
Myelodysplastic Syndromes with Isolated 20q Deletion: A New Clinical–Biological Entity?

 , , , add
Show full author list
, , , add
Show full author list
Abstract
:1. Introduction
2. Patients and Methods
2.1. Patients
2.2. Statistical Analysis
3. Results
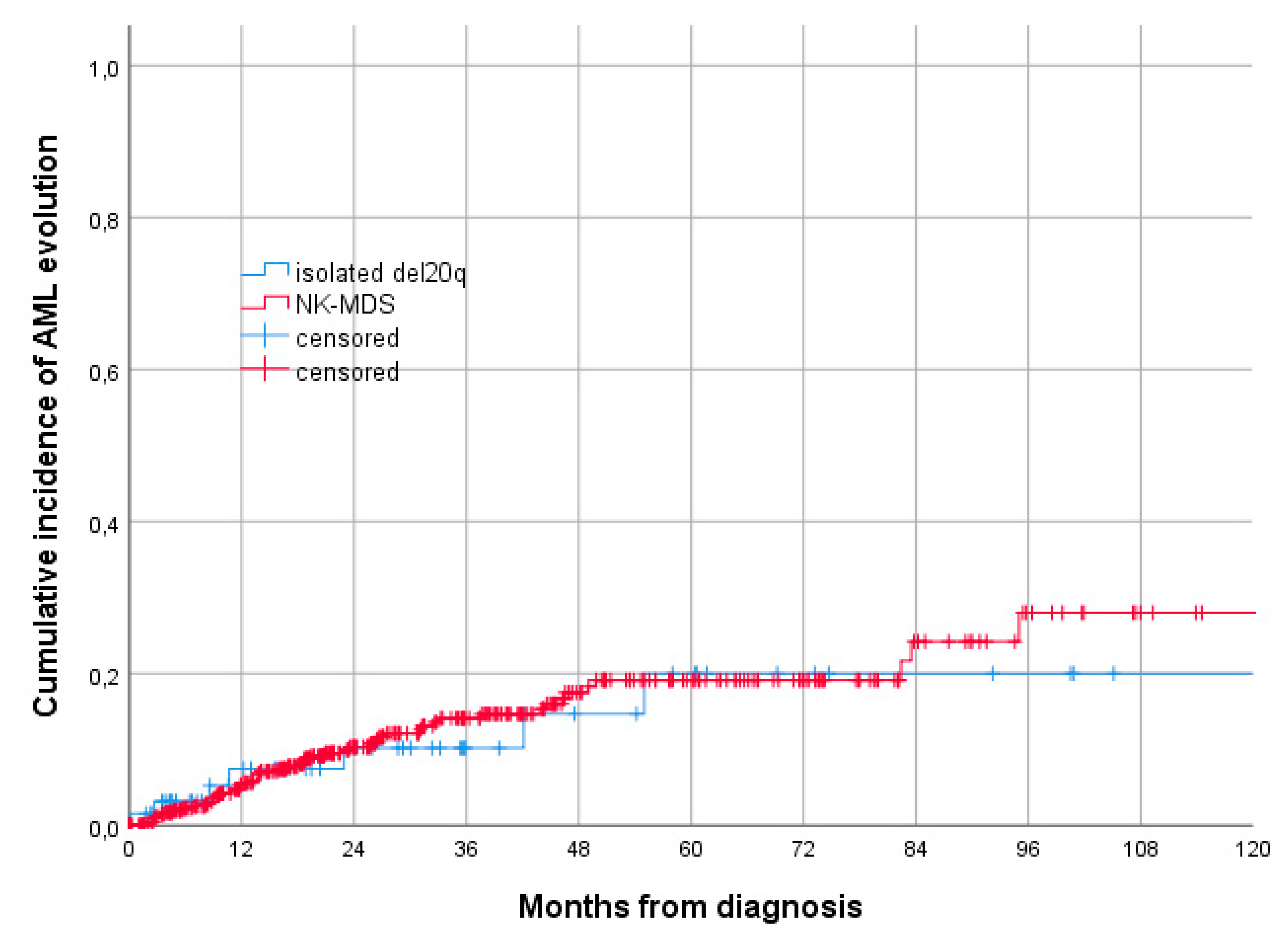
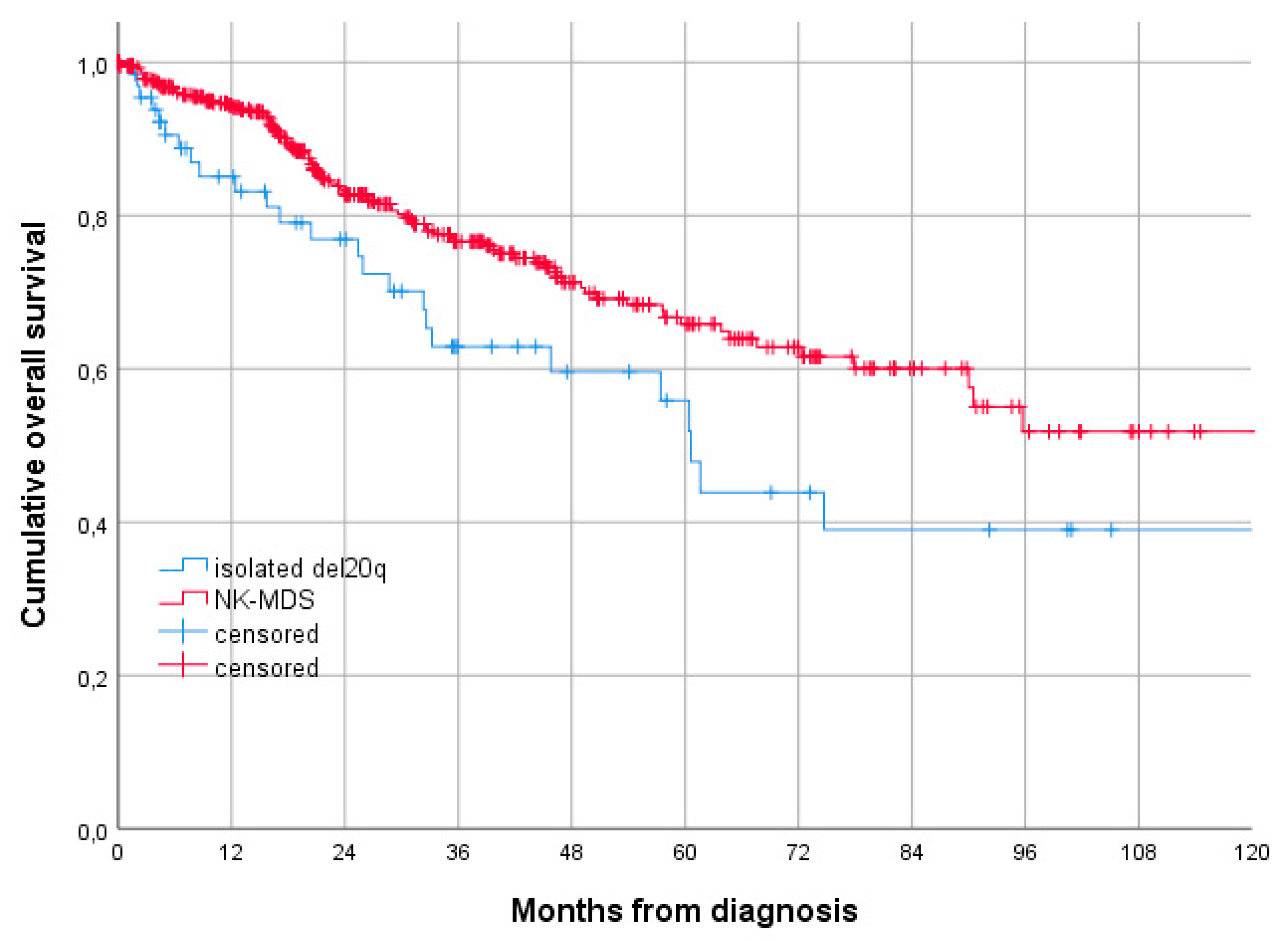
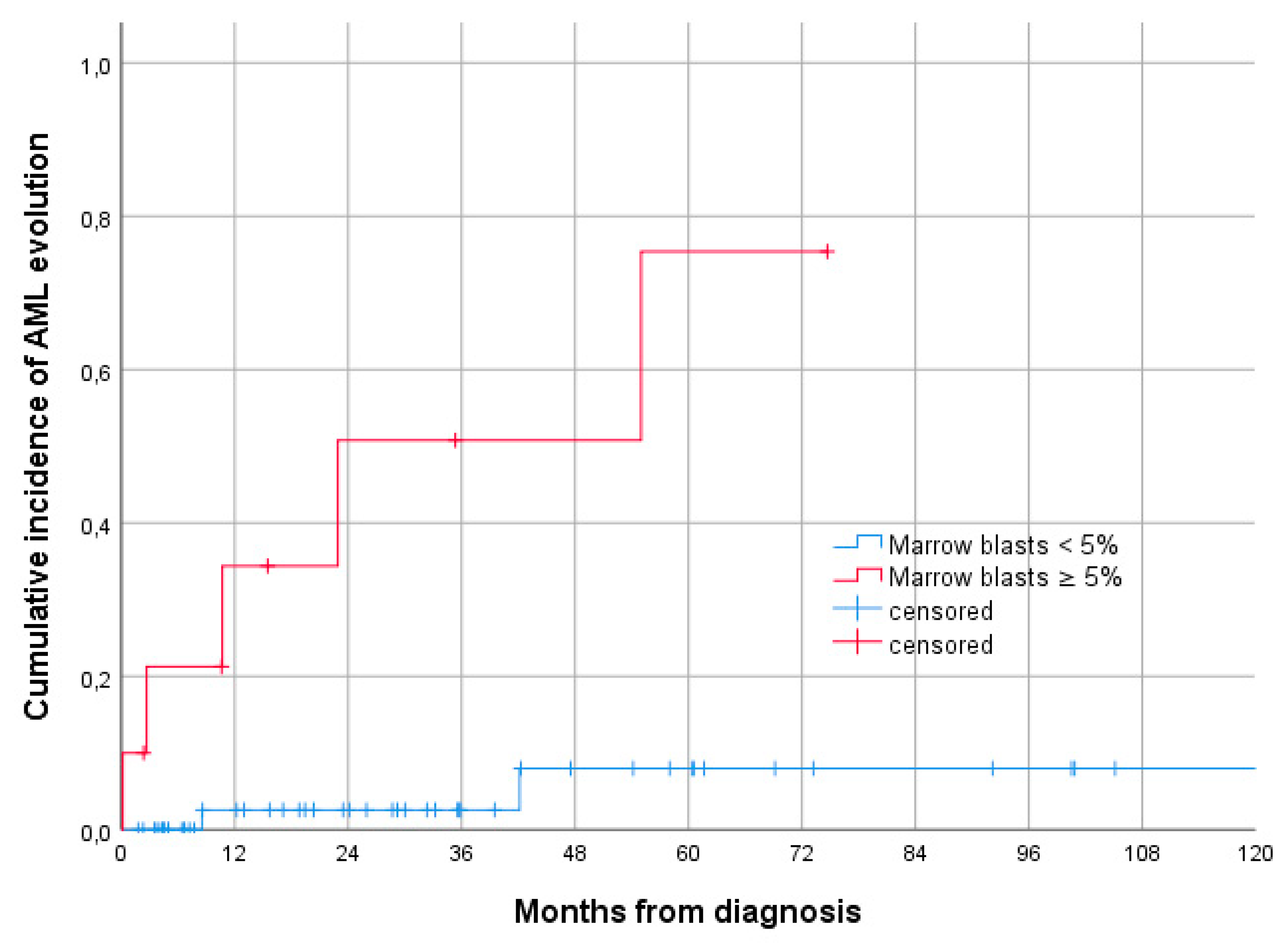
3.1. Baseline Characteristics and Comparison with NK-MDS
3.2. Treatment and Outcome of Patients with Isolated del 20q
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Fenaux, P.; Haase, D.; Sanz, G.F.; Santini, V.; Buske, C. ESMO Guidelines Working Group. Myelodysplastic syndromes: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann. Oncol. 2014, 25, 57–69. [Google Scholar] [CrossRef] [PubMed]
- Haase, D.; Germing, U.; Schanz, J.; Pfeilstöcker, M.; Nösslinger, T.; Hildebrandt, B.; Kundgen, A.; Lübbert, M.; Kunzmann, R.; Giagounidis, A.A.; et al. New insights into the prognostic impact of the karyotype in MDS and correlation with subtypes: Evidence from a core dataset of 2124 patients. Blood 2007, 110, 4385–4395. [Google Scholar] [CrossRef] [PubMed]
- Wattel, E.; Laï, J.L.; Hebbar, M.; Preudhomme, C.; Grahek, D.; Morel, P.; Bauters, F.; Fenaux, P. De novo myelodysplastic syndrome (MDS) with deletion of the long arm of chromosome 20: A subtype of MDS with distinct hematological and prognostic features? Leuk. Res. 1993, 17, 921–926. [Google Scholar] [CrossRef]
