
A Comprehensive Update on Retinal Vasculitis: Etiologies, Manifestations and Treatments

 , , ,
, , ,
Abstract
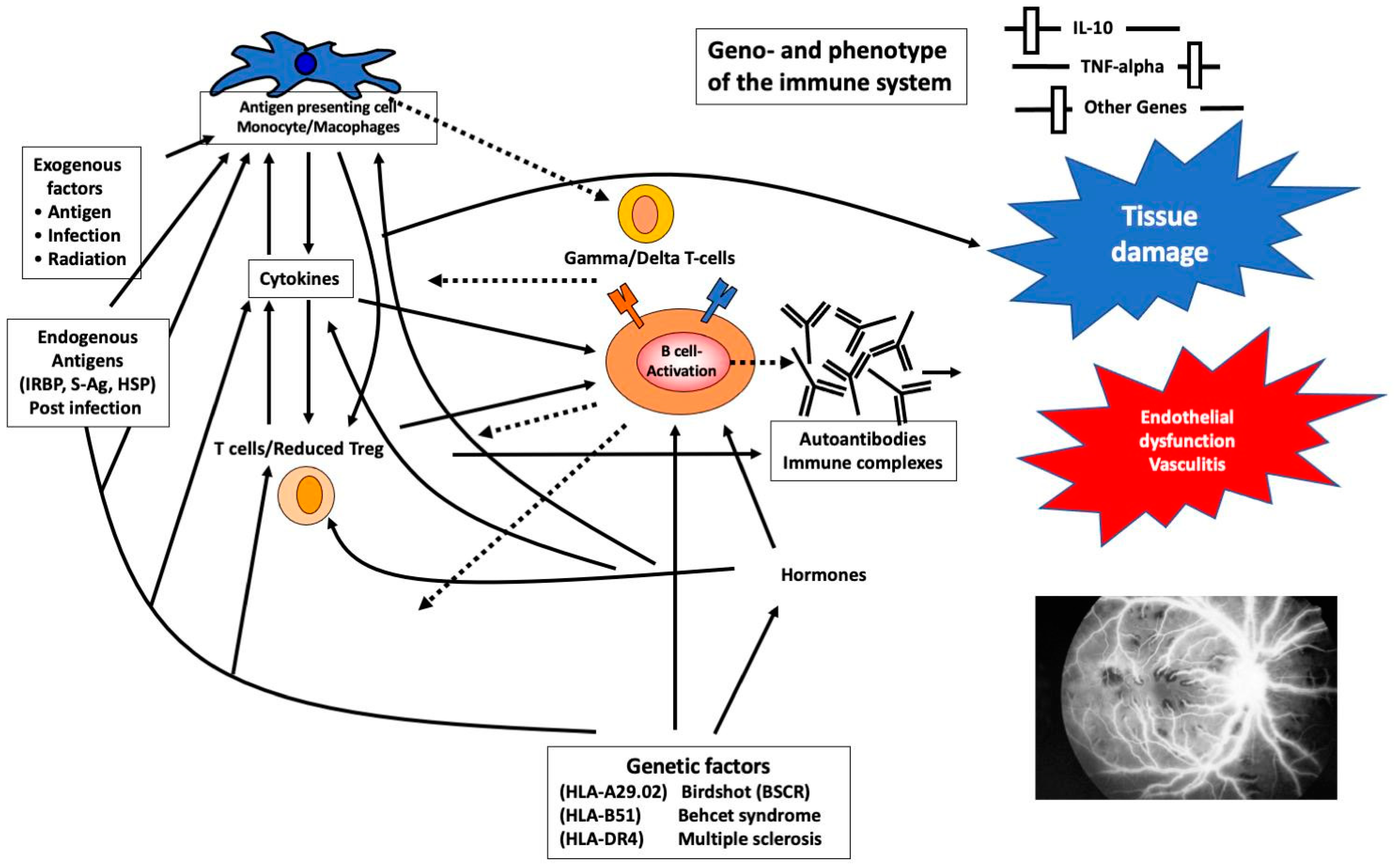
:
1. Introduction
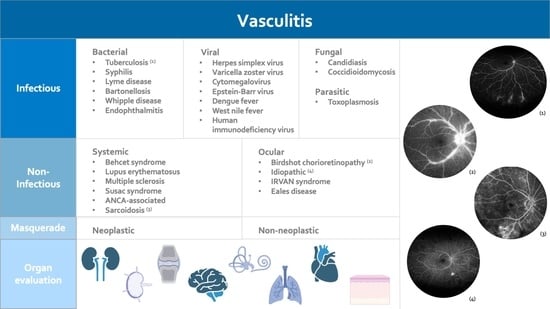
2. Infectious Retinal Vasculitis
2.1. Bacterial Retinal Vasculitis
2.1.1. Tubercular Retinal Vasculitis
2.1.2. Retinal Vasculitis Related to Syphilis
2.1.3. Retinal Vasculitis Associated with Lyme’s Disease
2.1.4. Bartonella-Related Retinal Vasculitis
2.1.5. Miscellaneous Bacterial Retinal Vasculitis
2.2. Viral Retinal Vasculitis
2.2.1. Herpes-Family Related Retinal Vasculitis
2.2.2. Dengue, West Nile Fever, and Other RNA Viruses
2.2.3. Human Immunodeficiency Virus
2.3. Fungal Retinal Vasculitis
2.3.1. Candidiasis
2.3.2. Coccidioidomycosis
2.4. Parasitic Retinal Vasculitis
2.4.1. Toxoplasma-Related Retinal Vasculitis
2.4.2. Miscellaneous Parasitic Retinal Vasculitis
3. Non-Infectious Retinal Vasculitis
3.1. Retinal Vasculitis with Systemic Involvement
3.1.1. Behcet’s Syndrome
3.1.2. Systemic Lupus Erythematosus
3.1.3. Uveitis Associated with Multiple Sclerosis
3.1.4. Susac’s Syndrome
3.1.5. ANCA-Associated Vasculitis
3.1.6. Sarcoidosis
3.2. Isolated Ocular Disorders (Vasculitis Limited to Ocular Manifestations)
3.2.1. Idiopathic Retinal Vasculitis
3.2.2. IRVAN Syndrome
3.2.3. Birdshot Chorioretinopathy (BSCR)
3.2.4. Eales’ Disease
3.3. Masquerade Syndromes
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Agarwal, A.; Karkhur, S.; Aggarwal, K.; Invernizzi, A.; Singh, R.; Dogra, M.R.; Gupta, V.; Gupta, A.; Do, D.V.; Nguyen, Q.D. Epidemiology and clinical features of inflammatory retinal vascular occlusions: Pooled data from two tertiary-referral institutions. Clin. Exp. Ophthalmol. 2018, 46, 62–74. [Google Scholar] [CrossRef] [PubMed]
- El-Asrar, A.M.A.; Herbort, C.P.; Tabbara, K.F. A clinical approach to the diagnosis of retinal vasculitis. Int. Ophthalmol. 2010, 30, 149–173. [Google Scholar] [CrossRef] [PubMed]
- Gascon, P.; Jarrot, P.A.; Matonti, F.; Kaplanski, G. Retinal vasculitis and systemic diseases. Rev. Med. Interne 2018, 39, 721–727. [Google Scholar] [CrossRef] [PubMed]
- Pelegrín, L.; Hernández-Rodríguez, J.; Espinosa, G.; Llorenç, V.; Sainz-De-La-Maza, M.; Fontenla, J.R.; Martínez, J.A.; Cid, M.C.; Adán, A. Characterization of isolated retinal vasculitis. Analysis of a cohort from a single center and literature review. Autoimmun. Rev. 2017, 16, 237–243. [Google Scholar] [CrossRef] [PubMed]
- Ebrahimiadib, N.; Maleki, A.; Fadakar, K.; Manhapra, A.; Ghassemi, F.; Foster, C.S. Vascular abnormalities in uveitis. Surv. Ophthalmol. 2021, 66, 653–667. [Google Scholar] [CrossRef]
- Agarwal, A.; Afridi, R.; Agrawal, R.; Do, D.V.; Gupta, V.; Nguyen, Q.D. Multimodal Imaging in Retinal Vasculitis. Ocul. Immunol. Inflamm. 2017, 25, 424–433. [Google Scholar] [CrossRef]
- Kurumkattil, R.; Trehan, H.S.; Tandel, K.; Sharma, V.K.; Dhar, S.K.; Mahapatra, T. Endogenous endophthalmitis secondary to Burkholderia cepacia: A rare presentation. Indian J. Ophthalmol. 2020, 68, 2283–2285. [Google Scholar] [CrossRef]
- Gupta, V.; Shoughy, S.S.; Mahajan, S.; Khairallah, M.; Rosenbaum, J.T.; Curi, A.; Tabbara, K.F. Clinics of ocular tuberculosis. Ocul. Immunol. Inflamm. 2015, 23, 14–24. [Google Scholar] [CrossRef]
- Agrawal, R.; Gunasekeran, D.V.; Grant, R.; Agarwal, A.; Kon, O.M.; Nguyen, Q.D.; Pavesio, C.; Gupta, V.; for the Collaborative Ocular Tuberculosis Study (COTS)-1 Study Group. Clinical Features and Outcomes of Patients with Tubercular Uveitis Treated with Antitubercular Therapy in the Collaborative Ocular Tuberculosis Study (COTS)-1. JAMA Ophthalmol. 2017, 135, 1318–1327. [Google Scholar] [CrossRef]
- Agarwal, A.; Mahajan, S.; Khairallah, M.; Mahendradas, P.; Gupta, A.; Gupta, V. Multimodal Imaging in Ocular Tuberculosis. Ocul. Immunol. Inflamm. 2017, 25, 134–145. [Google Scholar] [CrossRef]
- Agarwal, A.; Invernizzi, A.; Markan, A.; Testi, I.; Keane, P.A.; Agrawal, R.; Nguyen, Q.D.; Pavesio, C.; Gupta, V. Imaging in Tubercular Choroiditis: Current Concepts. Ocul. Immunol. Inflamm. 2020, 28, 1223–1238. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, A.; Handa, S.; Aggarwal, K.; Sharma, M.; Singh, R.; Sharma, A.; Agrawal, R.; Sharma, K.; Gupta, V. The Role of Dexamethasone Implant in the Management of Tubercular Uveitis. Ocul. Immunol. Inflamm. 2018, 26, 884–892. [Google Scholar] [CrossRef] [PubMed]
- Aggarwal, K.; Agarwal, A.; Sharma, A.; Sharma, K.; Gupta, V.; OCTA Study Group. Detection of type 1 choroidal neovascular membranes using optical coherence tomography angiography in tubercular posterior uveitis. Retina 2019, 39, 1595–1606. [Google Scholar] [CrossRef] [PubMed]
