
Hypertensive Heart Failure
 , , , and
, , , and
Abstract
:1. Introduction
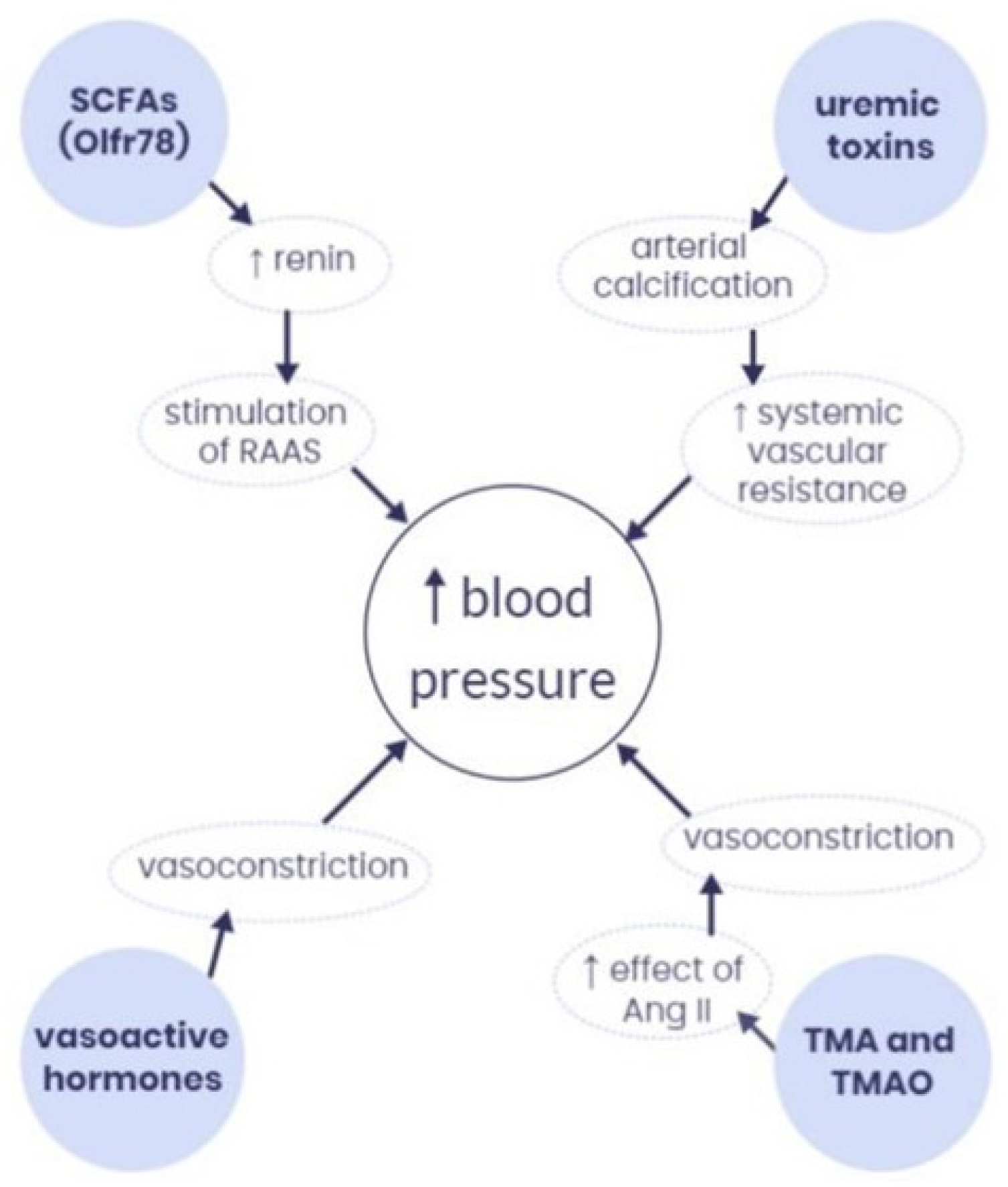
2. Pathophysiology of Hypertension
