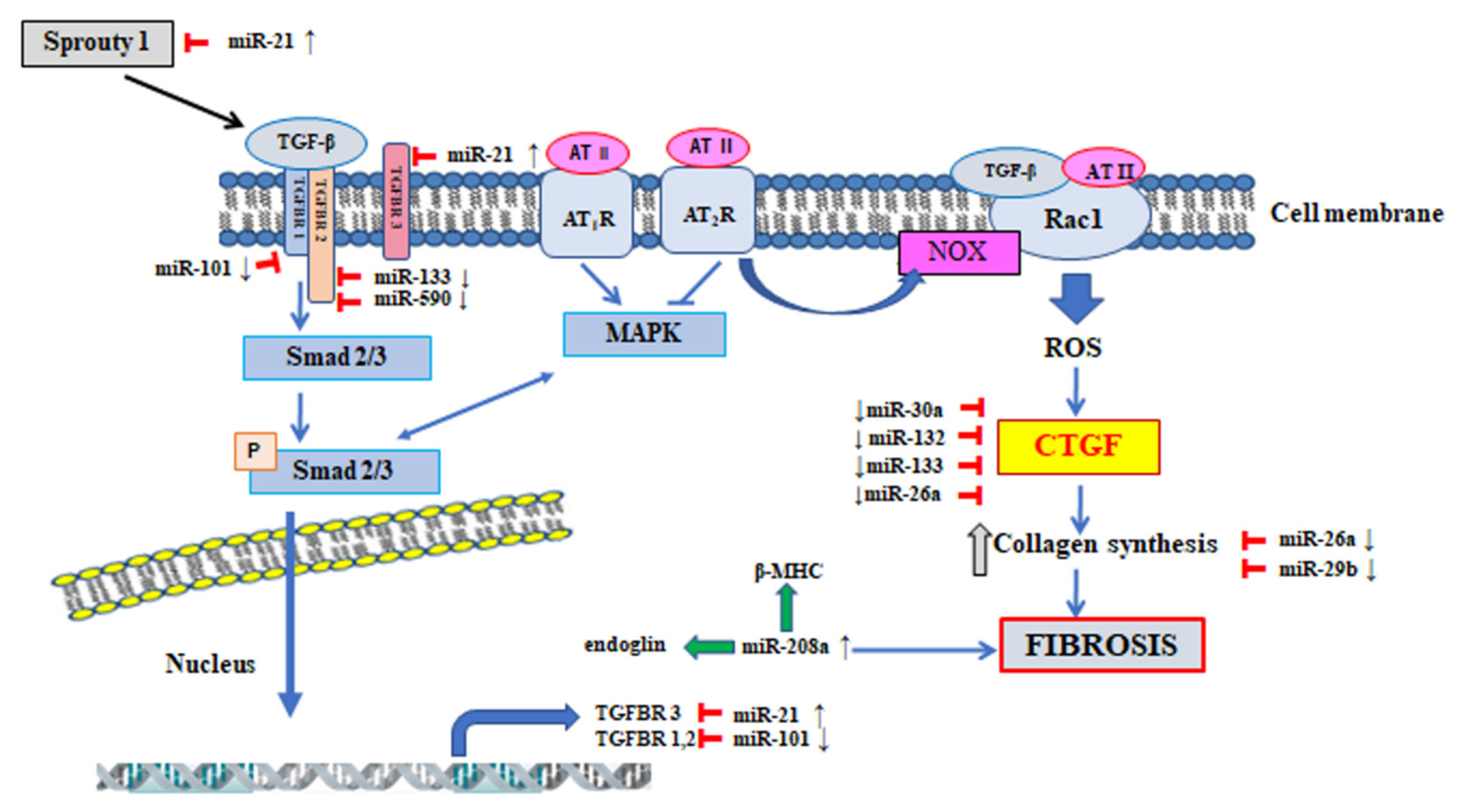
5. The Participation of MicroRNA in the Regulation of Signalling Pathway Involved in the Pathogenesis of Atrial Fibrillation
6. Conclusions and Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Sohns, C.; Marrouche, N.F. Atrial fibrillation and cardiac fibrosis. Eur. Heart J. 2020, 41, 1123–1131. [Google Scholar] [CrossRef] [PubMed]
- Schirone, L.; Forte, M.; Palmerio, S.; Yee, D.; Nocella, C.; Angelini, F.; Pagano, F.; Schiavon, S.; Bordin, A.; Carrizzo, A.; et al. A review of the molecular mechanisms underlying the development and progression of cardiac remodeling. Oxid. Med. Cell. Longev. 2017, 2017, 3920195. [Google Scholar] [CrossRef] [PubMed]
- Nattel, S. Molecular and cellular mechanisms of atrial fibrosis in atrial fibrillation. JACC Clin. Electrophysiol. 2017, 3, 425–435. [Google Scholar] [CrossRef] [PubMed]
- Nattel, S.; Harada, M. Atrial remodeling and atrial fibrillation: Recent advances and translational perspectives. J. Am. Coll. Cardiol. 2014, 63, 2335–2345. [Google Scholar] [CrossRef] [Green Version]
- Chen, L.Y.; Shen, W.K. Epidemiology of atrial fibrillation: A current perspective. Heart Rhythm 2007, 4, S1–S6. [Google Scholar] [CrossRef]
- Nakano, Y.; Niida, S.; Dote, K.; Takenaka, S.; Hirao, H.; Miura, F.; Ishida, M.; Shingu, T.; Sueda, T.; Yoshizumi, M.; et al. Matrix metalloproteinase-9 contributes to human atrial remodeling during atrial fibrillation. J. Am. Coll. Cardiol. 2004, 43, 818–825. [Google Scholar] [CrossRef] [Green Version]
- Wijesurendra, R.S.; Casadei, B. Atrial fibrillation: Effects beyond the atrium? Cardiovasc. Res. 2015, 105, 238–247. [Google Scholar] [CrossRef] [Green Version]
- Anné, W.; Willems, R.; Roskams, T.; Sergeant, P.; Herijgers, P.; Holemans, P.; Ector, H.; Heidbüchel, H. Matrix metalloproteinases and atrial remodeling in patients with mitral valve disease and atrial fibrillation. Cardiovasc. Res. 2005, 67, 655–666. [Google Scholar] [CrossRef] [Green Version]
- Thomas, L.; Abhayaratna, W.P. Left atrial reverse remodeling mechanisms, evaluation, and clinical significance. JACC Cardiovasc. Imaging 2017, 10, 65–77. [Google Scholar] [CrossRef]
- Li, C.Y.; Zhang, J.R.; Hu, W.N.; Li, S.N. Atrial fibrosis underlying atrial fibrillation (Review). Int. J. Mol. Med. 2021, 47, 9. [Google Scholar] [CrossRef]
- Lin, Y.K.; Chen, Y.A.; Lee, T.I.; Chen, Y.C.; Chen, S.A.; Chen, Y.J. Aging modulates the substrate and triggers remodeling in atrial fibrillation. Circ. J. 2018, 82, 1237–1244. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gullón, A.; Formiga, F.; Camafort, M.; Mostaza, J.M.; Díez-Manglano, J.; Cepeda, J.M.; Novo-Veleiro, I.; Pose, A.; Suárez Fernández, C.; NONAVASC Study Group. Vascular Risk Group of the Spanish Society of Internal Medicine. Baseline functional status as the strongest predictor of in-hospital mortality in elderly patients with non-valvular atrial fibrillation: Results of the NONAVASC registry. Eur. J. Intern. Med. 2018, 47, 69–74. [Google Scholar] [CrossRef]
- Allessie, M.; Ausma, J.; Schotten, U. Electrical, contractile and structural remodeling during atrial fibrillation. Cardiovasc. Res. 2002, 54, 230–246. [Google Scholar] [CrossRef]
- Kostin, S.; Klein, G.; Szalay, Z.; Hein, S.; Bauer, E.P.; Schaper, J. Structural correlate of atrial fibrillation in human patients. Cardiovasc. Res. 2002, 54, 361–379. [Google Scholar] [CrossRef] [Green Version]
- Andrade, J.; Khairy, P.; Dobrev, D.; Nattel, S. The clinical profile and pathophysiology of atrial fibrillation: Relationships among clinical features, epidemiology, and mechanisms. Circ. Res. 2014, 114, 1453–1468. [Google Scholar] [CrossRef]
- King, J.B.; Azadani, P.N.; Suksaranjit, P.; Bress, A.P.; Witt, D.M.; Han, F.T.; Chelu, M.G.; Silver, M.A.; Biskupiak, J.; Wilson, B.D.; et al. Left atrial fibrosis and risk of cerebrovascular and cardiovascular events in patients with atrial fibrillation. J. Am. Coll. Cardiol. 2017, 70, 1311–1321. [Google Scholar] [CrossRef] [PubMed]
- Zapolski, T.; Wysokiński, A. Atrial cardiomyopathy as a consequence of atrial fibrillation. Acta Cardiol. 2002, 57, 84–86. [Google Scholar] [PubMed]
- Gutierrez, C.; Blanchard, D.G. Diagnosis and Treatment of Atrial Fibrillation. Am. Fam. Phys. 2016, 94, 442–452. [Google Scholar]
- Fatimah, L. Paroxysmal supraventricular tachycardia and atrial fibrillation in the same patient: What seems to be the link? Biomed. J. Sci. Tech. Res. 2021, 37, 29314–29317. [Google Scholar] [CrossRef]