- Bacon, R.M.; Kugeler, K.J.; Mead, P.S.; Centers for Disease Control and Prevention (CDC). Surveillance for Lyme Disease—United States, 1992–2006; Morbidity and Mortality Weekly Report; CDC Surveillance Summaries: Washington, DC, USA, 2008; Volume 57, pp. 1–9. [Google Scholar]
- Christova, I.; Komitova, R. Clinical and epidemiological features of Lyme borreliosis in Bulgaria. Wien. Klin. Wochenschr. 2004, 116, 42–46. [Google Scholar] [CrossRef] [PubMed]
- Nelson, C.A.; Saha, S.; Kugeler, K.J.; DeLorey, M.J.; Shankar, M.B.; Hinckley, A.F.; Mead, P.S. Incidence of Clinician-Diagnosed Lyme Disease, United States, 2005–2010. Emerg. Infect. Dis. 2015, 21, 1625–1631. [Google Scholar] [CrossRef]
- Balcer, L.J.; Winterkorn, J.M.; Galetta, S.L. Neuro-ophthalmic manifestations of Lyme disease. J. Neuro-Ophthalmol. 1997, 17, 108–121. [Google Scholar] [CrossRef]
- Berglöff, J.; Gasser, R.; Feigl, B. Ophthalmic manifestations in Lyme borreliosis. A review. J. Neuro-Ophthalmol. 1994, 14, 15–20. [Google Scholar] [CrossRef]
- Sanchez, J.L. Clinical Manifestations and Treatment of Lyme Disease. Clin. Lab. Med. 2015, 35, 765–778. [Google Scholar] [CrossRef]
- Halperin, J.J. Chronic Lyme disease: Misconceptions and challenges for patient management. Infect. Drug Resist. 2015, 8, 119–128. [Google Scholar] [CrossRef] [Green Version]
- Reed, J.B.; Scales, D.K.; Wong, M.T.; Lattuada, C.P.; Dolan, M.J.; Schwab, I.R. Bartonella henselae neuroretinitis in cat scratch disease. Diagnosis, management, and sequelae. Ophthalmology 1998, 105, 459–466. [Google Scholar] [CrossRef]
- Biancardi, A.L.; Curi, A.L.L. Cat-scratch disease. Ocul. Immunol. Inflamm. 2014, 22, 148–154. [Google Scholar] [CrossRef]
- Labalette, P.; Bermond, D.; Dedes, V.; Savage, C. Cat-scratch disease neuroretinitis diagnosed by a polymerase chain reaction approach. Am. J. Ophthalmol. 2001, 132, 575–576. [Google Scholar] [CrossRef]
- Lee, A.G. Cat-scratch disease. Ophthalmology 1999, 106, 1. [Google Scholar] [CrossRef]
- Ormerod, L.D.; Dailey, J.P. Ocular manifestations of cat-scratch disease. Curr. Opin. Ophthalmol. 1999, 10, 209–216. [Google Scholar] [CrossRef] [PubMed]
- Kucharz, E.J.; Kramza, J.; Grosicka, A.; Pieczyrak, R. Clinical manifestations of Whipple’s disease mimicking rheumatic disorders. Reumatologia 2021, 59, 104–110. [Google Scholar] [CrossRef]
- Avila, M.P.; Jalkh, A.E.; Feldman, E.; Trempe, C.L.; Schepens, C.L. Manifestations of Whipple’s disease in the posterior segment of the eye. Arch. Ophthalmol. 1984, 102, 384–390. [Google Scholar] [CrossRef]
- Stanford, M.R.; Graham, E.M. Systemic associations of retinal vasculitis. Int. Ophthalmol. Clin. 1991, 31, 23–33. [Google Scholar] [CrossRef]
- Lee, J.H.; Agarwal, A.; Mahendradas, P.; Lee, C.S.; Gupta, V.; Pavesio, C.E.; Agrawal, R. Viral posterior uveitis. Surv. Ophthalmol. 2017, 62, 404–445. [Google Scholar] [CrossRef]
- Wensing, B.; de Groot-Mijnes, J.D.F.; Rothova, A. Necrotizing and nonnecrotizing variants of herpetic uveitis with posterior segment involvement. Arch. Ophthalmol. 2011, 129, 403–408. [Google Scholar] [CrossRef] [Green Version]
- Brydak-Godowska, J.; Szczepanik, S.; Ciszek, M.; Bialas, D.; Grzeszczyk, M.; Strzeleck, D.; Kecik, D. Bilateral acute retinal necrosis associated with neuroinfection in patient after renal transplantation. Med. Sci. Monit. Int. Med. J. Exp. Clin. Res. 2011, 17, CS99–CS102. [Google Scholar] [CrossRef]
- Kobayashi, T.; Sekar, P.; Meier, J.; Streit, J. Acute retinal necrosis in a patient with remote severe herpes simplex encephalitis. BMJ Case Rep. 2019, 12, e229137. [Google Scholar] [CrossRef]
- Spaide, R.F.; Byun, S.S. Recurrent Acute Retinal Necrosis. Retin. Cases Brief Rep. 2020. ahead of print. [Google Scholar] [CrossRef] [PubMed]
- Geng, S.; Ye, J.-J.; Zhao, J.L.; Li, T.S.; Han, Y. Cytomegalovirus retinitis associated with acquired immunodeficiency syndrome. Chin. Med. J. 2011, 124, 1134–1138. [Google Scholar] [PubMed]
- Ghani, E.; Noor, M.; Khalid, S. Cytomegalovirus Retinitis in Immunocompetent Young Male. J. Coll. Physicians Surg.-Pak. JCPSP 2017, 27, S137–S138. [Google Scholar] [PubMed]
- Invernizzi, A.; Agarwal, A.; Ravera, V.; Oldani, M.; Staurenghi, G.; Viola, F. Optical Coherence Tomography Findings in Cytomegalovirus Retinitis: A Longitudinal Study. Retina 2018, 38, 108–117. [Google Scholar] [CrossRef]
- Pathanapitoon, K.; Tesavibul, N.; Choopong, P.; Boonsopon, S.; Kongyai, N.; Ausayakhun, S.; Kunavisarut, P.; Rothova, A. Clinical manifestations of cytomegalovirus-associated posterior uveitis and panuveitis in patients without human immunodeficiency virus infection. JAMA Ophthalmol. 2013, 131, 638–645. [Google Scholar] [CrossRef] [Green Version]