- Braun, T.; de Botton, S.; Taksin, A.L.; Park, S.; Beyne-Rauzy, O.; Coiteux, V.; Sapena, R.; Lazareth, A.; Leroux, G.; Guenda, K.; et al. Characteristics and outcome of myelodysplastic syndromes (MDS) with isolated 20q deletion: A report on 62 cases. Leuk. Res. 2011, 35, 863–867. [Google Scholar] [CrossRef] [PubMed]
- Gupta, R.; Soupir, C.P.; Johari, V.; Hasserjian, R.P. Myelodysplastic syndrome with isolated deletion of chromosome 20q: An indolent disease with minimal morphological dysplasia and frequent thrombocytopenic presentation. Br. J. Haematol. 2007, 139, 265–268. [Google Scholar] [CrossRef] [PubMed]
- Soupir, C.P.; Vergilio, J.A.; Kelly, E.; Dal Cin, P.; Kuter, D.; Hasserjian, R.P. Identification of del(20q) in a subset of patients diagnosed with idiopathic thrombocytopenic purpura. Br. J. Haematol. 2008, 144, 794–805. [Google Scholar] [CrossRef] [PubMed]
- Mullier, F.; Daliphard, S.; Garand, R.; Dekeyser, M.; Cornet, Y.; Luquet, I.; Talmant, P.; Richebourg, S.; Jamar, M.; Dogné, J.M.; et al. Morphology, cytogenetics, and survival in myelodysplasia with del(20q) or ider(20q): A multicenter study. Ann. Hematol. 2012, 91, 203–213. [Google Scholar] [CrossRef] [PubMed]