- Bruins, P.; de Velthuis, H.; Yazdanbakhsh, A.P.; Jansen, P.G.; van Hardevelt, F.W.; de Beaumont, E.M.; Wildevuur, C.R.; Eijsman, L.; Trouwborst, A.; Hack, C.E. Activation of the complement system during and after cardiopulmonary bypass surgery: Postsurgery activation involves C-reactive protein and is associated with postoperative arrhythmia. Circulation 1997, 96, 3542–3548. [Google Scholar] [CrossRef]
- Zhang, H.; Li, J.; Chen, X.; Wu, N.; Xie, W.; Tang, H.; Li, C.; Wu, L.; Xiang, Y.; Zhong, L.; et al. Association of systemic inflammation score with atrial fibrillation: A case-control study with propensity score matching. Heart Lung Circ. 2018, 27, 489–496. [Google Scholar] [CrossRef]
- Boos, C.J.; Anderson, R.A.; Lip, G.Y. Is atrial fibrillation an inflammatory disorder? Eur. Heart J. 2006, 27, 136–149. [Google Scholar] [CrossRef] [Green Version]
- Zhou, X.; Dudley, S.C., Jr. Evidence for inflammation as a driver of atrial fibrillation. Front. Cardiovasc. Med. 2020, 7, 62. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.F.; Chen, Y.J.; Lin, Y.J.; Chen, S.A. Inflammation and the pathogenesis of atrial fibrillation. Nat. Rev. Cardiol. 2015, 12, 230–243. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Solus, J.; Chen, Q.; Rho, H.R.; Milne, G.; Stein, M.; Darbar, D. The role of inflammation and oxidative stress in atrial fibrillation. Heart Rhythm 2010, 7, 438–444. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jalife, J. Mechanisms of persistent atrial fibrillation. Curr. Opin. Cardiol. 2014, 29, 20–27. [Google Scholar] [CrossRef] [PubMed]
- Jabati, S.; Fareed, J.; Liles, J.; Otto, A.; Hoppensteadt, D.; Bontekoe, J.; Phan, T.; Walborn, A.; Syed, M. Biomarkers of inflammation, thrombogenesis, and collagen turnover in patients with atrial fibrillation. Clin. Appl. Thromb. Hemost. 2018, 24, 718–723. [Google Scholar] [CrossRef]
- Hijazi, Z.; Aulin, J.; Andersson, U.; Alexander, J.H.; Gersh, B.; Granger, C.B.; Hanna, M.; Horowitz, J.; Hylek, E.M.; Lopes, R.D.; et al. Biomarkers of inflammation and risk of cardiovascular events in anticoagulated patients with atrial fibrillation. Heart 2016, 102, 508–517. [Google Scholar] [CrossRef] [Green Version]
- Li, T.; Sun, Z.L.; Xie, Q.Y. Meta-analysis identifies serum C-reactive protein as an indicator of atrial fibrillation risk after coronary artery bypass graft. Am. J. Ther. 2016, 23, e1586–e1596. [Google Scholar] [CrossRef]
- Jiang, Z.; Dai, L.; Song, Z.; Li, H.; Shu, M. Association between C-reactive protein and atrial fibrillation recurrence after catheter ablation: A meta-analysis. Clin. Cardiol. 2013, 36, 548–554. [Google Scholar] [CrossRef]
- Boldt, A.; Wetzel, U.; Weigl, J.; Garbade, J.; Lauschke, J.; Hindricks, G.; Kottkamp, H.; Gummert, J.F.; Dhein, S. Expression of angiotensin II receptors in human left and right atrial tissue in atrial fibrillation with and without underlying mitral valve disease. J. Am. Coll. Cardiol. 2003, 42, 1785–1792. [Google Scholar] [CrossRef] [Green Version]
- Goette, A.; Staack, T.; Röcken, C.; Arndt, M.; Geller, J.C.; Huth, C.; Ansorge, S.; Klein, H.U.; Lendeckel, U. Increased expression of extracellular signal-regulated kinase and angiotensin-converting enzyme in human atria during atrial fibrillation. J. Am. Coll. Cardiol. 2000, 35, 1669–1677. [Google Scholar] [CrossRef] [Green Version]
- Xiao, H.D.; Fuchs, S.; Campbell, D.J.; Lewis, W.; Dudley, S.C., Jr.; Kasi, V.S.; Hoit, B.D.; Keshelava, G.; Zhao, H.; Capecchi, M.R.; et al. Mice with cardiac-restricted angiotensin-converting enzyme (ACE) have atrial enlargement, cardiac arrhythmia, and sudden death. Am. J. Pathol. 2004, 165, 1019–1032. [Google Scholar] [CrossRef] [Green Version]
- Putnam, K.; Shoemaker, R.; Yiannikouris, F.; Cassis, L.A. The renin-angiotensin system: A target of and contributor to dyslipidemias, altered glucose homeostasis, and hypertension of the metabolic syndrome. Am. J. Physiol. Heart Circ. Physiol. 2012, 302, H1219–H1230. [Google Scholar] [CrossRef] [Green Version]
- Spät, A.; Hunyady, L. Control of aldosterone secretion: A model for convergence in cellular signaling pathways. Physiol. Rev. 2004, 84, 489–539. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tomaschitz, A.; Pilz, S.; Ritz, E.; Obermayer-Pietsch, B.; Pieber, T.R. Aldosterone and arterial hypertension. Nat. Rev. Endocrinol. 2010, 6, 83–93. [Google Scholar] [CrossRef] [PubMed]