- Wong, M.H.Y.; Cheung, G.C.M.; Chee, S.P. Posterior segment findings of ocular cytomegalovirus infection in immunocompetent patients. Graefes Arch. Clin. Exp. Ophthalmol. 2014, 252, 1811–1816. [Google Scholar] [CrossRef]
- Abroug, N.; Khairallah, M.; Zina, S.; Ksiaa, I.; Ben Amor, H.; Attia, S.; Jelliti, B.; Khochtali, S. Ocular Manifestations of Emerging Arthropod-Borne Infectious Diseases. J. Curr. Ophthalmol. 2021, 33, 227–235. [Google Scholar] [CrossRef]
- Agarwal, L.; Agrawal, N. Retinal Vasculitis with Macular Infarction: A Dengue-related Ophthalmic Complication. Int. Med. Case Rep. J. 2020, 13, 363–366. [Google Scholar] [CrossRef]
- Merle, H.; Donnio, A.; Jean-Charles, A.; Guyomarch, J.; Hage, R.; Najioullah, F.; Césaire, R.; Cabié, A. Ocular manifestations of emerging arboviruses: Dengue fever, Chikungunya, Zika virus, West Nile virus, and yellow fever. J. Fr. Ophtalmol. 2018, 41, e235–e243. [Google Scholar] [CrossRef]
- Nah, G.; Tan, M.; Teoh, S.; Chong, C.H. Maculopathy associated with Dengue fever in a military pilot. Aviat. Space Environ. Med. 2007, 78, 1064–1067. [Google Scholar] [CrossRef] [PubMed]
- Yip, V.C.H.; Sanjay, S.; Koh, Y.T. Ophthalmic complications of dengue Fever: A systematic review. Ophthalmol. Ther. 2012, 1, 2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pleyer, U.; Klauß, V.; Wilking, H.; Nentwich, M.M. Tropical ophthalmology: Intraocular inflammation caused by “new” infectious pathogens and travel-related infections. Ophthalmol. Z. Dtsch. Ophthalmol. Ges. 2016, 113, 35–46. [Google Scholar] [CrossRef]
- Agarwal, A.; Choudhary, T.; Gupta, V. Optical coherence tomography angiography features of bilateral retinopathy associated with Chikungunya fever. Indian J. Ophthalmol. 2018, 66, 142–145. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, A.; Aggarwal, K.; Gupta, V. Infectious uveitis: An Asian perspective. Eye 2019, 33, 50–65. [Google Scholar] [CrossRef]
- Drake-Casanova, P.; Moreno-Arrones, J.P.; Gorroño Echebarría, M.; Dapena-Sevilla, I.; Pareja-Esteban, J.; Vleming-Pinilla, E. HIV retinal vasculitis. Arch. Soc. Esp. Oftalmol. 2010, 85, 32–34. [Google Scholar] [CrossRef]
- Moschos, M.M.; Guex-Crosier, Y. Retinal vasculitis and cystoid macular edema after body tattooing: A case report. Klin. Mon. Augenheilkd. 2004, 221, 424–426. [Google Scholar] [CrossRef]
- Invernizzi, A.; Symes, R.; Miserocchi, E.; Cozzi, M.; Cereda, M.; Fogliato, G.; Staurenghi, G.; Cimino, L.; McCluskey, P. Spectral domain optical coherence tomography findings in endogenous candida endophthalmitis and their clinical relevance. Retina 2018, 38, 1011–1018. [Google Scholar] [CrossRef]
- Zhuang, H.; Ding, X.; Gao, F.; Zhang, T.; Ni, Y.; Chang, Q.; Xu, G. Optical coherence tomography features of retinal lesions in Chinese patients with endogenous Candida endophthalmitis. BMC Ophthalmol. 2020, 20, 52. [Google Scholar] [CrossRef] [Green Version]
- Vinekar, A.; Avadhani, K.; Maralusiddappa, P.; Prabhu, V.M.D.; Mahendradas, P.; Indumathi, V.A. Retinal vasculitis as an early indicator of systemic candidal abscesses in a premature infant. J. Am. Assoc. Pediatric Ophthalmol. Strabismus 2011, 15, 96–97. [Google Scholar] [CrossRef]
- Nordstrom, B.; Sherpa, N.; Marshall, M.; Chawla, A.; Heidari, A.; Johnson, R. Coccidioidomycosis Chorioretinitis. J. Investig. Med. High Impact Case Rep. 2019, 7, 2324709619881561. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quinlan, E.J.; Gill, V. Initial Presentation of Disseminated Coccidioidomycosis with Ocular Lesions. Case Rep. Infect. Dis. 2020, 2020, 1305193. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holland, G.N. Ocular toxoplasmosis: A global reassessment. Part I: Epidemiology and course of disease. Am. J. Ophthalmol. 2003, 136, 973–988. [Google Scholar] [CrossRef] [PubMed]
- Antoniazzi, E.; Guagliano, R.; Meroni, V.; Pezzotta, S.; Bianchi, P.E. Ocular impairment of toxoplasmosis. Parassitologia 2008, 50, 35–36. [Google Scholar]
- Maenz, M.; Schlüter, D.; Liesenfeld, O.; Schares, G.; Gross, U.; Pleyer, U. Ocular toxoplasmosis past, present and new aspects of an old disease. Prog. Retin. Eye Res. 2014, 39, 77–106. [Google Scholar] [CrossRef]
- Friedmann, C.T.; Knox, D.L. Variations in recurrent active toxoplasmic retinochoroiditis. Arch. Ophthalmol. 1969, 81, 481–493. [Google Scholar] [CrossRef]
- Goldenberg, D.; Goldstein, M.; Loewenstein, A.; Habot-Wilner, Z. Vitreal, retinal, and choroidal findings in active and scarred toxoplasmosis lesions: A prospective study by spectral-domain optical coherence tomography. Graefes Arch. Clin. Exp. Ophthalmol. 2013, 251, 2037–2045. [Google Scholar] [CrossRef]