- Huh, J.; Tiu, R.V.; Gondek, L.P.; O’Keefe, C.L.; Jasek, M.; Makishima, H.; Jankowska, A.M.; Jiang, Y.; Verma, A.; Theil, K.S.; et al. Characterization of chromosome arm 20q abnormalities in myeloid malignancies using genome-wide single nucleotide polymorphism array analysis. Genes Chromosomes Cancer 2010, 49, 390–399. [Google Scholar] [CrossRef] [PubMed]
- Okada, M.; Suto, Y.; Hirai, M.; Shiseki, M.; Usami, A.; Okajima, K.; Teramura, M.; Mori, N.; Motoji, T. Microarray CGH analyses of chromosomal 20q deletions in patients with hematopoietic malignancies. Cancer Genet. 2012, 205, 18–24. [Google Scholar] [CrossRef] [PubMed]
- Clarke, M.; Dumon, S.; Ward, C.; Jäger, R.; Freeman, S.; Dawood, B.; Sheriff, L.; Lorvellec, M.; Kralovics, R.; Frampton, J.; et al. MYBL2 haploinsufficiency increases susceptibility to age-related haematopoietic neoplasia. Leukemia 2013, 27, 661–670. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perna, F.; Gurvich, N.; Hoya-Arias, R.; Abdel-Wahab, O.; Levine, R.L.; Asai, T.; Voza, F.; Menendez, S.; Wang, L.; Liu, F.; et al. Depletion of L3MBTL1 promotes the erythroid differentiation of human hematopoietic progenitor cells: Possible role in 20q- polycythemia vera. Blood 2010, 116, 2812–2821. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stoner, S.A.; Yan, M.; Liu, K.T.; Arimoto, K.I.; Shima, T.; Wang, H.Y.; Johnson, D.T.; Bejar, R.; Jamieson, C.; Guan, K.L.; et al. Hippo kinase loss contributes to del(20q) hematologic malignancies through chronic innate immune activation. Blood 2019, 134, 1730–1744. [Google Scholar] [CrossRef] [PubMed]