- López-Andrés, N.; Martin-Fernandez, B.; Rossignol, P.; Zannad, F.; Lahera, V.; Fortuno, M.A.; Cachofeiro, V.; Díez, J. A role for cardiotrophin-1 in myocardial remodeling induced by aldosterone. Am. J. Physiol. Heart Circ. Physiol. 2011, 301, H2372–H2382. [Google Scholar] [CrossRef] [Green Version]
- Dartsch, T.; Fischer, R.; Gapelyuk, A.; Weiergraeber, M.; Ladage, D.; Schneider, T.; Schirdewan, A.; Reuter, H.; Mueller-Ehmsen, J.; Zobel, C. Aldosterone induces electrical remodeling independent of hypertension. Int. J. Cardiol. 2013, 164, 170–178. [Google Scholar] [CrossRef] [PubMed]
- Qin, W.; Rudolph, A.E.; Bond, B.R.; Rocha, R.; Blomme, E.A.; Goellner, J.J.; Funder, J.W.; McMahon, E.G. Transgenic model of aldosterone-driven cardiac hypertrophy and heart failure. Circ. Res. 2003, 93, 69–76. [Google Scholar] [CrossRef] [Green Version]
- Tsybouleva, N.; Zhang, L.; Chen, S.; Patel, R.; Lutucuta, S.; Nemoto, S.; DeFreitas, G.; Entman, M.; Carabello, B.A.; Roberts, R.; et al. Aldosterone, through novel signaling proteins, is a fundamental molecular bridge between the genetic defect and the cardiac phenotype of hypertrophic cardiomyopathy. Circulation 2004, 109, 1284–1291. [Google Scholar] [CrossRef]
- Hiroki, J.; Shimokawa, H.; Higashi, M.; Morikawa, K.; Kandabashi, T.; Kawamura, N.; Kubota, T.; Ichiki, T.; Amano, M.; Kaibuchi, K.; et al. Inflammatory stimuli upregulate Rho-kinase in human coronary vascular smooth muscle cells. J. Mol. Cell Cardiol. 2004, 37, 537–546. [Google Scholar] [CrossRef] [PubMed]
- Shimokawa, H.; Satoh, K. 2015 ATVB Plenary Lecture: Translational research on Rho-kinase in cardiovascular medicine. Arterioscler. Thromb. Vasc. Biol. 2015, 35, 1756–1769. [Google Scholar] [CrossRef] [PubMed]
- Iwashima, F.; Yoshimoto, T.; Minami, I.; Sakurada, M.; Hirono, Y.; Hirata, Y. Aldosterone induces superoxide generation via Rac1 activation in endothelial cells. Endocrinology 2008, 149, 1009–1014. [Google Scholar] [CrossRef] [PubMed]
- Vahebi, S.; Kobayashi, T.; Warren, C.M.; de Tombe, P.P.; Solaro, R.J. Functional effects of Rho-kinase-dependent phosphorylation of specific sites on cardiac troponin. Circ. Res. 2005, 96, 740–747. [Google Scholar] [CrossRef] [Green Version]
- Li, D.; Shinagawa, K.; Pang, L.; Leung, T.K.; Cardin, S.; Wang, Z.; Nattel, S. Effects of angiotensin-converting enzyme inhibition on the development of the atrial fibrillation substrate in dogs with ventricular tachypacing-induced congestive heart failure. Circulation 2001, 104, 2608–2614. [Google Scholar] [CrossRef] [Green Version]
- Leask, A. Potential therapeutic targets for cardiac fibrosis: TGFbeta, angiotensin, endothelin, CCN2, and PDGF, partners in fibroblast activation. Circ. Res. 2010, 106, 1675–1680. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Conway, S.J.; Molkentin, J.D. Periostin as a heterofunctional regulator of cardiac development and disease. Curr. Genom. 2008, 9, 548–555. [Google Scholar] [CrossRef] [Green Version]
- Bornstein, P. Matricellular proteins: An overview. J. Cell Commun. Signal. 2009, 3, 163–165. [Google Scholar] [CrossRef] [Green Version]
- Heger, J.; Warga, B.; Meyering, B.; Abdallah, Y.; Schlüter, K.D.; Piper, H.M.; Euler, G. TGFβ receptor activation enhances cardiac apoptosis via SMAD activation and concomitant NO release. J. Cell. Physiol. 2011, 226, 2683–2690. [Google Scholar] [CrossRef]
- Rodríguez-Vita, J.; Sánchez-López, E.; Esteban, V.; Rupérez, M.; Egido, J.; Ruiz-Ortega, M. Angiotensin II activates the Smad pathway in vascular smooth muscle cells by a transforming growth factor-beta-independent mechanism. Circulation 2005, 111, 2509–2517. [Google Scholar] [CrossRef] [Green Version]
- Mehta, P.K.; Griendling, K.K. Angiotensin II cell signaling: Physiological and pathological effects in the cardiovascular system. Am. J. Physiol. Cell Physiol. 2007, 292, C82–C97. [Google Scholar] [CrossRef]
- Sciarretta, S.; Paneni, F.; Palano, F.; Chin, D.; Tocci, G.; Rubattu, S.; Volpe, M. Role of the renin-angiotensin-aldosterone system and inflammatory processes in the development and progression of diastolic dysfunction. Clin. Sci. 2009, 116, 467–477. [Google Scholar] [CrossRef] [Green Version]
- Richter, K.; Kietzmann, T. Reactive oxygen species and fibrosis: Further evidence of a significant liaison. Cell Tissue Res. 2016, 365, 591–605. [Google Scholar] [CrossRef] [Green Version]