- Ozgonul, C.; Besirli, C.G. Recent Developments in the Diagnosis and Treatment of Ocular Toxoplasmosis. Ophthalmic Res. 2017, 57, 1–12. [Google Scholar] [CrossRef]
- Vasconcelos-Santos, D.V. Ocular manifestations of systemic disease: Toxoplasmosis. Curr. Opin. Ophthalmol. 2012, 23, 543–550. [Google Scholar] [CrossRef]
- Rothova, A.; Meenken, C.; Buitenhuis, H.J.; Brinkman, C.; Baarsma, G.; Boen-Tan, T.; de Jong, P.; Klaassen-Broekema, N.; Schweitzer, C.; Timmerman, Z.; et al. Therapy for ocular toxoplasmosis. Am. J. Ophthalmol. 1993, 115, 517–523. [Google Scholar] [CrossRef] [Green Version]
- Baharivand, N.; Mahdavifard, A.; Fouladi, R.F. Intravitreal clindamycin plus dexamethasone versus classic oral therapy in toxoplasmic retinochoroiditis: A prospective randomized clinical trial. Int. Ophthalmol. 2013, 33, 39–46. [Google Scholar] [CrossRef] [PubMed]
- Bhaleeya, S.D.; Davis, J. Imaging retinal vascular changes in uveitis. Int. Ophthalmol. Clin. 2012, 52, 83–96. [Google Scholar] [CrossRef] [PubMed]
- Taylor, S.R.J.; Singh, J.; Menezo, V.; Wakefield, D.; McCluskey, P.; Lightman, S. Behçet disease: Visual prognosis and factors influencing the development of visual loss. Am. J. Ophthalmol. 2011, 152, 1059–1066. [Google Scholar] [CrossRef]
- Kahloun, R.; Ben Yahia, S.; Mbarek, S.; Attia, S.; Zaouali, S.; Khairallah, M. Macular involvement in patients with Behçet’s uveitis. J. Ophthalmic Inflamm. Infect. 2012, 2, 121–124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mamo, J.G. The rate of visual loss in Behçet’s disease. Arch. Ophthalmol. 1970, 84, 451–452. [Google Scholar] [CrossRef] [PubMed]
- Hazirolan, D.; Stübiger, N.; Pleyer, U. Light on the horizont: Biologicals in Behçet uveitis. Acta Ophthalmol. 2013, 91, 297–306. [Google Scholar] [CrossRef] [PubMed]
- Tugal-Tutkun, I.; Özdal, P.C. Behçet’s disease uveitis: Is there a need for new emerging drugs? Expert Opin. Emerg. Drugs 2020, 25, 531–547. [Google Scholar] [CrossRef]
- Posarelli, C.; Maglionico, M.N.; Talarico, R.; Covello, G.; Figus, M. Behçet’s syndrome and ocular involvement: Changes over time. Clin. Exp. Rheumatol. 2020, 38 (Suppl. S127), 86–93. [Google Scholar]
- Pleyer, U.; Neri, P.; Deuter, C. New pharmacotherapy options for noninfectious posterior uveitis. Int. Ophthalmol. 2021, 41, 2265–2281. [Google Scholar] [CrossRef]
- Pleyer, U.; Algharably, E.A.H.; Feist, E.; Kreutz, R. Small molecules as therapy for uveitis: A selected perspective of new and developing agents. Expert Opin. Pharmacother. 2017, 18, 1311–1323. [Google Scholar] [CrossRef]
- Hatemi, G.; Christensen, R.; Bang, D.; Bodaghi, B.; Celik, A.F.; Fortune, F.; Gaudric, J.; Gul, A.; Kötter, I.; Leccese, P.; et al. 2018 update of the EULAR recommendations for the management of Behçet’s syndrome. Ann. Rheum. Dis. 2018, 77, 808–818. [Google Scholar] [CrossRef] [PubMed]
- Kötter, I.; Günaydin, I.; Zierhut, M.; Stübiger, N. The use of interferon alpha in Behçet disease: Review of the literature. Semin. Arthritis Rheum. 2004, 33, 320–335. [Google Scholar] [CrossRef] [PubMed]
- Celiker, H.; Kazokoglu, H.; Direskeneli, H. Conventional immunosuppressive therapy in severe Behcet’s Uveitis: The switch rate to the biological agents. BMC Ophthalmol. 2018, 18, 261. [Google Scholar] [CrossRef]
- Sertoglu, E.; Omma, A.; Yucel, C.; Colak, S.; Sandıkcı, S.C.; Ozgurtas, T. The relationship of serum VEGF and sVEGFR-1 levels with vascular involvement in patients with Behçet’s disease. Scand. J. Clin. Lab. Investig. 2018, 78, 443–449. [Google Scholar] [CrossRef] [PubMed]
- Asherson, R.A.; Merry, P.; Acheson, J.F.; Harris, E.N.; Hughes, G.R. Antiphospholipid antibodies: A risk factor for occlusive ocular vascular disease in systemic lupus erythematosus and the ‘primary’ antiphospholipid syndrome. Ann. Rheum. Dis. 1989, 48, 358–361. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barile-Fabris, L.; Hernández-Cabrera, M.F.; Barragan-Garfias, J.A. Vasculitis in systemic lupus erythematosus. Curr. Rheumatol. Rep. 2014, 16, 440. [Google Scholar] [CrossRef]
- Lin, Y.C.; Wang, A.G.; Yen, M.Y. Systemic lupus erythematosus-associated optic neuritis: Clinical experience and literature review. Acta Ophthalmol. 2009, 87, 204–210. [Google Scholar] [CrossRef]
- Klinkhoff, A.V.; Beattie, C.W.; Chalmers, A. Retinopathy in systemic lupus erythematosus: Relationship to disease activity. Arthritis Rheum. 1986, 29, 1152–1156. [Google Scholar] [CrossRef]
