
Elexacaftor/Tezacaftor/Ivacaftor in Patients with Cystic Fibrosis Homozygous for the F508del Mutation and Advanced Lung Disease: A 48-Week Observational Study

 , ,
, ,  , , , and
, , , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Study Design and Outcome Measures
2.2. Study Population
2.3. Methods
2.4. Analysis
3. Results
3.1. Changes Following ETI Treatment
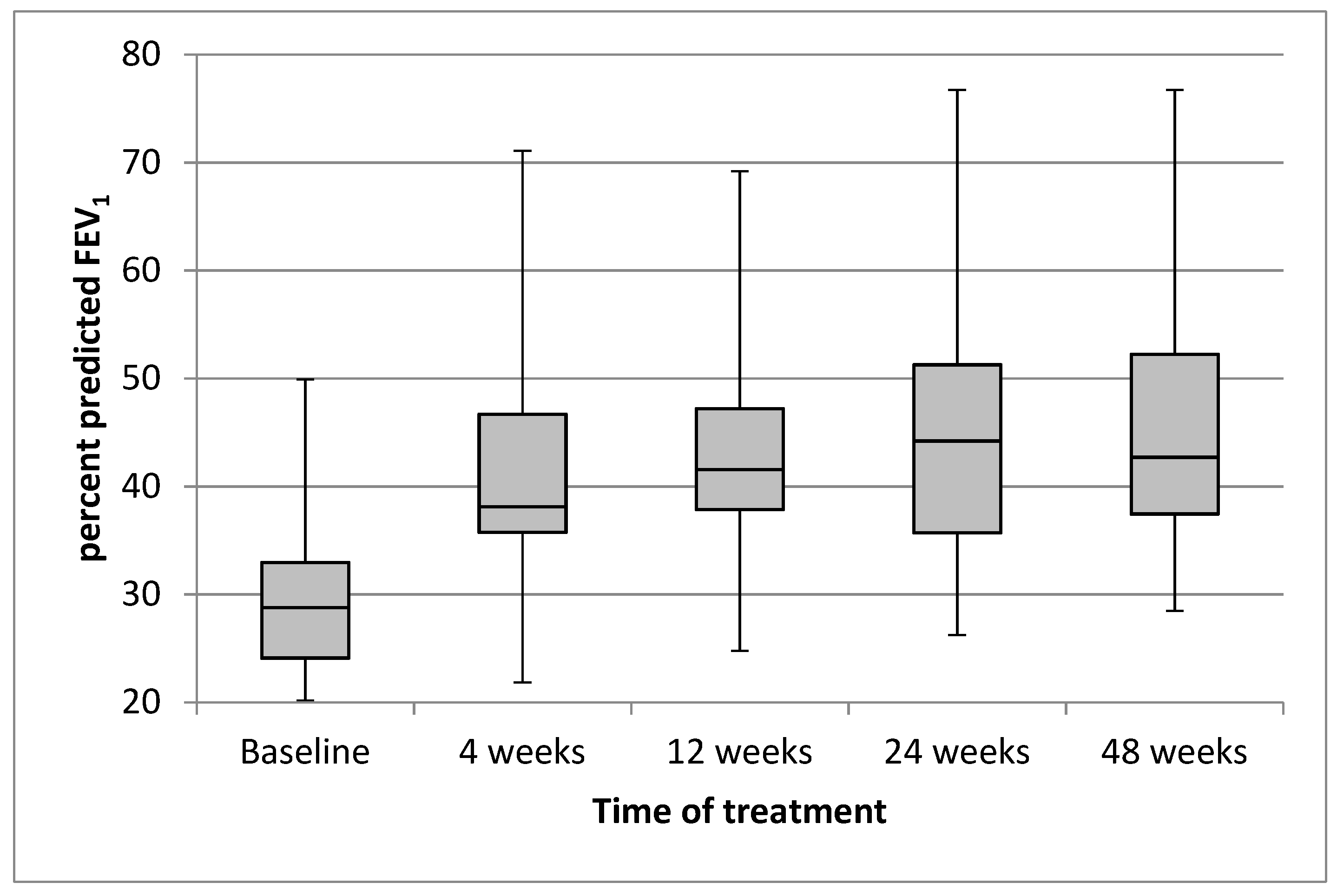
3.1.1. Lung Function
3.1.2. BMI
3.1.3. Sweat Chloride
3.1.4. Rate of Pulmonary Exacerbations
3.1.5. CFQ-R Respiratory Domain
3.1.6. Treatment-Related Adverse Events
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Mall, M.A.; Mayer-Hamblett, N.; Rowe, S.M. Cystic Fibrosis: Emergence of Highly Effective Targeted Therapeutics and Potential Clinical Implications. Am. J. Respir. Crit. Care Med. 2020, 201, 1193–1208. [Google Scholar] [CrossRef] [PubMed]
- Konstan, M.W.; McKone, E.F.; Moss, R.B.; Marigowda, G.; Tian, S.; Waltz, D.; Huang, X.; Lubarsky, B.; Rubin, J.; Millar, S.J.; et al. Assessment of safety and efficacy of long-term treatment with combination lumacaftor and ivacaftor therapy in patients with cystic fibrosis homozygous for the F508del-CFTR mutation (PROGRESS): A phase 3, extension study. Lancet Respir. Med. 2017, 5, 107–118. [Google Scholar] [CrossRef]
- Wainwright, C.E.; Elborn, J.S.; Ramsey, B.W.; Marigowda, G.; Huang, X.; Cipolli, M.; Colombo, C.; Davies, J.C.; de Boeck, K.; Flume, P.A.; et al. Lumacaftor-Ivacaftor in Patients with Cystic Fibrosis Homozygous for F508del CFTR. N. Engl. J. Med. 2015, 373, 220–231. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taylor-Cousar, J.L.; Jain, M.; Barto, T.L.; Haddad, T.; Atkinson, J.; Tian, S.; Tang, R.; Marigowda, G.; Waltz, D.; Pilewski, J.; et al. Lumacaftor/ivacaftor in patients with cystic fibrosis and advanced lung disease homozygous for F508del-CFTR. J. Cyst. Fibros. 2018, 17, 228–235. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McColley, S.A.; Konstan, M.W.; Ramsey, B.W.; Stuart Elborn, J.; Boyle, M.P.; Wainwright, C.E.; Waltz, D.; Vera Llonch, M.; Marigowda, G.; Jiang, J.G.; et al. Lumacaftor/Ivacaftor reduces pulmonary exacerbations in patients irrespective of initial changes in FEV1. J. Cyst. Fibros. 2019, 18, 94–101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Flume, P.A.; Biner, R.F.; Downey, D.G.; Brown, C.; Jain, M.; Fischer, R.; De Boeck, K.; Sawicki, G.S.; Chang, P.; Paz-Diaz, H.; et al. Long-term safety and efficacy of tezacaftor-ivacaftor in individuals with cystic fibrosis aged 12 years or older who are homozygous or heterozygous for F508del CFTR (EXTEND): An open-label extension study. Lancet Respir. Med. 2021, 9, 733–746. [Google Scholar] [CrossRef]