- Su, F.; Zhang, W.; Chen, Y.; Ma, L.; Zhang, H.; Wang, F. Significance of hypoxia-inducible factor-1α expression with atrial fibrosis in rats induced with isoproterenol. Exp. Ther. Med. 2014, 8, 1677–1682. [Google Scholar] [CrossRef] [Green Version]
- Tuuminen, R.; Nykanen, A.I.; Krebs, R.; Saronen, J.; Pajusola, K.; Keranen, M.A.I.; Koskinen, P.K.; Alitalo, K.; Lemstrom, K.B. PDGF-A, -C, and -D but not PDGF-B increase TGF-β-1 and chronic rejection in rat cardiac allografts. Arterioscler. Thromb. Vasc. Biol. 2009, 29, 691–698. [Google Scholar] [CrossRef] [Green Version]
- Zhao, T.; Zhao, W.; Chen, Y.; Li, V.S.; Meng, W.; Sun, Y. Platelet-derived growth factor-D promotes fibrogenesis of cardiac fibroblasts. Am. J. Physiol. Heart Circ. Physiol. 2013, 304, H1719–H1726. [Google Scholar] [CrossRef] [Green Version]
- Gallini, R.; Lindblom, P.; Bondjers, C.; Betsholtz, C.; Andrae, J. PDGF-A and PDGF-B induces cardiac fibrosis in transgenic mice. Exp. Cell Res. 2016, 349, 282–290. [Google Scholar] [CrossRef]
- Babapoor-Farrokhran, S.; Gill, D.; Alzubi, J.; Mainigi, S.K. Atrial fibrillation: The role of hypoxia-inducible factor-1-regulated cytokines. Mol. Cell. Biochem. 2021, 476, 2283–2293. [Google Scholar] [CrossRef] [PubMed]
- de Winter, P.; Leoni, P.; Abraham, D. Connective tissue growth factor: Structure-function relationships of a mosaic, multifunctional protein. Growth Factors 2008, 26, 80–91. [Google Scholar] [CrossRef] [PubMed]
- Holbourn, K.P.; Acharya, K.R.; Perbal, B. The CCN family of proteins: Structure-function relationships. Trends Biochem. Sci. 2008, 33, 461–473. [Google Scholar] [CrossRef] [PubMed]
- Oliver, N.; Sternlicht, M.; Gerritsen, K.; Goldschmeding, R. Could aging human skin use a connective tissue growth factor boost to increase collagen content? J. Investig. Dermatol. 2010, 130, 338–341. [Google Scholar] [CrossRef] [Green Version]
- Gore-Hyer, E.; Shegogue, D.; Markiewicz, M.; Lo, S.; Hazen-Martin, D.; Greene, E.L.; Grotendorst, G.; Trojanowska, M. TGF-beta and CTGF have overlapping and distinct fibrogenic effects on human renal cells. Am. J. Physiol. Renal Physiol. 2002, 283, F707–F716. [Google Scholar] [CrossRef] [Green Version]
- Grotendorst, G.R.; Rahmanie, H.; Duncan, M.R. Combinatorial signaling pathways determine fibroblast proliferation and myofibroblast differentiation. FASEB J. 2004, 18, 469–479. [Google Scholar] [CrossRef] [Green Version]
- Lee, C.H.; Shah, B.; Moioli, E.K.; Mao, J.J. CTGF directs fibroblast differentiation from human mesenchymal stem/stromal cells and defines connective tissue healing in a rodent injury model. J. Clin. Investig. 2010, 120, 3340–3349. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de las Heras, N.; Ruiz-Ortega, M.; Miana, M.; Rupérez, M.; Sanz-Rosa, D.; Aragoncillo, P.; Mezzano, S.; Cachofeiro, V.; Egido, J.; Lahera, V. Interactions between aldosterone and connective tissue growth factor in vascular and renal damage in spontaneously hypertensive rats. J. Hypertens. 2007, 25, 629–638. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Huang, Y.; Chen, X.; Liu, J.; Lu, Y.; Bu, L.; Xia, L.; Xiao, W.; Chen, M.; Nie, Q.; et al. The role of CTGF in the diabetic rat retina and its relationship with VEGF and TGF-β(2), elucidated by treatment with CTGFsiRNA. Acta Ophthalmol. 2010, 88, 652–659. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.Y.; Xiao, L.; Peng, Y.M.; Duan, S.B.; Liu, H.; Liu, Y.H.; Ling, G.H.; Yuan, F.; Chen, J.X.; Fu, X.; et al. Inhibition effect of small interfering RNA of connective tissue growth factor on the expression of vascular endothelial growth factor and connective tissue growth factor in cultured human peritoneal mesothelial cells. Chin. Med. J. 2007, 120, 231–236. [Google Scholar] [CrossRef]
- Lipson, K.E.; Wong, C.; Teng, Y.; Spong, S. CTGF is a central mediator of tissue remodeling and fibrosis and its inhibition can reverse the process of fibrosis. Fibrogenesis Tissue Repair. 2012, 5, S24. [Google Scholar] [CrossRef] [Green Version]
- Tsai, C.T.; Lai, L.P.; Kuo, K.T.; Hwang, J.J.; Hsieh, C.S.; Hsu, K.L.; Tseng, C.D.; Tseng, Y.Z.; Chiang, F.T.; Lin, J.L. Angiotensin II activates signal transducer and activators of transcription 3 via Rac1 in atrial myocytes and fibroblasts: Implication for the therapeutic effect of statin in atrial structural remodeling. Circulation 2008, 117, 344–355. [Google Scholar] [CrossRef] [Green Version]