- Chen, S.Y.; Suaya, J.A.; Li, Q.; Galindo, C.M.; Misurski, D.; Burstin, S.; Levin, M.J. Incidence of herpes zoster in patients with altered immune function. Infection 2014, 42, 325–334. [Google Scholar] [CrossRef] [Green Version]
- Dougal, M.A.; Evans, L.S.; McClellan, K.R.; Robinson, J. Central retinal artery occlusion in systemic lupus erythematosus. Ann. Ophthalmol. 1983, 15, 38–40. [Google Scholar]
- Belmont, H.M.; Abramson, S.B.; Lie, J.T. Pathology and pathogenesis of vascular injury in systemic lupus erythematosus. Interactions of inflammatory cells and activated endothelium. Arthritis Rheum. 1996, 39, 9–22. [Google Scholar] [CrossRef] [PubMed]
- Ali Dhirani, N.; Ahluwalia, V.; Somani, S. Case of combination therapy to treat lupus retinal vasculitis refractory to steroids. Can. J. Ophthalmol. 2017, 52, e13–e15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frischer, J.M.; Bramow, S.; Dal-Bianco, A.; Lucchinetti, C.F.; Rauschka, H.; Schmidbauer, M.; Laursen, H.; Sorensen, P.S.; Lassmann, H. The relation between inflammation and neurodegeneration in multiple sclerosis brains. Brain J. Neurol. 2009, 132 Pt 5, 1175–1189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gil, H.; Fery-Blanco, C.; Schwartz, C.; Meaux-Ruault, N.; Tisserand, G.L.; Delbosc, B.; Magy-Bertrand, N. Contribution of cerebral magnetic resonance imaging to etiological investigation of uveitis. Rev. Med. Interne 2014, 35, 790–793. [Google Scholar] [CrossRef] [PubMed]
- Thompson, A.J.; Banwell, B.L.; Barkhof, F.; Carroll, W.M.; Coetzee, T.; Comi, G.; Correale, J.; Fazekas, F.; Filippi, M.; Freedman, M.S.; et al. Diagnosis of multiple sclerosis: 2017 revisions of the McDonald criteria. Lancet Neurol. 2018, 17, 162–173. [Google Scholar] [CrossRef]
- Olsen, T.G.; Frederiksen, J. The association between multiple sclerosis and uveitis. Surv. Ophthalmol. 2017, 62, 89–95. [Google Scholar] [CrossRef]
- Toosy, A.T.; Mason, D.F.; Miller, D.H. Optic neuritis. Lancet Neurol. 2014, 13, 83–99. [Google Scholar] [CrossRef]
- Barnes, D.; McDonald, W.I. The ocular manifestations of multiple sclerosis. 2. Abnormalities of eye movements. J. Neurol. Neurosurg. Psychiatry 1992, 55, 863–868. [Google Scholar] [CrossRef] [Green Version]
- Tang, W.M.; Pulido, J.S.; Eckels, D.D.; Han, D.P.; Mieler, W.F.; Pierce, K. The association of HLA-DR15 and intermediate uveitis. Am. J. Ophthalmol. 1997, 123, 70–75. [Google Scholar] [CrossRef]
- Bloch-Michel, E.; Nussenblatt, R.B. International Uveitis Study Group recommendations for the evaluation of intraocular inflammatory disease. Am. J. Ophthalmol. 1987, 103, 234–235. [Google Scholar] [CrossRef]
- Donaldson, M.J.; Pulido, J.S.; Herman, D.C.; Diehl, N.; Hodge, D. Pars planitis: A 20-year study of incidence, clinical features, and outcomes. Am. J. Ophthalmol. 2007, 144, 812–817. [Google Scholar] [CrossRef] [PubMed]
- Standardization of Uveitis Nomenclature (SUN) Working Group. Classification Criteria for Multiple Sclerosis-Associated Intermediate Uveitis. Am. J. Ophthalmol. 2021, 228, 72–79. [Google Scholar] [CrossRef] [PubMed]
- Shugaiv, E.; Tüzün, E.; Kürtüncü, M.; Kıyat-Atamer, A.; Çoban, A.; Akman-Demir, G.; Tugal-Tutkun, I.; Eraksoy, M. Uveitis as a prognostic factor in multiple sclerosis. Mult. Scler. J. 2015, 21, 105–107. [Google Scholar] [CrossRef] [PubMed]
- Velazquez-Villoria, D.; Macia-Badia, C.; Segura-García, A.; Idoate, S.P.; Arcos-Algaba, G.; Velez-Escola, L.; García-Arumí, J. Efficacy of immunomodulatory therapy with interferon-β or glatiramer acetate on multiple sclerosis-associated uveitis. Arch. Soc. Espanola Oftalmol. 2017, 92, 273–279. [Google Scholar] [CrossRef]
- Hedayatfar, A.; Falavarjani, K.G.; Soheilian, M.; Sadr, N.E.; Modarres, M.; Parvaresh, M.M.; Naseripour, M.; Rohani, M.; Almasi, M.; Chee, S.-P. Mycophenolate Mofetil for the Treatment of Multiple Sclerosis-associated Uveitis. Ocul. Immunol. Inflamm. 2017, 25, 308–314. [Google Scholar] [CrossRef]
- Roemer, S.; Bissig, A.; Rocca, A.; Du Pasquier, R.; Guex-Crosier, Y. Efficacy of Natalizumab in Intermediate Uveitis Related to Multiple Sclerosis: A Case Report. Klin. Mon. Augenheilkd. 2018, 235, 476–477. [Google Scholar] [CrossRef]
- Willis, M.D.; Pickersgill, T.P.; Robertson, N.P.; Lee, R.W.J.; Dick, A.D.; Carreño, E. Alemtuzumab-induced remission of multiple sclerosis-associated uveitis. Int. Ophthalmol. 2017, 37, 1229–1233. [Google Scholar] [CrossRef]
- Lopalco, G.; Schiraldi, S.; Venerito, V.; Guerriero, S.; Iannone, F. Effectiveness and safety profile of anakinra in a HLA-B27 positive patient with multiple sclerosis-associated uveitis. Mult. Scler. Relat. Disord. 2020, 42, 102152. [Google Scholar] [CrossRef] [PubMed]