- Taylor-Cousar, J.L.; Munck, A.; McKone, E.F.; van der Ent, C.K.; Moeller, A.; Simard, C.; Wang, L.T.; Ingenito, E.P.; McKee, C.; Lu, Y.; et al. Tezacaftor-Ivacaftor in Patients with Cystic Fibrosis Homozygous for F508del. N. Engl. J. Med. 2017, 377, 2013–2023. [Google Scholar] [CrossRef]
- Heijerman, H.G.M.; McKone, E.F.; Downey, D.G.; Van Braeckel, E.; Rowe, S.M.; Tullis, E.; Mall, M.A.; Welter, J.J.; Ramsey, B.W.; McKee, C.M.; et al. Efficacy and safety of the elexacaftor plus tezacaftor plus ivacaftor combination regimen in people with cystic fibrosis homozygous for the F508del mutation: A double-blind, randomised, phase 3 trial. Lancet 2019, 394, 1940–1948, Erratum in Lancet 2020, 395, 1694. [Google Scholar] [CrossRef]
- Middleton, P.G.; Mall, M.A.; Drevinek, P.; Lands, L.C.; McKone, E.F.; Polineni, D.; Ramsey, B.W.; Taylor-Cousar, J.L.; Tullis, E.; Vermeulen, F.; et al. Elexacaftor-Tezacaftor-Ivacaftor for Cystic Fibrosis with a Single F508del Allele. N. Engl. J. Med. 2019, 381, 1809–1819. [Google Scholar] [CrossRef]
- Griese, M.; Costa, S.; Linnemann, R.W.; Mall, M.A.; McKone, E.F.; Polineni, D.; Quon, B.S.; Ringshausen, F.C.; Taylor-Cousar, J.L.; Withers, N.J.; et al. Safety and Efficacy of Elexacaftor/Tezacaftor/Ivacaftor for 24 Weeks or Longer in People with Cystic Fibrosis and One or More F508del Alleles: Interim Results of an Open-Label Phase 3 Clinical Trial. Am. J. Respir. Crit. Care Med. 2021, 203, 381–385. [Google Scholar] [CrossRef]
- Ramos, K.J.; Pilewski, J.M.; Taylor-Cousar, J.L. Challenges in the use of highly effective modulator treatment for cystic fibrosis. J. Cyst. Fibros. 2021, 20, 381–387. [Google Scholar] [CrossRef] [PubMed]
- Davies, J.C.; Wainwright, C.E.; Canny, G.J.; Chilvers, M.A.; Howenstine, M.S.; Munck, A.; Mainz, J.G.; Rodriguez, S.; Li, H.; Yen, K.; et al. Efficacy and safety of ivacaftor in patients aged 6 to 11 years with cystic fibrosis with a G551D mutation. Am. J. Respir. Crit. Care Med. 2013, 187, 1219–1225. [Google Scholar] [CrossRef] [Green Version]
- Sutharsan, S.; McKone, E.F.; Downey, D.G.; Duckers, J.; MacGregor, G.; Tullis, E.; Van Braeckel, E.; Wainwright, C.E.; Watson, D.; Ahluwalia, N.; et al. Efficacy and safety of elexacaftor plus tezacaftor plus ivacaftor versus tezacaftor plus ivacaftor in people with cystic fibrosis homozygous for F508del-CFTR: A 24-week, multicentre, randomised, double-blind, active-controlled, phase 3b trial. Lancet Respir. Med. 2021. [Google Scholar] [CrossRef]