- Babapoor-Farrokhran, S.; Rasekhi, R.T.; Gill, D.; Alzubi, J.; Mainigi, S.K. How transforming growth factor contributes to atrial fibrillation? Life Sci. 2021, 266, 118823. [Google Scholar] [CrossRef]
- Adam, O.; Frost, G.; Custodis, F.; Sussman, M.A.; Schäfers, H.J.; Böhm, M.; Laufs, U. Role of Rac1 GTPase activation in atrial fibrillation. J. Am. Coll. Cardiol. 2007, 50, 359–367. [Google Scholar] [CrossRef] [Green Version]
- Dudley, S.C., Jr.; Hoch, N.E.; McCann, L.A.; Honeycutt, C.; Diamandopoulos, L.; Fukai, T.; Harrison, D.G.; Dikalov, S.I.; Langberg, J. Atrial fibrillation increases production of superoxide by the left atrium and left atrial appendage: Role of the NADPH and xanthine oxidases. Circulation 2005, 112, 1266–1273. [Google Scholar] [CrossRef] [Green Version]
- Liao, J.K. Rac1 and connective tissue growth factor. JACC 2010, 5, 481–482. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Habibi, J.; Whaley-Connell, A.; Qazi, M.A.; Hayden, M.R.; Cooper, S.A.; Tramontano, A.; Thyfault, J.; Stump, C.; Ferrario, C.; Muniyappa, R.; et al. Rosuvastatin, a 3-hydroxy-3-methylglutaryl coenzyme a reductase inhibitor, decreases cardiac oxidative stress and remodeling in Ren2 transgenic rats. Endocrinology 2007, 148, 2181–2188. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takemoto, M.; Node, K.; Nakagami, H.; Liao, Y.; Grimm, M.; Takemoto, Y.; Kitakaze, M.; Liao, J.K. Statins as antioxidant therapy for preventing cardiac myocyte hypertrophy. J. Clin. Investig. 2001, 108, 1429–1437. [Google Scholar] [CrossRef] [PubMed]
- Huxley, R.R.; Lopez, F.L.; MacLehose, R.F.; Eckfeldt, J.H.; Couper, D.; Leiendecker-Foster, C.; Hoogeveen, R.C.; Chen, L.Y.; Soliman, E.Z.; Agarwal, S.K.; et al. Novel association between plasma matrix metalloproteinase-9 and risk of incident atrial fibrillation in a case-cohort study: The Atherosclerosis Risk in Communities study. PLoS ONE 2013, 8, e59052. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spinale, F.G. Myocardial matrix remodeling and the matrix metalloproteinases: Influence on cardiac form and function. Physiol. Rev. 2007, 87, 1285–1342. [Google Scholar] [CrossRef] [PubMed]
- Fan, D.; Takawale, A.; Lee, J.; Kassiri, Z. Cardiac fibroblasts, fibrosis and extracellular matrix remodeling in heart disease. Fibrogenesis Tissue Repair. 2012, 5, 15. [Google Scholar] [CrossRef] [Green Version]
- Doxakis, A.; Polyanthi, K.; Androniki, T.; Savvas, P.; Eleni, Z.; Roubini, L.; Nikolaos, R. Targeting metalloproteinases in cardiac remodeling. J. Cardiovasc. Med. Cardiol. 2019, 6, 51–60. [Google Scholar] [CrossRef] [Green Version]
- Reese-Petersen, A.L.; Olesen, M.S.; Karsdal, M.A.; Svendsen, J.H.; Genovese, F. Atrial fibrillation and cardiac fibrosis: A review on the potential of extracellular matrix proteins as biomarkers. Matrix Biol. 2020, 91–92, 188–203. [Google Scholar] [CrossRef]
- Dzeshka, M.S.; Lip, G.Y.; Snezhitskiy, V.; Shantsila, E. Cardiac fibrosis in patients with atrial fibrillation: Mechanisms and clinical implications. J. Am. Coll. Cardiol. 2015, 66, 943–959. [Google Scholar] [CrossRef] [Green Version]
- Spinale, F.G. Matrix metalloproteinases: Regulation and dysregulation in the failing heart. Circ. Res. 2002, 90, 520–530. [Google Scholar] [CrossRef] [Green Version]
- McManus, D.D.; Shaikh, A.Y.; Abhishek, F.; Vasan, R.S. Atrial fibrillation and heart failure parallels: Lessons for atrial fibrillation prevention. Crit. Pathw. Cardiol. 2011, 10, 46–51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Diwan, A.; Dibbs, Z.; Nemoto, S.; DeFreitas, G.; Carabello, B.A.; Sivasubramanian, N.; Wilson, E.M.; Spinale, F.G.; Mann, D.L. Targeted overexpression of noncleavable and secreted forms of tumor necrosis factor provokes disparate cardiac phenotypes. Circulation 2004, 109, 262–268. [Google Scholar] [CrossRef] [Green Version]
- Arpino, V.; Brock, M.; Gill, S.E. The role of TIMPs in regulation of extracellular matrix proteolysis. Matrix Biol. 2015, 44–46, 247–254. [Google Scholar] [CrossRef] [PubMed]