- Martín, I.; Villamón, E.; Abellán, R.; Calasanz, M.J.; Irigoyen, A.; Sanz, G.; Such, E.; Mora, E.; Gutiérrez, M.; Collado, R.; et al. Myelodysplastic Syndromes (GESMD). Myelodysplastic syndromes with 20q deletion: Incidence, prognostic value and impact on response to azacitidine of ASXL1 chromosomal deletion and genetic mutations. Br. J. Haematol. 2021, 194, 708–717. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| MDS with del(20q) | MDS with NK | p | |
|---|---|---|---|
| M/F, n (%) | 50/19 (72.5/27.5) | 276/226 (54.9/45.1) | 0.006 |
| Median age, years (IQR) | 76.0 (68.9–81.5) | 72.1 (63.2–73.3) | 0.020 |
| WHO classification, n (%): | |||
| MDS with unilineage dysplasia | 15 (21.7) | 173 (34.5) | |
| MDS with multilineage dysplasia | 40 (58.1) | 138 (27.5) | |
| MDS with sideroblasts | 3 (4.3) | 24 (4.8) | <0.001 |
| MDS with excess of blasts 1 | 6 (8.7) | 76 (15.1) | |
| MDS with excess of blasts 2 | 4 (5.8) | 66 (13.1) | |
| MDS-Unclassifiable | 1 (1.4) | 25 (5.0) | |
| Marrow blasts, n° (%): | |||
| <5% | 58 (84.0) | 335 (66.7) | 0.004 |
| ≥5% | 11 (16.0) | 167 (33.3) | |
| Median Hb, g/dL (IQR) | 10.2 (9.0–12.0) | 10 (8.7–11.6) | 0.849 |
| Median WBC, ×109/L (IQR) | 3.7 (2.3–5.5) | 3.6 (2.7–5.5) | 0.920 |