- Hauser, S.L.; Cree, B.A.C. Treatment of Multiple Sclerosis: A Review. Am. J. Med. 2020, 133, 1380–1390.e2. [Google Scholar] [CrossRef]
- Kemanetzoglou, E.; Andreadou, E. CNS Demyelination with TNF-α Blockers. Curr. Neurol. Neurosci. Rep. 2017, 17, 36. [Google Scholar] [CrossRef] [Green Version]
- Susac, J.O. Susac’s syndrome: The triad of microangiopathy of the brain and retina with hearing loss in young women. Neurology 1994, 44, 591–593. [Google Scholar] [CrossRef] [PubMed]
- Dörr, J.; Ringelstein, M.; Duning, T.; Kleffner, I. Update on Susac syndrome: New insights in brain and retinal imaging and treatment options. J. Alzheimers Dis. 2014, 42 (Suppl. S3), S99–S108. [Google Scholar] [CrossRef] [PubMed]
- Magro, C.M.; Poe, J.C.; Lubow, M.; Susac, J.O. Susac syndrome: An organ-specific autoimmune endotheliopathy syndrome associated with anti-endothelial cell antibodies. Am. J. Clin. Pathol. 2011, 136, 903–912. [Google Scholar] [CrossRef] [PubMed]
- Rennebohm, R.M.; Asdaghi, N.; Srivastava, S.; Gertner, E. Guidelines for treatment of Susac syndrome—An update. Int. J. Stroke Off. J. Int. Stroke Soc. 2020, 15, 484–494. [Google Scholar] [CrossRef] [PubMed]
- Dörr, J.; Krautwald, S.; Wildemann, B.; Jarius, S.; Ringelstein, M.; Duning, T.; Aktas, O.; Ringelstein, E.B.; Paul, F.; Kleffner, I. Characteristics of Susac syndrome: A review of all reported cases. Nat. Rev. Neurol. 2013, 9, 307–316. [Google Scholar] [CrossRef] [PubMed]
- Bateman, N.D.; Johnson, I.J.; Gibbin, K.P. Susac’s syndrome: A rare cause of fluctuating sensorineural hearing loss. J. Laryngol. Otol. 1997, 111, 1072–1074. [Google Scholar] [CrossRef] [PubMed]
- Johnson, M.W.; Thomley, M.L.; Huang, S.S.; Gass, J.D. Idiopathic recurrent branch retinal arterial occlusion. Natural history and laboratory evaluation. Ophthalmology 1994, 101, 480–489. [Google Scholar] [CrossRef]
- Egan, R.A.; Hills, W.L.; Susac, J.O. Gass plaques and fluorescein leakage in Susac Syndrome. J. Neurol. Sci. 2010, 299, 97–100. [Google Scholar] [CrossRef]
- Bagaglia, S.A.; Passani, F.; Oliverio, G.W.; Inferrera, L.; Menna, F.; Meduri, A.; Mazzotta, C. Multimodal Imaging in Susac Syndrome: A Case Report and Literature Review. Int. J. Environ. Res. Public Health 2021, 18, 3435. [Google Scholar] [CrossRef]
- Kronbichler, A.; Lee, K.H.; Denicolò, S.; Choi, D.; Lee, H.; Ahn, D.; Kim, K.H.; Lee, J.H.; Kim, H.; Hwang, M.; et al. Immunopathogenesis of ANCA-Associated Vasculitis. Int. J. Mol. Sci. 2020, 21, 7319. [Google Scholar] [CrossRef]
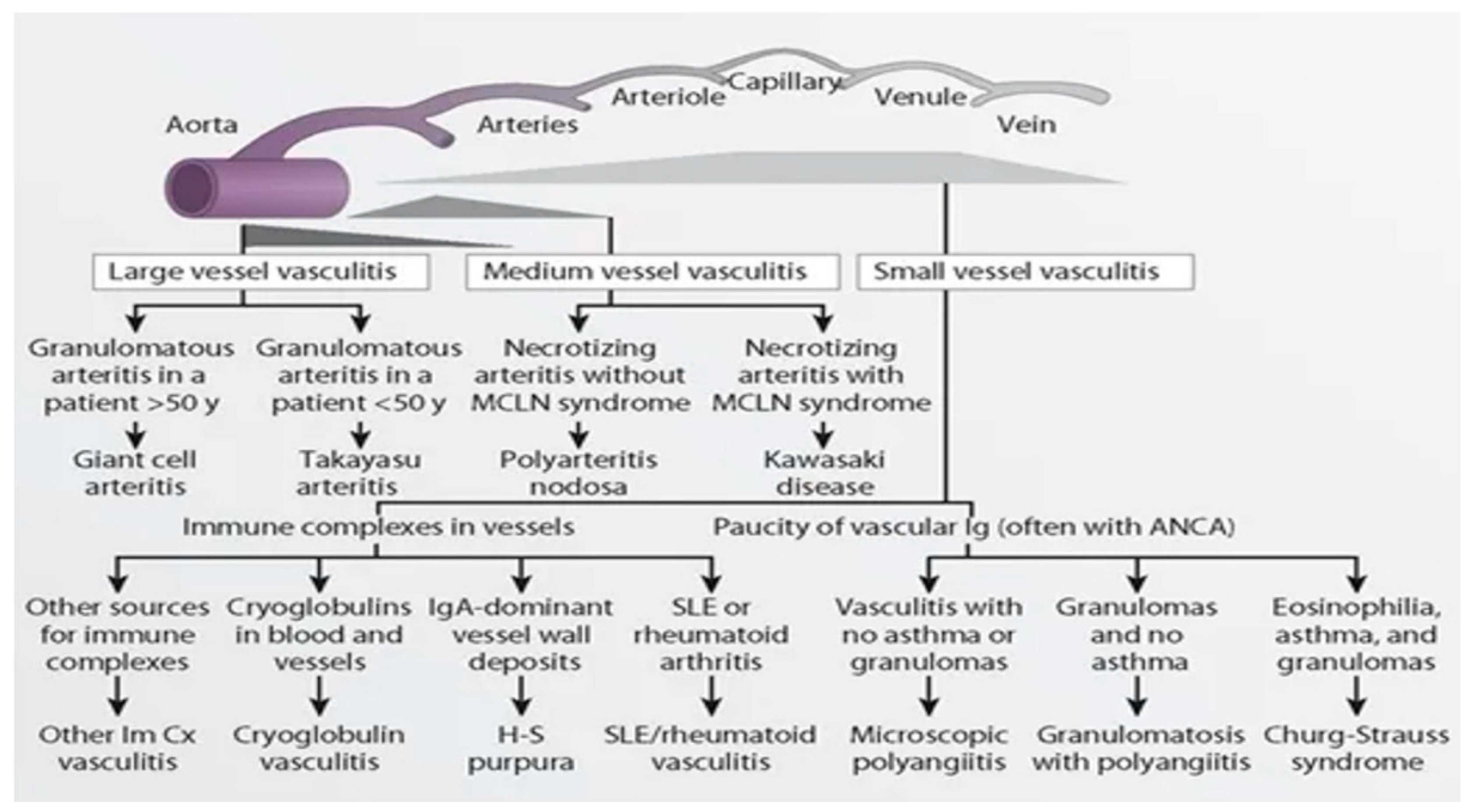
- Jennette, J.C.; Falk, R.J.; Bacon, P.A.; Basu, N.; Cid, M.C.; Ferrario, F.; Flores-Suarez, L.F.; Gross, W.L.; Guillevin, L.; Hagen, E.C.; et al. 2012 revised International Chapel Hill Consensus Conference Nomenclature of Vasculitides. Arthritis Rheum. 2013, 65, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Watts, R.A.; Mahr, A.; Mohammad, A.J.; Gatenby, P.; Basu, N.; Flores-Suárez, L.F. Classification, epidemiology and clinical subgrouping of antineutrophil cytoplasmic antibody (ANCA)-associated vasculitis. Nephrol. Dial. Transplant. 2015, 30 (Suppl. S1), i14–i22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pakrou, N.; Selva, D.; Leibovitch, I. Wegener’s granulomatosis: Ophthalmic manifestations and management. Semin. Arthritis Rheum. 2006, 35, 284–292. [Google Scholar] [CrossRef] [PubMed]
- Watkins, A.S.; Kempen, J.H.; Choi, D.; Liesegang, T.L.; Pujari, S.S.; Newcomb, C.; Nussenblatt, R.B.; Rosenbaum, J.T.; Thorne, J.E.; Foster, C.S.; et al. Ocular disease in patients with ANCA-positive vasculitis. J. Ocul. Biol. Dis. Inform. 2009, 3, 12–19. [Google Scholar] [CrossRef] [Green Version]
- Macarie, S.S.; Kadar, A. Eye involvement in ANCA positive vasculitis. Rom. J. Ophthalmol. 2020, 64, 3–7. [Google Scholar] [CrossRef]
- Hellmich, B.; Flossmann, O.; Gross, W.L.; Bacon, P.; Cohen-Tervaert, J.W.; Guillevin, L.; Jayne, D.; Mahr, A.; Merkel, P.A.; Raspe, H.; et al. EULAR recommendations for conducting clinical studies and/or clinical trials in systemic vasculitis: Focus on anti-neutrophil cytoplasm antibody-associated vasculitis. Ann. Rheum. Dis. 2007, 66, 605–617. [Google Scholar] [CrossRef] [Green Version]
- Sève, P.; Pacheco, Y.; Durupt, F.; Jamilloux, Y.; Gerfaud-Valentin, M.; Isaac, S.; Boussel, L.; Calender, A.; Androdias, G.; Valeyre, D.; et al. Sarcoidosis: A Clinical Overview from Symptoms to Diagnosis. Cells 2021, 10, 766. [Google Scholar] [CrossRef]
- Mochizuki, M.; Smith, J.R.; Takase, H.; Kaburaki, T.; Acharya, N.R.; Rao, N.A. Revised criteria of International Workshop on Ocular Sarcoidosis (IWOS) for the diagnosis of ocular sarcoidosis. Br. J. Ophthalmol. 2019, 103, 1418–1422. [Google Scholar] [CrossRef]