- Polyakova, V.; Miyagawa, S.; Szalay, Z.; Risteli, J.; Kostin, S. Atrial extracellular matrix remodelling in patients with atrial fibrillation. J. Cell. Mol. Med. 2008, 12, 189–208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tarone, G.; Balligand, J.L.; Bauersachs, J.; Clerk, A.; De Windt, L.; Heymans, S.; Hilfiker-Kleiner, D.; Hirsch, E.; Iaccarino, G.; Knöll, R.; et al. Targeting myocardial remodelling to develop novel therapies for heart failure: A position paper from the Working Group on Myocardial Function of the European Society of Cardiology. Eur. J. Heart Fail. 2014, 16, 494–508. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spallarossa, P.; Altieri, P.; Garibaldi, S.; Ghigliotti, G.; Barisione, C.; Manca, V.; Fabbi, P.; Ballestrero, A.; Brunelli, C.; Barsotti, A. Matrix metalloproteinase-2 and -9 are induced differently by doxorubicin in H9c2 cells: The role of MAP kinases and NAD(P)H oxidase. Cardiovasc. Res. 2006, 69, 736–745. [Google Scholar] [CrossRef]
- DeLeon-Pennell, K.Y.; Meschiari, C.A.; Jung, M.; Lindsey, M.L. Matrix metalloproteinases in myocardial infarction and heart failure. Prog. Mol. Biol. Transl. Sci. 2017, 147, 75–100. [Google Scholar] [CrossRef] [Green Version]
- Nambi, V.; Morrison, A.C.; Hoogeveen, R.C.; Coresh, J.; Miles, S.; Rhodes, C.E.; Sharrett, A.R.; Boerwinkle, B.E.; Ballantyne, C.M. Matrix metalloproteinase-1 and tissue inhibitors do not predict incident coronary artery disease in the atherosclerosis risk in communities (ARIC) study. Tex. Heart Inst. J. 2008, 35, 388–394. [Google Scholar]
- Welsh, P.; Whincup, P.H.; Papacosta, O.; Wannamethee, S.G.; Lennon, L.; Thomson, A.; Rumley, A.; Lowe, G.D. Serum matrix metalloproteinase-9 and coronary heart disease: A prospective study in middle-aged men. QJM 2008, 101, 785–791. [Google Scholar] [CrossRef] [Green Version]
- Kakkar, R.; Lee, R.T. Intramyocardial fibroblast myocyte communication. Circ. Res. 2010, 106, 47–57. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.Y.; McTiernan, C.F.; Feldman, A.M. Interplay of matrix metalloproteinases, tissue inhibitors of metalloproteinases and their regulators in cardiac matrix remodeling. Cardiovasc. Res. 2000, 46, 214–224. [Google Scholar] [CrossRef] [Green Version]
- Baum, J.; Duffy, H.S. Fibroblasts and myofibroblasts: What are we talking about? J. Cardiovasc. Pharmacol. 2011, 57, 376–379. [Google Scholar] [CrossRef]
- Khalil, H.; Kanisicak, O.; Prasad, V.; Correll, R.N.; Fu, X.; Schips, T.; Vagnozzi, R.J.; Liu, R.; Huynh, T.; Lee, S.J.; et al. Fibroblast-specific TGF-β-Smad 2/3 signaling underlies cardiac fibrosis. J. Clin. Investig. 2017, 127, 3770–3783. [Google Scholar] [CrossRef] [PubMed]
- Evans, R.A.; Tian, Y.C.; Steadman, R.; Phillips, A.O. TGF-beta1-mediated fibroblast—Myofibroblast terminal differentiation—The role of Smad proteins. Exp. Cell Res. 2003, 282, 90–100. [Google Scholar] [CrossRef]
- Nakajima, H.; Nakajima, H.O.; Salcher, O.; Ditti, A.S.; Dembowsky, K.; Jing, S.; Field, L.J. Atrial but not ventricular fibrosis in mice expressing a mutant transforming growth factor-beta(1) transgene in the heart. Circ. Res. 2000, 86, 571–579. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Camelliti, P.; Borg, T.K.; Kohl, P. Structural and functional characterisation of cardiac fibroblasts. Cardiovasc. Res. 2005, 65, 40–51. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Cui, G.; Esmailian, F.; Plunkett, M.; Marelli, D.; Ardehali, A.; Odim, J.; Laks, H.; Sen, L. Atrial extracellular matrix remodeling and the maintenance of atrial fibrillation. Circulation 2004, 109, 363–368. [Google Scholar] [CrossRef] [Green Version]
- Bartel, D.P. MicroRNAs: Genomics, biogenesis, mechanism, and function. Cell 2004, 116, 281–297. [Google Scholar] [CrossRef] [Green Version]
- Satoh, J.; Tabunoki, H. Comprehensive analysis of human microRNA target networks. BioData Min. 2011, 4, 17. [Google Scholar] [CrossRef] [Green Version]
- Grenda, A.; Budzyński, M.; Filip, A.A. Biogenesis of microRNAs and their role in the development and course of selected hematologic disorders. Postepy Hig. Med. Dosw. 2013, 67, 174–185. [Google Scholar] [CrossRef]
- Jiang, X.; Tsitsiou, E.; Herrick, S.E.; Lindsay, M.A. MicroRNAs and regulation of fibrosis. FEBS J. 2010, 277, 2015–2021. [Google Scholar] [CrossRef]
- Colpaert, R.M.W.; Calore, M. MicroRNAs in cardiac diseases. Cells 2019, 8, 737. [Google Scholar] [CrossRef] [Green Version]
- Sygitowicz, G.; Tomaniak, M.; Błaszczyk, O.; Kołtowski, Ł.; Filipiak, K.J.; Sitkiewicz, D. Circulating microribonucleic acids: miR-1, miR-21 and miR-208a in patients with symptomatic heart failure: Preliminary results. Arch. Cardiovasc. Dis. 2015, 108, 634–642. [Google Scholar] [CrossRef] [Green Version]