- O’Shea, K.M.; O’Carroll, O.M.; Carroll, C.; Grogan, B.; Connolly, A.; O’Shaughnessy, L.; Nicholson, T.T.; Gallagher, C.G.; McKone, E.F. Efficacy of elexacaftor/tezacaftor/ivacaftor in patients with cystic fibrosis and advanced lung disease. Eur. Respir. J. 2021, 57, 2003079. [Google Scholar] [CrossRef] [PubMed]
- Burgel, P.R.; Durieu, I.; Chiron, R.; Ramel, S.; Danner-Boucher, I.; Prevotat, A.; Grenet, D.; Marguet, C.; Reynaud-Gaubert, M.; Macey, J.; et al. Rapid Improvement after Starting Elexacaftor-Tezacaftor-Ivacaftor in Patients with Cystic Fibrosis and Advanced Pulmonary Disease. Am. J. Respir. Crit. Care Med. 2021, 204, 64–73. [Google Scholar] [CrossRef] [PubMed]
- Bermingham, B.; Rueschhoff, A.; Ratti, G.; Nesmith, A.; Goodwin, D.; Gray, S.; Flume, P.; Solomon, G.M.; Cohen, L.; Garcia, B. Short-term effect of elexacaftor-tezacaftor-ivacaftor on lung functions and transplant planning in cystic fibrosis patients with advanced lung disease. J. Cyst. Fibros. 2021, 20, 768–771. [Google Scholar] [CrossRef]
- Djavid, A.R.; Thompson, A.E.; Irace, A.L.; Gusman, E.; Altman, K.; DiMango, E.A.; Keating, C.L. Efficacy of Elexacaftor/Tezacaftor/Ivacaftor in Advanced Cystic Fibrosis Lung Disease. Ann. Am. Thorac. Soc. 2021, 18, 1924–1927. [Google Scholar] [CrossRef]
- Nichols, D.P.; Paynter, A.C.; Heltshe, S.L.; Donaldson, S.H.; Frederick, C.A.; Freedman, S.D.; Gelfond, D.; Hoffman, L.R.; Kelly, A.; Narkewicz, M.R.; et al. Clinical Effectiveness of Elexacaftor/Tezacftor/Ivacaftor in People with Cystic Fibrosis. Am. J. Respir. Crit. Care Med. 2021. [Google Scholar] [CrossRef]
- Burgel, P.R.; Da Silva, J.; Paillasseur, J.L.; Martin, C. Optimism with Caution: Elexacaftor-Tezacaftor-Ivacaftor in Patients with Advanced Pulmonary Disease. Am. J. Respir. Crit. Care Med. 2021, 204, 372–374. [Google Scholar] [CrossRef]
- Graeber, S.Y.; Vitzthum, C.; Pallenberg, S.T.; Naehrlich, L.; Stahl, M.; Rohrbach, A.; Drescher, M.; Minso, R.; Ringshausen, F.C.; Rueckes-Nilges, C.; et al. Effects of Elexacaftor/Tezacaftor/Ivacaftor Therapy on CFTR Function in Patients with Cystic Fibrosis and One or Two F508del Alleles. Am. J. Respir. Crit. Care Med. 2021. [Google Scholar] [CrossRef]
- Keogh, R.H.; Cosgriff, R.; Andrinopoulou, E.R.; Brownlee, K.G.; Carr, S.B.; Diaz-Ordaz, K.; Granger, E.; Jewell, N.P.; Lewin, A.; Leyrat, C.; et al. Projecting the impact of triple CFTR modulator therapy on intravenous antibiotic requirements in cystic fibrosis using patient registry data combined with treatment effects from randomised trials. Thorax 2021. [Google Scholar] [CrossRef]