| Median PLTS, ×109/L (IQR) | 92 (51–133) | 135 (75–230) | <0.001 |
| IPSS risk score, n (%): | |||
| Low/Intermediate-1 | 66 (95.6) | 431 (85.8) | 0.023 |
| Intermediate-2/High | 3 (4.4) | 71 (14.2) | |
| Median ferritin, mg/dL (IQR) | 213 (78–385) | 193 (96–421) | 0.741 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Campagna, A.; De Benedittis, D.; Fianchi, L.; Scalzulli, E.; Rizzo, L.; Niscola, P.; Piccioni, A.L.; Di Veroli, A.; Mancini, S.; Villivà, N.; et al. Myelodysplastic Syndromes with Isolated 20q Deletion: A New Clinical–Biological Entity? J. Clin. Med. 2022, 11, 2596. https://0-doi-org.brum.beds.ac.uk/10.3390/jcm11092596
Campagna A, De Benedittis D, Fianchi L, Scalzulli E, Rizzo L, Niscola P, Piccioni AL, Di Veroli A, Mancini S, Villivà N, et al. Myelodysplastic Syndromes with Isolated 20q Deletion: A New Clinical–Biological Entity? Journal of Clinical Medicine. 2022; 11(9):2596. https://0-doi-org.brum.beds.ac.uk/10.3390/jcm11092596
Chicago/Turabian StyleCampagna, Alessia, Daniela De Benedittis, Luana Fianchi, Emilia Scalzulli, Lorenzo Rizzo, Pasquale Niscola, Anna Lina Piccioni, Ambra Di Veroli, Stefano Mancini, Nicoletta Villivà, and et al. 2022. "Myelodysplastic Syndromes with Isolated 20q Deletion: A New Clinical–Biological Entity?" Journal of Clinical Medicine 11, no. 9: 2596. https://0-doi-org.brum.beds.ac.uk/10.3390/jcm11092596