- Tomaniak, M.; Sygitowicz, G.; Filipiak, K.J.; Błaszczyk, O.; Kołtowski, Ł.; Gasecka, A.; Kochanowski, J.; Sitkiewicz, D. Dysregulations of miRNAs and galectin-3 cause left ventricle dilatation in systolic heart failure patients. Pol. Heart J. 2018, 76, 1012–1014. [Google Scholar] [CrossRef] [Green Version]
- Tomaniak, M.; Sygitowicz, G.; Błaszczyk, O.; Kołtowski, Ł.; Puchta, D.; Malesa, K.; Kochanowski, J.; Sitkiewicz, D.; Filipiak, K.J. miR-1, miR-21 and galectin-3 in hypertensive patients with symptomatic heart failure and left ventricle hypertrophy. Pol. Heart J. 2018, 76, 1009–1011. [Google Scholar] [CrossRef]
- Kumari, R.; Kumar, S.; Kant, R. Role of circulating miRNAs in the pathophysiology of CVD: As a potential biomarker. Gene Rep. 2018, 13, 146–150. [Google Scholar] [CrossRef]
- Weckbach, L.T.; Grabmaier, U.; Clauss, S.; Wakili, R. MicroRNAs as a diagnostic tool for heart failure and atrial fibrillation. Curr. Opin. Pharmacol. 2016, 27, 24–30. [Google Scholar] [CrossRef] [PubMed]
- Chen, F.; Yang, J.; Li, Y.; Wang, H. Circulating microRNAs as novel biomarkers for heart failure. Hellenic J. Cardiol. 2018, 59, 209–214. [Google Scholar] [CrossRef] [PubMed]
- Luo, X.; Yang, B.; Nattel, S. MicroRNAs and atrial fibrillation: Mechanisms and translational potential. Nat. Rev. Cardiol. 2015, 12, 80–90. [Google Scholar] [CrossRef] [PubMed]
- Bohm, A.; Vachalcova, M.; Snopek, P.; Bacharova, L.; Komarova, D.; Hatala, R. Molecular mechanisms, diagnostic aspects and therapeutic opportunities of micro ribonucleic acids in atrial fibrillation. Int. J. Mol. Sci. 2020, 21, 2742. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Komal, S.; Yin, J.J.; Wang, S.H.; Huang, C.Z.; Tao, H.L.; Dong, J.Z.; Han, S.N.; Zhang, L.R. MicroRNAs: Emerging biomarkers for atrial fibrillation. J. Cardiol. 2019, 74, 475–482. [Google Scholar] [CrossRef] [PubMed]
- van den Berg, N.W.E.; Kawasaki, M.; Berger, W.R.; Neefs, J.; Meulendijks, E.; Tijsen, A.J.; de Groot, J.R. MicroRNAs in atrial fibrillation: From expression signatures to functional implications. Cardiovasc. Drugs Ther. 2017, 31, 345–365. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Ponnusamy, M.; Liu, C.; Gao, J.; Wang, K.; Li, P. MicroRNA as a therapeutic target in cardiac remodeling. Biomed Res. Int. 2017, 2017, 1278436. [Google Scholar] [CrossRef] [Green Version]
- Wakili, R.; Voigt, N.; Kääb, S.; Dobrev, D.; Nattel, S. Recent advances in the molecular pathophysiology of atrial fibrillation. J. Clin. Investig. 2011, 121, 2955–2968. [Google Scholar] [CrossRef] [Green Version]
- Qiao, G.; Xia, D.; Cheng, Z.; Zhang, G. miR-132 in atrial fibrillation directly targets connective tissue growth factor. Mol. Med. Rep. 2017, 16, 4143–4150. [Google Scholar] [CrossRef]
- Adam, O.; Löhfelm, B.; Thum, T.; Gupta, S.K.; Puhl, S.L.; Schäfers, H.J.; Böhm, M.; Laufs, U. Role of miR-21 in the pathogenesis of atrial fibrosis. Basic Res. Cardiol. 2012, 107, 278. [Google Scholar] [CrossRef] [PubMed]
- Liang, H.; Zhang, C.; Ban, T.; Liu, Y.; Mei, L.; Piao, X.; Zhao, D.; Lu, Y.; Chu, W.; Yang, B. A novel reciprocal loop between microRNA-21 and TGFβRIII is involved in cardiac fibrosis. Int. J. Biochem. Cell Biol. 2012, 44, 2152–2160. [Google Scholar] [CrossRef] [PubMed]
- Thum, T.; Gross, C.; Fiedler, J.; Fischer, T.; Kissler, S.; Bussen, M.; Galuppo, P.; Just, S.; Rottbauer, W.; Frantz, S.; et al. MicroRNA-21 contributes to myocardial disease by stimulating MAP kinase signalling in fibroblasts. Nature 2008, 456, 980–984. [Google Scholar] [CrossRef]
- Huang, Z.; Chen, X.J.; Qian, C.; Dong, Q.; Ding, D.; Wu, Q.F.; Li, J.; Wang, H.F.; Li, W.H.; Xie, Q.; et al. Signal transducer and activator of transcription 3/microRNA-21 feedback loop contributes to atrial fibrillation by promoting atrial fibrosis in a rat sterile pericarditis model. Circ. Arrhythm. Electrophysiol. 2016, 9, e003396. [Google Scholar] [CrossRef] [PubMed]
- Shyu, K.G.; Wang, B.W.; Cheng, W.P.; Lo, H.M. MicroRNA-208a increases myocardial endoglin expression and myocardial fibrosis in acute myocardial infarction. Can. J. Cardiol. 2015, 31, 679–690. [Google Scholar] [CrossRef] [PubMed]
- Canón, S.; Caballero, R.; Herraiz-Martínez, A.; Pérez-Hernández, M.; López, B.; Atienza, F.; Jalife, J.; Hove-Madsen, L.; Delpón, E.; Bernad, A. miR-208b upregulation interferes with calcium handling in HL-1 atrial myocytes: Implications in human chronic atrial fibrillation. J. Mol. Cell. Cardiol. 2016, 99, 162–173. [Google Scholar] [CrossRef]