- Carnovale, V.; Iacotucci, P.; Terlizzi, V.; Colangelo, C.; Medio, P.; Ferrillo, L.; De Gregorio, F.; Francalanci, M.; Taccetti, G.; Buonaurio, S.; et al. Effectiveness and safety of elexacaftor/tezacaftor/ivacaftor in patients with cystic fibrosis and advanced lung disease with the F508del/minimal function genotype. Respir. Med. 2021, 189, 106646. [Google Scholar] [CrossRef] [PubMed]
- Salvatore, D.; Terlizzi, V.; Francalanci, M.; Taccetti, G.; Messore, B.; Biglia, C.; Pisi, G.; Calderazzo, M.A.; Caloiero, M.; Pizzamiglio, G.; et al. Ivacaftor improves lung disease in patients with advanced CF carrying CFTR mutations that confer residual function. Respir. Med. 2020, 171, 106073. [Google Scholar] [CrossRef] [PubMed]
- Bonhoure, A.; Boudreau, V.; Litvin, M.; Colomba, J.; Bergeron, C.; Mailhot, M.; Tremblay, F.; Lavoie, A.; Rabasa-Lhoret, R. Overweight, obesity and significant weight gain in adult patients with cystic fibrosis association with lung function and cardiometabolic risk factors. Clin. Nutr. 2020, 39, 2910–2916. [Google Scholar] [CrossRef] [PubMed]
- Gramegna, A.; Aliberti, S.; Contarini, M.; Savi, D.; Sotgiu, G.; Majo, F.; Saderi, L.; Lucidi, V.; Amati, F.; Pappalettera, M.; et al. Overweight and obesity in adults with cystic fibrosis: An Italian multicenter cohort study. J. Cyst. Fibros. 2021, 21, 111–114. [Google Scholar] [CrossRef] [PubMed]
- Gabel, M.E.; Fox, C.K.; Grimes, R.; Lowman, J.D.; McDonald, C.M.; Stallings, V.A.; Michel, S.H. Overweight and Cystic Fibrosis: An Unexpected Challenge. Pediatr. Pulmonol. 2021. [Google Scholar] [CrossRef]
- Shteinberg, M.; Taylor-Cousar, J.L. Impact of CFTR modulator use on outcomes in people with severe cystic fibrosis lung disease. Eur. Respir. Rev. 2020, 29, 190112. [Google Scholar] [CrossRef] [Green Version]
- Hisert, K.B.; Heltshe, S.L.; Pope, C.; Jorth, P.; Wu, X.; Edwards, R.M.; Radey, M.; Accurso, F.J.; Wolter, D.J.; Cooke, G.; et al. Restoring cystic fibrosis transmembrane conductance regulator function reduces airway bacteria and inflammation in people with cystic fibrosis and chronic lung infections. Am. J. Respir. Crit. Care Med. 2017, 195, 1617–1628. [Google Scholar] [CrossRef]
- Harris, J.K.; Wagner, B.D.; Zemanick, E.T.; Robertson, C.E.; Stevens, M.J.; Heltshe, S.L.; Rowe, S.M.; Sagel, S.D. Changes in Airway Microbiome and Inflammation with Ivacaftor Treatment in Patients with Cystic Fibrosis and the G551D Mutation. Ann. Am. Thorac. Soc. 2020, 17, 212–220. [Google Scholar] [CrossRef]
- Graeber, S.Y.; Boutin, S.; Wielpütz, M.O.; Joachim, C.; Frey, D.L.; Wege, S.; Sommerburg, O.; Kauczor, H.U.; Stahl, M.; Dalpke, A.H.; et al. Effects of Lumacaftor-Ivacaftor on Lung Clearance Index, Magnetic Resonance Imaging, and Airway Microbiome in F508del Homozygous Patients with Cystic Fibrosis. Ann. Am. Thorac. Soc. 2021, 18, 971–980. [Google Scholar] [CrossRef]