- Cerquaglia, A.; Iaccheri, B.; Fiore, T.; Fruttini, D.; Belli, F.B.; Khairallah, M.; Lupidi, M.; Cagini, C. New Insights On Ocular Sarcoidosis: An Optical Coherence Tomography Angiography Study. Ocul. Immunol. Inflamm. 2019, 27, 1057–1066. [Google Scholar] [CrossRef]
- Liu, D.; Birnbaum, A.D. Update on sarcoidosis. Curr. Opin. Ophthalmol. 2015, 26, 512–516. [Google Scholar] [CrossRef]
- Desai, A.; Chaon, B.; Berkenstock, M. Neurosarcoidosis and Ocular Inflammation: A Case Series and Literature Review. J. Neuro-Ophthalmol. 2021, 41, e259–e266. [Google Scholar] [CrossRef] [PubMed]
- Cunningham, E.T.; Pavesio, C.E.; Goldstein, D.A.; Forooghian, F.; Zierhut, M. Multiple Sclerosis-Associated Uveitis. Ocul. Immunol. Inflamm. 2017, 25, 299–301. [Google Scholar] [CrossRef] [PubMed]
- Lin, M.Y.; Wang, Q.; Newman, N.J.; Dattilo, M. An unusual presentation of neurosarcoidosis: Concurrent optic perineuritis and optic neuritis. Taiwan J. Ophthalmol. 2021, 11, 104–107. [Google Scholar] [CrossRef] [PubMed]
- Momtchilova, M.; Pelosse, B.; Ngoma, E.; Laroche, L. Branch retinal vein occlusion and sarcoidosis in a child: A case report. J. Fr. Ophtalmol. 2011, 34, 243–247. [Google Scholar] [CrossRef] [PubMed]
- Takase, H.; Acharya, N.R.; Babu, K.; Bodaghi, B.; Khairallah, M.; McCluskey, P.J.; Tesavibul, N.; Thorne, J.E.; Tugal-Tutkun, I.; Yamamoto, J.H.; et al. Recommendations for the management of ocular sarcoidosis from the International Workshop on Ocular Sarcoidosis. Br. J. Ophthalmol. 2021, 105, 1515–1519. [Google Scholar] [CrossRef]
- Dammacco, R.; Biswas, J.; Kivelä, T.T.; Zito, F.A.; Leone, P.; Mavilio, A.; Sisto, D.; Alessio, G.; Dammacco, F. Ocular sarcoidosis: Clinical experience and recent pathogenetic and therapeutic advancements. Int. Ophthalmol. 2020, 40, 3453–3467. [Google Scholar] [CrossRef]
- Massicotte, E.; Hassanaly, S.; Bélair, M.L.; Oliver, K.; Fortin, E. Long-term outcomes in a series of idiopathic retinal vasculitis, aneurysms, and neuroretinitis (IRVAN) syndrome. Can. J. Ophthalmol. 2018, 53, 435–440. [Google Scholar] [CrossRef]
- Pichi, F.; Ciardella, A.P. Imaging in the diagnosis and management of idiopathic retinal vasculitis, aneurysms, and neuroretinitis (IRVAN). Int. Ophthalmol. Clin. 2012, 52, 275–282. [Google Scholar] [CrossRef]
- Standardization of Uveitis Nomenclature (SUN) Working Group. Classification Criteria for Birdshot Chorioretinitis. Am. J. Ophthalmol. 2021, 228, 65–71. [Google Scholar] [CrossRef]
- Kuiper, J.; Rothova, A.; de Boer, J.; Radstake, T. The immunopathogenesis of birdshot chorioretinopathy; a bird of many feathers. Prog. Retin. Eye Res. 2015, 44, 99–110. [Google Scholar] [CrossRef]
- Forte, R.; Saleh, M.; Aptel, F.; Chiquet, C. Evaluation of photoreceptors, retinal capillary plexuses, and choriocapillaris in patients with birdshot chorioretinopathy. Retina 2020, 40, 977–988. [Google Scholar] [CrossRef] [PubMed]
- Testi, I.; Ajamil-Rodanes, S.; AlBloushi, A.F.; Pavesio, C. Peripheral Capillary Non-perfusion in Birdshot Retinochoroiditis: A Novel Finding on Ultra-widefield Fluorescein Angiography. Ocul. Immunol. Inflamm. 2020, 28, 1192–1195. [Google Scholar] [CrossRef] [PubMed]
- Agrawal, R.; Joachim, N.; Li, L.J.; Lee, J.Y.; Agarwal, A.; Sim, D.A.; Keane, P.A.; Liew, G.; Pavesio, C.E. Assessment of retinal vascular calibres as a biomarker of disease activity in birdshot chorioretinopathy. Acta Ophthalmol. 2017, 95, e113–e118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gupta, P.; Biswas, J. Further evidence of the association of latent Mycobacterium tuberculosis in Eales’ disease. Int. Ophthalmol. 2021, 41, 901–906. [Google Scholar] [CrossRef]
- López, S.M.; Medina, S.M.; López, F.M. Eales’ disease: Epidemiology, diagnostic and therapeutic concepts. Int. J. Retin. Vitr. 2022, 8, 3. [Google Scholar] [CrossRef]
- Raizada, K.; Tripathy, K. Eales Disease. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2022. Available online: http://0-www-ncbi-nlm-nih-gov.brum.beds.ac.uk/books/NBK559121/ (accessed on 4 February 2022).
- Grange, L.K.; Kouchouk, A.; Dalal, M.D.; Vitale, S.; Nussenblatt, R.B.; Chan, C.-C.; Sen, H.N. Neoplastic masquerade syndromes in patients with uveitis. Am. J. Ophthalmol. 2014, 157, 526–531. [Google Scholar] [CrossRef] [Green Version]
- Biewald, E.; Rating, P.; Bechrakis, N.E.; Lommatzsch, A.P. Uveitis Masquerade Syndrome: Typical Symptoms and Presentations. Klin. Mon. Augenheilkd 2020, 237, 614–620. [Google Scholar] [CrossRef]