- Zhao, X.; Wang, K.; Liao, Y.; Zeng, Q.; Li, Y.; Hu, F.; Liu, Y.; Meng, K.; Qian, C.; Zhang, Q. MicroRNA-101a inhibits cardiac fibrosis induced by hypoxia via targeting TGF? RI on cardiac fibroblasts. Cell. Physiol. Biochem. 2015, 35, 213–226. [Google Scholar] [CrossRef]
- Lu, Y.; Zhang, Y.; Wang, N.; Pan, Z.; Gao, X.; Zhang, F.; Zhang, Y.; Shan, H.; Luo, X.; Bai, Y. MicroRNA-328 contributes to adverse electrical remodeling in atrial fibrillation. Circulation 2010, 122, 2378–2387. [Google Scholar] [CrossRef] [PubMed]
- Duisters, R.F.; Tijsen, A.J.; Schroen, B.; Leenders, J.J.; Lentink, V.; van der Made, I.; Herias, V.; van Leeuwen, R.E.; Schellings, M.W.; Barenbrug, P. miR-133 and miR-30 regulate connective tissue growth factor: Implications for a role of microRNAs in myocardial matrix remodeling. Circ. Res. 2009, 104, 170–178, 6p following 178. [Google Scholar] [CrossRef] [Green Version]
- Yuan, C.T.; Li, X.X.; Cheng, Q.J.; Wang, Y.H.; Wang, J.H.; Liu, C.L. MiR-30a regulates the atrial fibrillation-induced myocardial fibrosis by targeting snail 1. Int. J. Clin. Exp. Pathol. 2015, 8, 15527–15536. [Google Scholar]
- Shan, H.; Zhang, Y.; Lu, Y.; Zhang, Y.; Pan, Z.; Cai, B.; Wang, N.; Li, X.; Feng, T.; Hong, Y. Downregulation of miR-133 and miR-590 contributes to nicotine-induced atrial remodelling in canines. Cardiovasc. Res. 2009, 83, 465–472. [Google Scholar] [CrossRef] [PubMed]
- Wei, C.; Kim, I.K.; Kumar, S.; Jayasinghe, S.; Hong, N.; Castoldi, G.; Catalucci, D.; Jones, W.K.; Gupta, S. NF-κB mediated miR-26a regulation in cardiac fibrosis. J. Cell. Physiol. 2013, 228, 1433–1442. [Google Scholar] [CrossRef]
- Dawson, K.; Wakili, R.; Ordög, B.; Clauss, S.; Chen, Y.; Iwasaki, Y.; Voigt, N.; Qi, X.Y.; Sinner, M.F.; Dobrev, D. MicroRNA29: A mechanistic contributor and potential biomarker in atrial fibrillation. Circulation 2013, 127, 1466–1475, 1475e1-28. [Google Scholar] [CrossRef] [Green Version]
- Liu, H.; Qin, H.; Chen, G.X.; Liang, M.Y.; Rong, J.; Yao, J.P.; Wu, Z.K. Comparative expression profiles of microRNA in left and right atrial appendages from patients with rheumatic mitral valve disease exhibiting sinus rhythm or atrial fibrillation. J. Transl. Med. 2014, 12, 90. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harada, M.; Luo, X.; Qi, X.Y.; Tadevosyan, A.; Maguy, A.; Ordog, B.; Ledoux, J.; Kato, T.; Naud, P.; Voigt, N. Transient receptor potential canonical-3 channel-dependent fibroblast regulation in atrial fibrillation. Circulation 2012, 126, 2051–2064. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| miRNA | Effect | Regulation in Fibrosis | Signal Pathway | Target Genes | References |
|---|---|---|---|---|---|
| miR-21 | profibrotic | up | TGF-β-Smad 3 ERK-MAPK | TβR-III | [118,119,120,121] |
| Spry1 | |||||
| STAT3 | |||||
| miR-208a/b | profibrotic | up | TGF-β | Endoglin | [115,122,123] |
| β-MHC | |||||
| Sox5 | |||||
| Sox6 | |||||
| THRAP1 | |||||
| miR-101 | antifibrotic | down | TGF-β | TβR-I | [124,125] |
| miR-30a | antifibrotic | down | CTGF | CTGF | [126,127] |
| Snail 1 | |||||
| Periostin | |||||
| miR-133 | antifibrotic | down | CTGF | CTGF | [126,128] |
| TGF-β | |||||
| TβR-II | |||||
| miR-590 | antifibrotic | down | TGF-β | TGF-β | [128] |
| TβR-II | |||||
| miR-132 | antifibrotic | down | CTGF | CTGF | [117] |
| miR-26a | antifibrotic | down | CTGF | CTGF | [129] |
| PI3K-AKT | COL1 | ||||
| miR-29b | antifibrotic | down | ERK | COL1α1 | [130] |
| COL1α2 | |||||
| COL3α1 | |||||
| Elastin | |||||
| Fibronectin |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sygitowicz, G.; Maciejak-Jastrzębska, A.; Sitkiewicz, D. A Review of the Molecular Mechanisms Underlying Cardiac Fibrosis and Atrial Fibrillation. J. Clin. Med. 2021, 10, 4430. https://0-doi-org.brum.beds.ac.uk/10.3390/jcm10194430
Sygitowicz G, Maciejak-Jastrzębska A, Sitkiewicz D. A Review of the Molecular Mechanisms Underlying Cardiac Fibrosis and Atrial Fibrillation. Journal of Clinical Medicine. 2021; 10(19):4430. https://0-doi-org.brum.beds.ac.uk/10.3390/jcm10194430
Chicago/Turabian StyleSygitowicz, Grażyna, Agata Maciejak-Jastrzębska, and Dariusz Sitkiewicz. 2021. "A Review of the Molecular Mechanisms Underlying Cardiac Fibrosis and Atrial Fibrillation" Journal of Clinical Medicine 10, no. 19: 4430. https://0-doi-org.brum.beds.ac.uk/10.3390/jcm10194430