- Barry, P.J.; Mall, M.A.; Álvarez, A.; Colombo, C.; de Winter-de Groot, K.M.; Fajac, I.; McBennett, K.A.; McKone, E.F.; Ramsey, B.W.; Sutharsan, S.; et al. Triple Therapy for Cystic Fibrosis F508del-Gating and -Residual Function Genotypes. N. Engl. J. Med. 2021, 385, 815–825. [Google Scholar] [CrossRef] [PubMed]
- Stanojevic, S.; Vukovojac, K.; Sykes, J.; Ratjen, F.; Tullis, E.; Stephenson, A.L. Projecting the impact of delayed access to elexacaftor/tezacaftor/ivacaftor for people with cystic fibrosis. J. Cyst. Fibros. 2021, 20, 243–249. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Demographics | |
|---|---|
| Patients | 26 |
| Age, years (median, range) | 31.1 (20.8–48.3) |
| Sex, female (n, %) | 12 (46.2) |
| FEV1 (% predicted, mean, SD) | 29.9 (8.4) |
| FVC (% predicted, mean, SD) | 50.9 (11.6) |
| FEF25–75 (% predicted, mean, SD) | 12.2 (6.7) |
| Sweat chloride (mmol/L, mean, SD) | 77.5 (35.3) |
| BMI (kg/m2, mean, SD) | 20.9 (2.2) |
| Microbiology | |
| Staphylococcus aureus (%) | 9 (34.6) |
| Pseudomonas aeruginosa (%) | 19 (73.1) |
| Burkholderia cepacia complex (%) | 2 (7.7) |
| Pancreatic insufficiency (n, %) | 26 (100) |
| CFRD (n, %) | 11 (42.3) |
| Concomitant medications (n, %) | |
| Bronchodilators | 26 (100.0) |
| Dornase alpha | 18 (70.6) |
| Hypertonic saline | 15 (58.8) |
| Inhaled antibiotics | 23 (88.2) |
| Oxygen | 5 (17.6) |
| Azithromycin | 13 (50.0) |
| Variable | After 4 Weeks | After 12 Weeks | After 24 Weeks | After 48 Weeks |
|---|---|---|---|---|
| FEV1 (% Predicted) | 12.06 (8.54, 15.57) ‡ | 13.22 (9.47, 16.98) ‡ | 15.32 (11.3, 19.34) ‡ | 14.48 (10.64, 18.32) ‡ |
| FVC (% Predicted) | 13.08 (8.54, 17.62) ‡ | 14.59 (9.69, 19.49) ‡ | 18.89(14.20, 23.59) ‡ | 18.50(13.64, 23.35) ‡ |
| Variable | Baseline | After 4 Weeks | After 12 Weeks | After 24 Weeks | After 48 Weeks |
|---|---|---|---|---|---|
| Sweat chloride (mmol/L) a | 77.5 (35.3) | 35.9 (20.1) ‡ | 35.4(16.8) ‡ | 29.0 (11.0) ‡ | 29.2 (19.5) ‡ |
| BMI (kg/m2) a | 20.9 (2.2) | 21.1 (3.1) ‡ | 22.4 (2.2) ‡ | 23.1 (2.3) ‡ | 23.0 (2.2) ‡ |
| Reference | Year | Number of Patients | Follow Up Period | ppFEV1 (%) | SCC (mmol/L) | Weight (Kg) and BMI (kg/m2) | PEx Requiring Hospitalisations |
|---|---|---|---|---|---|---|---|
| Carnovale V et al. | 26 | 48 weeks | Absolute change in ppFEV1: 14.48% (10.64 to 18.32) | 77.5 ± 35.3 vs. 29.2 ± 19.5 | BMI: 20.9 ± 2.2 vs. 23.0 ± 2.2 | 97% lower rate of PEx | |
| Heijerman, H. et al. [8] | 2019 | 107 ETI group: 55 | 4 weeks | Absolute change in ppFEV1: 10.4% (8.6 to 12.2) | Absolute change: −43.4 mmol/L (−46.9 to 40.0) | Mean increase in BMI: 0.60 Kg/m2 | No data |