- Rothova, A.; Berendschot, T.T.J.M.; Probst, K.; van Kooij, B.; Baarsma, G.S. Birdshot chorioretinopathy: Long-term manifestations and visual prognosis. Ophthalmology 2004, 111, 954–959. [Google Scholar] [CrossRef]
- Sonne, S.J.; Shieh, W.S.; Srivastava, S.K.; Smith, B.T. Lymphoma masquerading as occlusive retinal vasculitis: A case study. Am. J. Ophthalmol. Case Rep. 2020, 19, 100777. [Google Scholar] [CrossRef]
- Gangaputra, S.; Kodati, S.; Kim, M.; Aranow, M.; Sen, H.N. Multimodal Imaging in Masquerade Syndromes. Ocul. Immunol. Inflamm. 2017, 25, 160–168. [Google Scholar] [CrossRef]
- Katoch, D.; Bansal, R.; Nijhawan, R.; Gupta, A. Primary intraocular central nervous system lymphoma masquerading as diffuse retinal vasculitis. BMJ Case Rep. 2013, 2013, bcr2013009354. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brown, S.M.; Jampol, L.M.; Cantrill, H.L. Intraocular lymphoma presenting as retinal vasculitis. Surv. Ophthalmol. 1994, 39, 133–140. [Google Scholar] [CrossRef]
- Say, E.A.T.; Knupp, C.L.; Gertsch, K.R.; Chavala, S.H. Metastatic B-cell lymphoma masquerading as infectious retinitis and vasculitis. Oncol. Lett. 2012, 3, 1245–1248. [Google Scholar] [CrossRef] [PubMed]
- Steel, D.H.; Mahomed, I.; Sheffield, E. Unilateral choroidal melanoma with bilateral retinal vasculitis. Br. J. Ophthalmol. 1996, 80, 850–851. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manusow, J.S.; Khoja, L.; Pesin, N.; Joshua, A.M.; Mandelcorn, E.D. Retinal vasculitis and ocular vitreous metastasis following complete response to PD-1 inhibition in a patient with metastatic cutaneous melanoma. J. Immunother. Cancer 2014, 2, 41. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Diao, T.; Han, L.; Tao, Y.; Yu, L. Association of Meniere’s disease and retinal vascular calibre: A prospective observational study in China. BMJ Open 2018, 8, e022069. [Google Scholar] [CrossRef]
- Papavasileiou, E.; Sobrin, L.; Papaliodis, G.N. Ocular ischemic syndrome presenting as retinal vasculitis in a patient with moyamoya syndrome. Retin. Cases Brief Rep. 2015, 9, 170–172. [Google Scholar] [CrossRef]
- Tang, L.J.; Gu, C.L.; Zhang, P. Intraocular lymphoma. Int. J. Ophthalmol. 2017, 10, 1301–1307. [Google Scholar] [CrossRef]


















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
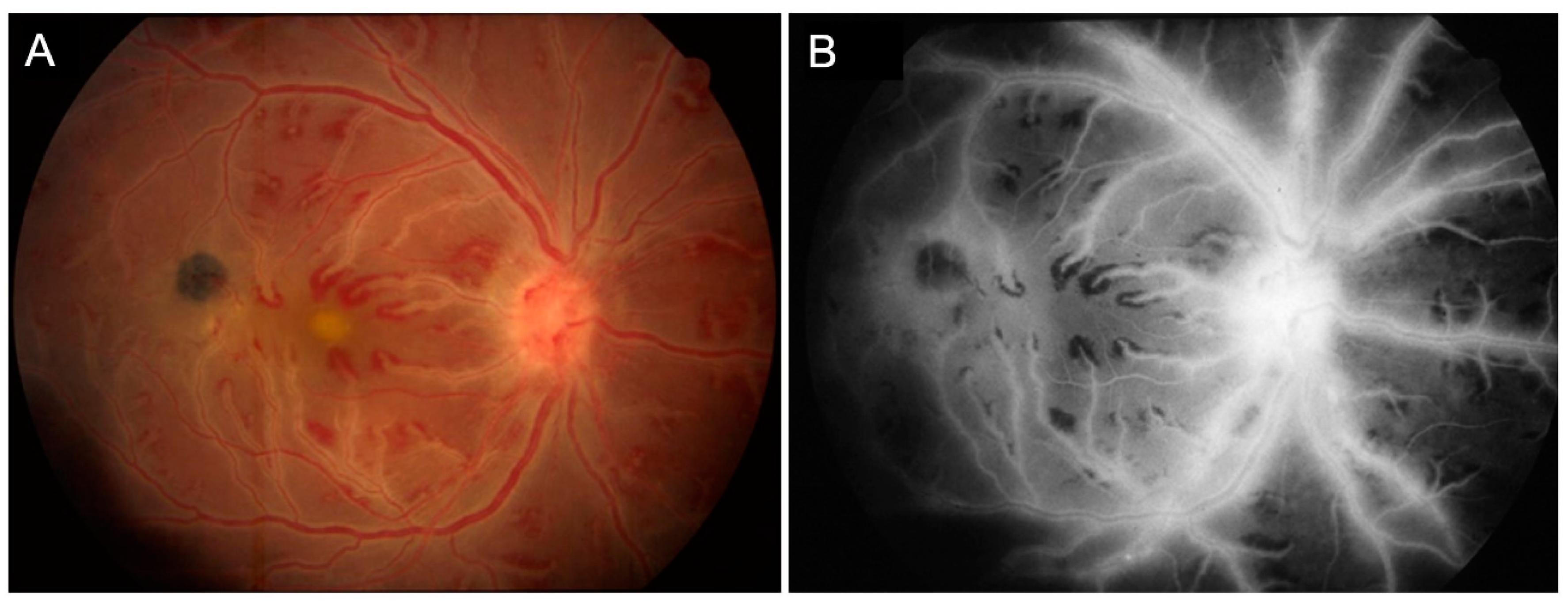{kind=link}
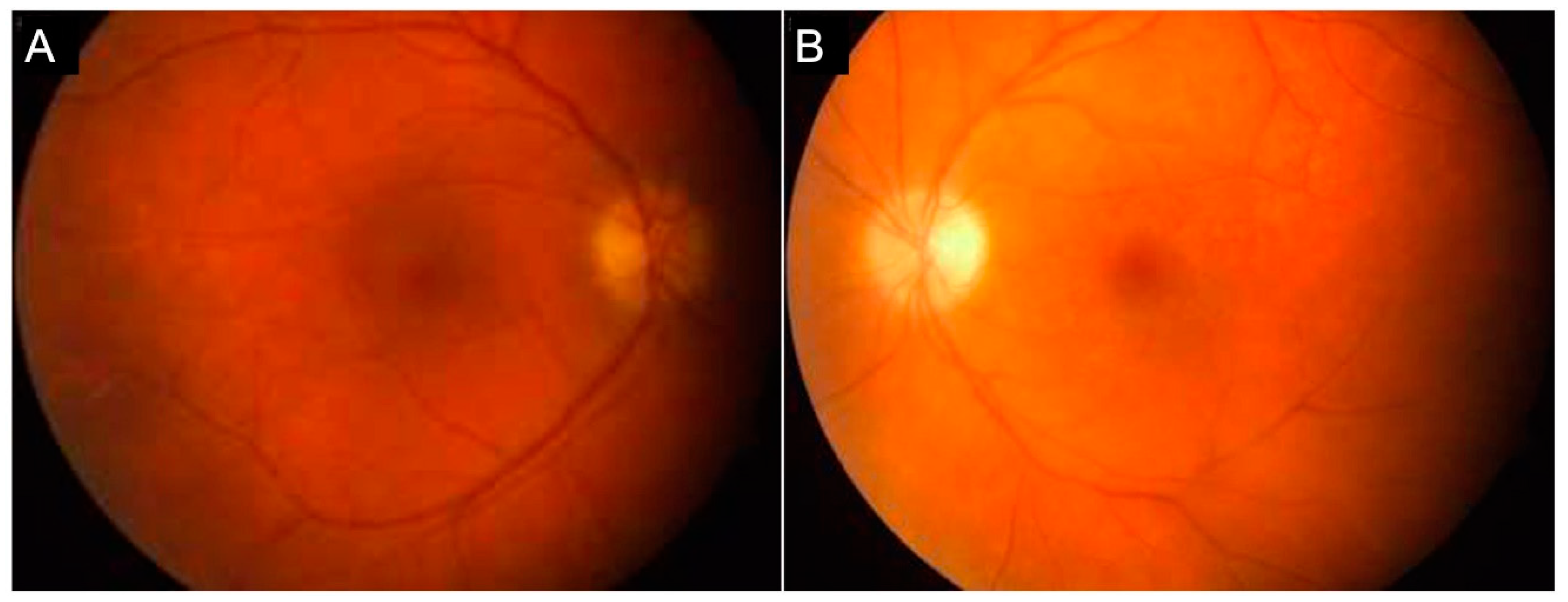{kind=link}
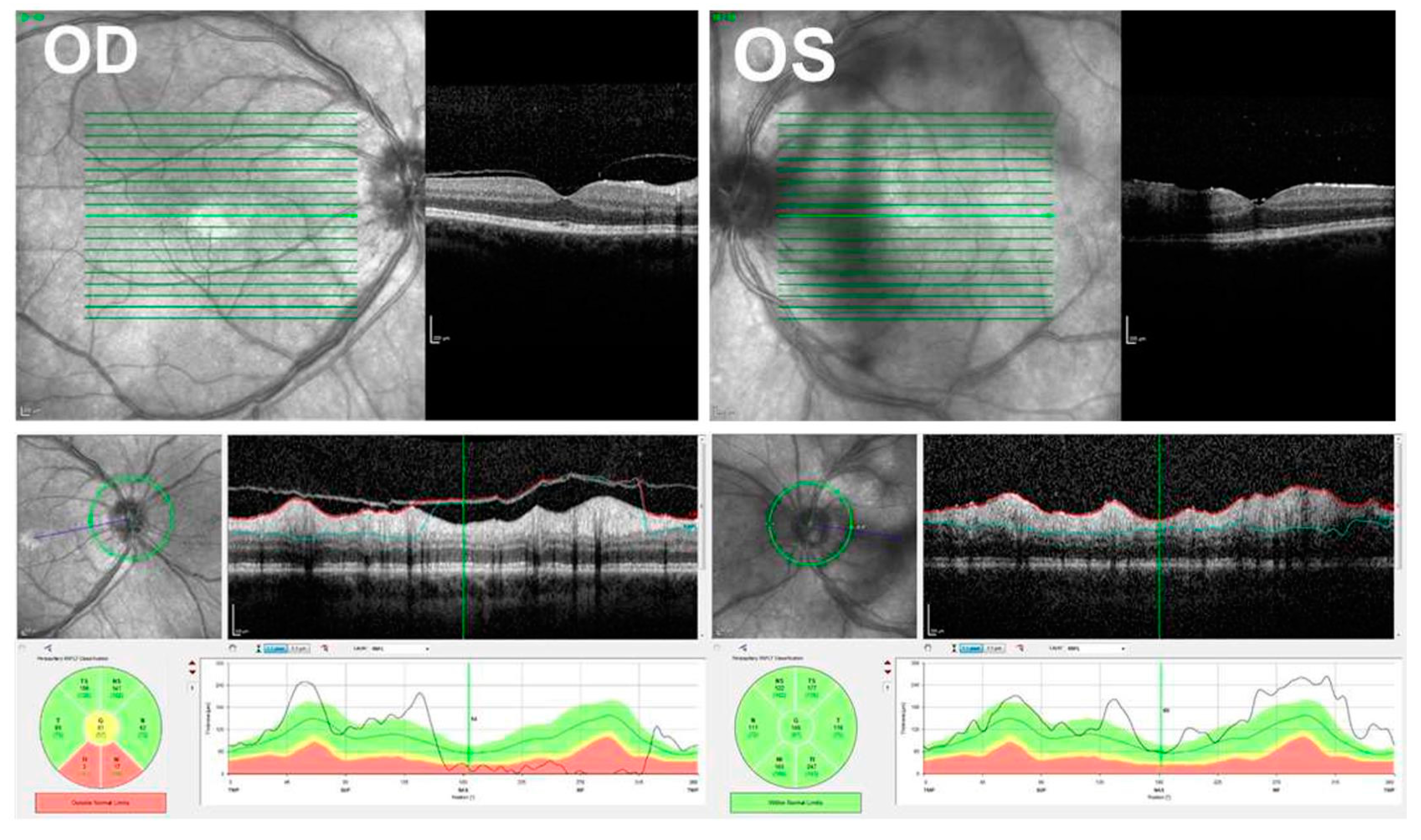{kind=link}
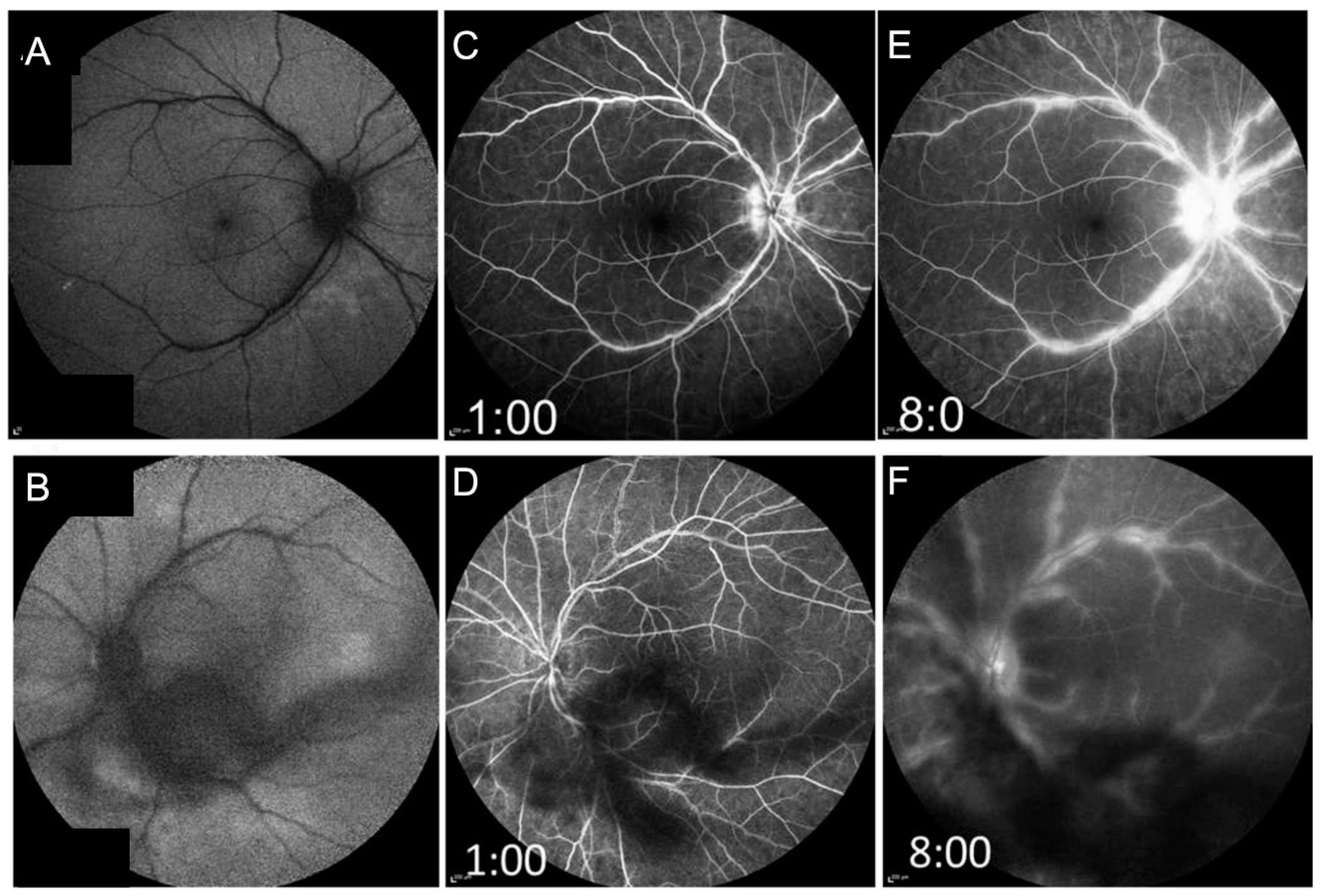{kind=link}
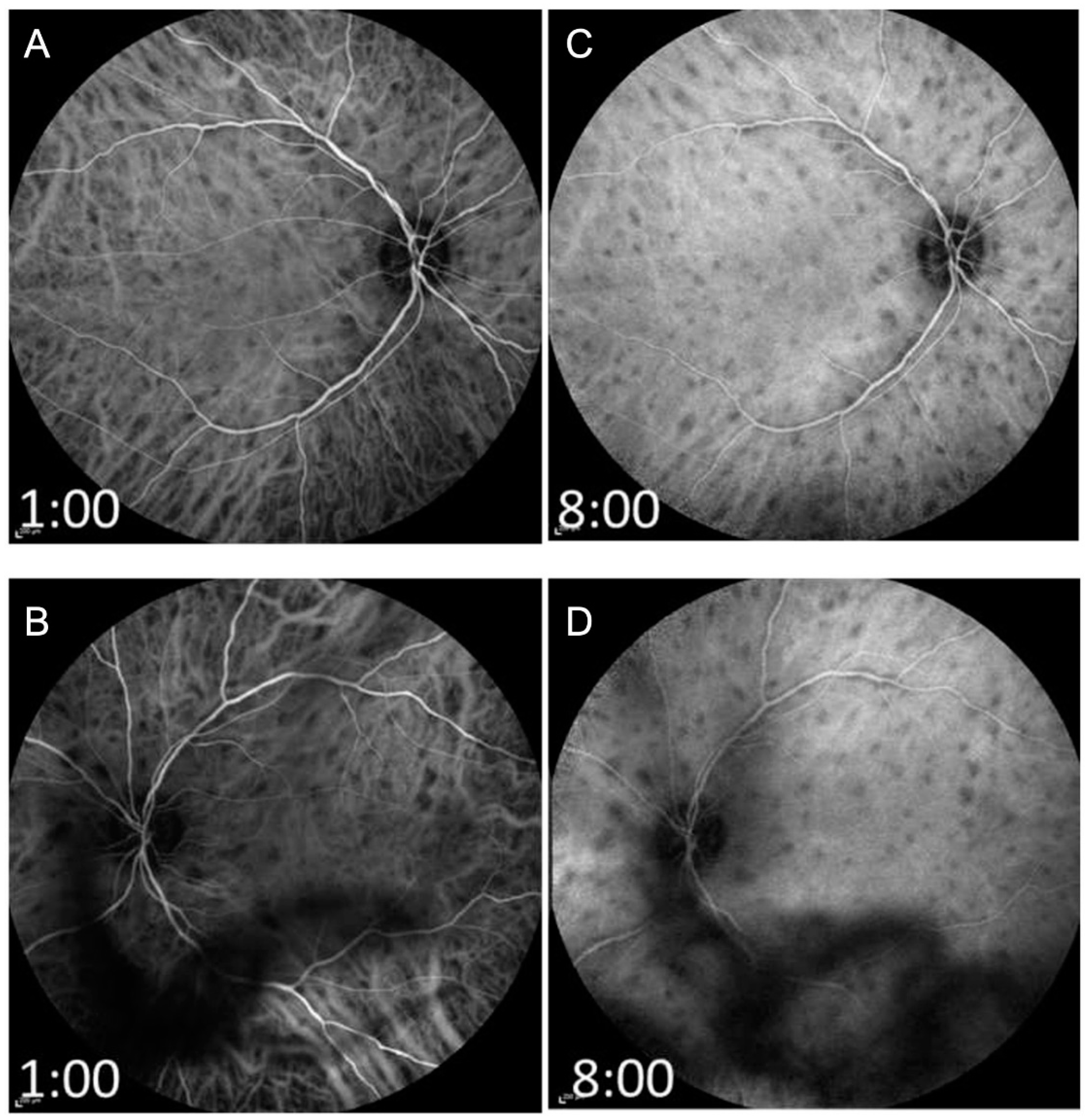{kind=link}
| Infectious Disorders |
|---|
| Bacterial disorders Borreliosis (Borellia burgdorferi) Brucellosis Cat scratch disease (Bartonella henselae) Endophthalmitis Syphilis Tuberculosis Whipple’s disease |
| Viral disorders Acquired immunodeficiency syndrome Acute retinal necrosis Cytomegalovirus Retinitis Chikungunya Dengue fever Hepatitis Human T cell lymphoma virus type 1 Rift Valley fever West Nile Virus |
| Parasitic disorders Mediterranean spotted fever Rickettsia disorders Rocky Mountain spotted fever Toxoplasmosis |
| Noninfectious disorders |
| Systemic inflammatory diseases Behçet’s disease Churg-Strauss syndrome Crohn’s disease Dermatomyositis Granulomatosis with polyangiitis (GPA) HLA-B27-associated uveitis Multiple sclerosis Sarcoidosis Systemic lupus erythematosus Relapsing polychondritis Sjögren’s syndrome Polymyositis Postvaccination Rheumatoid arthritis Susac’s syndrome Takayasu’s disease |
| Isolated ocular disorders Acute multifocal hemorrhagic retinal vasculitis Birdshot chorioretinopathy Frosted branch angiitis (IRVAN) Idiopathic recurrent branch retinal arterial occlusion Intermediate Uveitis |
| Masquerade Syndromes Leukemia Ocular lymphoma (B- T-cell) Paraneoplastic syndromes |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Agarwal, A.; Rübsam, A.; zur Bonsen, L.; Pichi, F.; Neri, P.; Pleyer, U. A Comprehensive Update on Retinal Vasculitis: Etiologies, Manifestations and Treatments. J. Clin. Med. 2022, 11, 2525. https://0-doi-org.brum.beds.ac.uk/10.3390/jcm11092525
Agarwal A, Rübsam A, zur Bonsen L, Pichi F, Neri P, Pleyer U. A Comprehensive Update on Retinal Vasculitis: Etiologies, Manifestations and Treatments. Journal of Clinical Medicine. 2022; 11(9):2525. https://0-doi-org.brum.beds.ac.uk/10.3390/jcm11092525
Chicago/Turabian StyleAgarwal, Aniruddha, Anne Rübsam, Lynn zur Bonsen, Francesco Pichi, Piergiorgio Neri, and Uwe Pleyer. 2022. "A Comprehensive Update on Retinal Vasculitis: Etiologies, Manifestations and Treatments" Journal of Clinical Medicine 11, no. 9: 2525. https://0-doi-org.brum.beds.ac.uk/10.3390/jcm11092525