| O’Shea KM et al. [14] | 2020 | 14 (57% F508del homozygous) | ppFEV1: 26.4 ± 4.2 days SSC: 64 ± 84 days BMI: 62 ± 35 days PEX:4.9 ± 1.94 months | 27.3 ± 7.3% basal vs. 36.3 ± 16.5% after treatment | 104.9 ± 15.04 vs. 53.6 ± 23.3 (only 11 patients) | BMI: 20.7 ± 3.6 vs. 22.1 ± 3.4 kg/m2 | 0.28 ± 0.17 PEx per month in 12 months prior vs. 0.04 ± 0.07 PEx per month during follow-up period |
| Burgel PR et al. [15] | 2021 | 236 (40.8% F508del homozygous) | 3 months | Mean change in ppFEV1: 15.1% | No data | Mean increase in weight: 4.2 Kg | No data |
| Bermingham B et al. [16] | 2021 | 50 (64% F508del homozygous) | ppFEV1: 39.1 ± 24.8 days | Mean change in ppFEV1: 7.9% | No data | No data | No data |
| Djavid AR et al. [17] | 2021 | 22 (9% F508del homozygous)) | 12–15 months | Mean change in ppFEV1 in 16/22 patients: 7.6% | No data | Mean increase in BMI: 2.0 Kg/m2 | Decrease of 2.38 PEx per patient |
| Nichols DP et al. [18] | 2021 | 487 (48.5% F508del homozygous) | 6 months | Mean change in ppFEV1: 9.76 | 88.0 ± 18.4 vs. 45.7 ± 21.2 | BMI: 23.1 ± 4.0 vs. 24.5 ± 4.6 | No data |
| Sutharsan S et al. [13] | 2021 | 175 ETI group: 87 | 24 weeks | Absolute change in ppFEV1: 11.2% (9.8 to 12.6) | Absolute change: −46.2 mmol/L (−48.7 to 43.7) | Absolute change in BMI: 1.59 Kg/m2 | No data |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Carnovale, V.; Iacotucci, P.; Terlizzi, V.; Colangelo, C.; Ferrillo, L.; Pepe, A.; Francalanci, M.; Taccetti, G.; Buonaurio, S.; Celardo, A.; et al. Elexacaftor/Tezacaftor/Ivacaftor in Patients with Cystic Fibrosis Homozygous for the F508del Mutation and Advanced Lung Disease: A 48-Week Observational Study. J. Clin. Med. 2022, 11, 1021. https://0-doi-org.brum.beds.ac.uk/10.3390/jcm11041021
Carnovale V, Iacotucci P, Terlizzi V, Colangelo C, Ferrillo L, Pepe A, Francalanci M, Taccetti G, Buonaurio S, Celardo A, et al. Elexacaftor/Tezacaftor/Ivacaftor in Patients with Cystic Fibrosis Homozygous for the F508del Mutation and Advanced Lung Disease: A 48-Week Observational Study. Journal of Clinical Medicine. 2022; 11(4):1021. https://0-doi-org.brum.beds.ac.uk/10.3390/jcm11041021
Chicago/Turabian StyleCarnovale, Vincenzo, Paola Iacotucci, Vito Terlizzi, Carmela Colangelo, Lorenza Ferrillo, Angela Pepe, Michela Francalanci, Giovanni Taccetti, Serena Buonaurio, Assunta Celardo, and et al. 2022. "Elexacaftor/Tezacaftor/Ivacaftor in Patients with Cystic Fibrosis Homozygous for the F508del Mutation and Advanced Lung Disease: A 48-Week Observational Study" Journal of Clinical Medicine 11, no. 4: 1021. https://0-doi-org.brum.beds.ac.uk/10.3390/jcm11041021