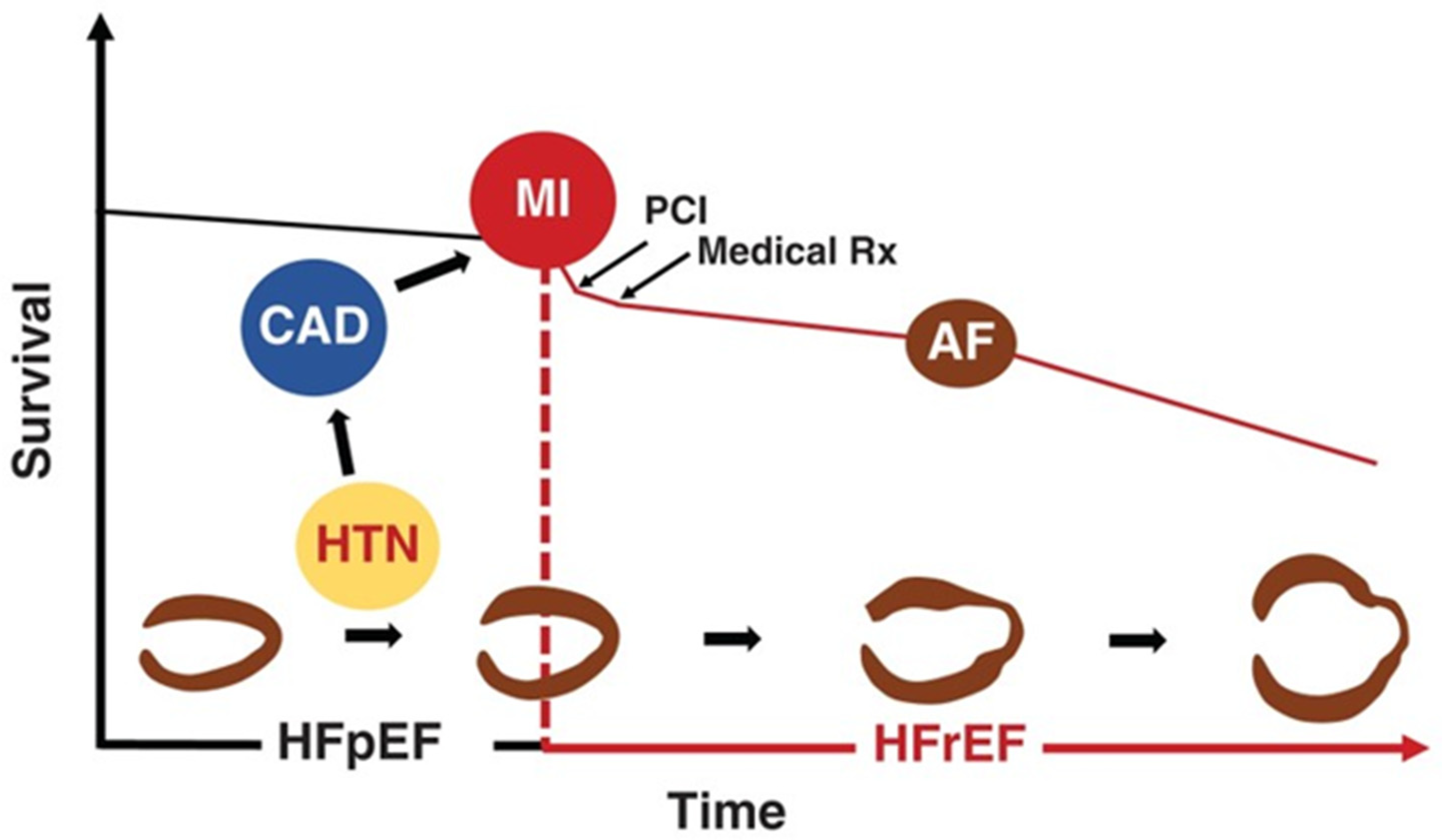
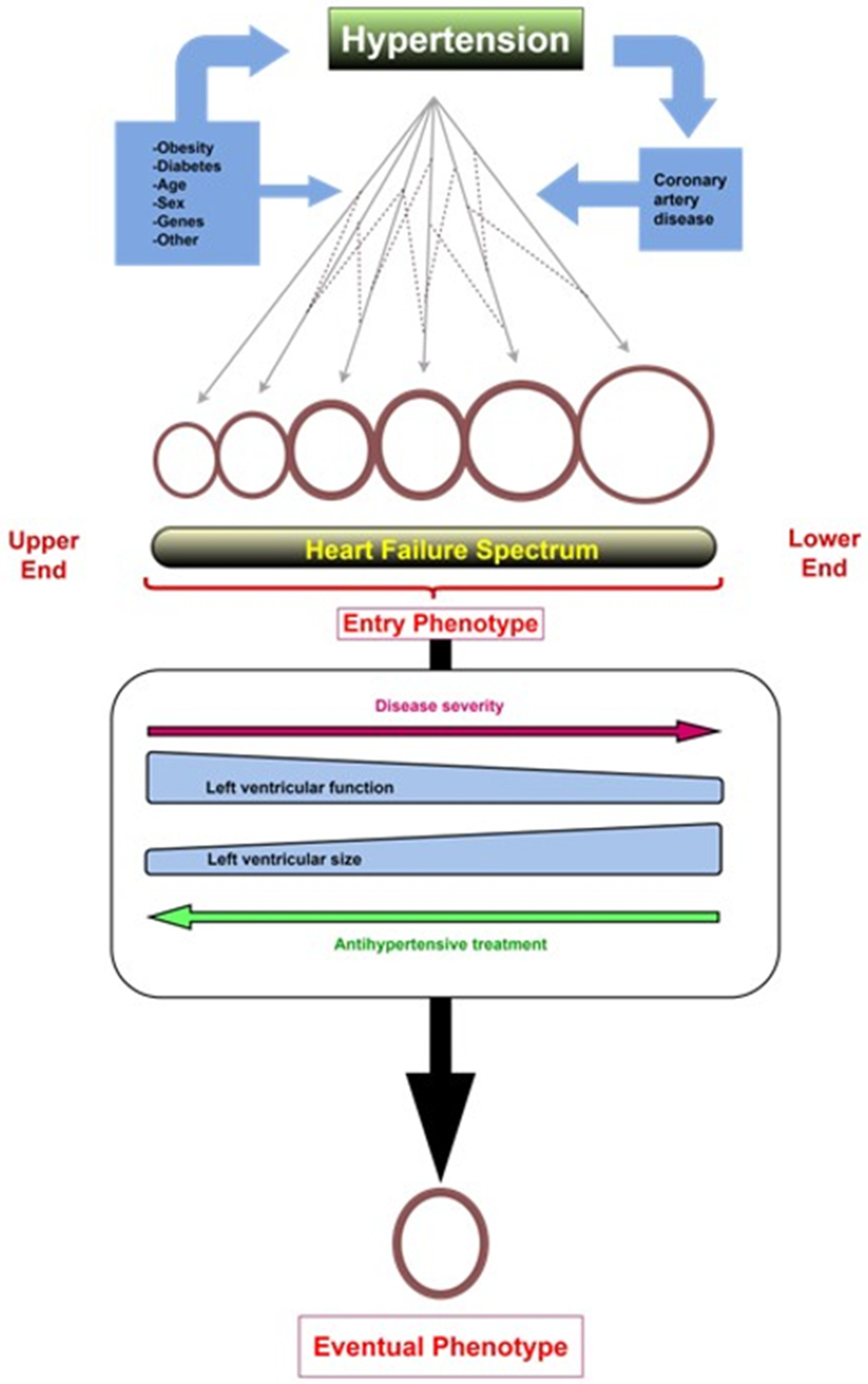
3. From Hypertension to Hypertensive Heart Failure
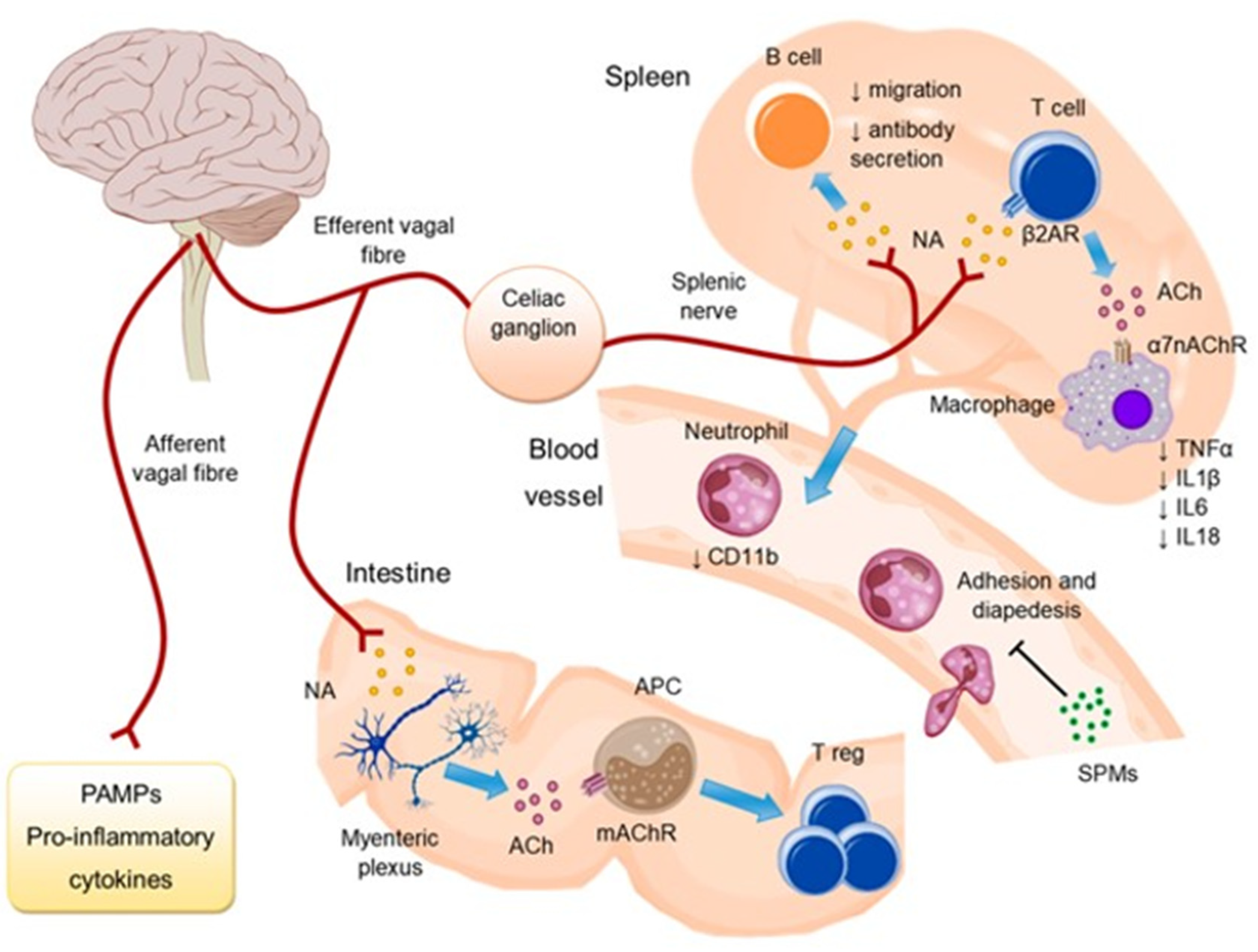
4. Cardiac Autonomic Imbalance
5. Treatment of Hypertensive Heart Failure
5.1. Blood Pressure Targets
5.1.1. Target Average Blood Pressure
5.1.2. Time in Therapeutic Range
5.2. Medications
5.2.1. RAAS Inhibitors (RAASi)
5.2.2. Sodium Glucose Cotransporter 2 Inhibitors (SGLT-2i)
5.2.3. Beta Adrenergic Receptor Blockers (BBs)
5.2.4. Diuretics
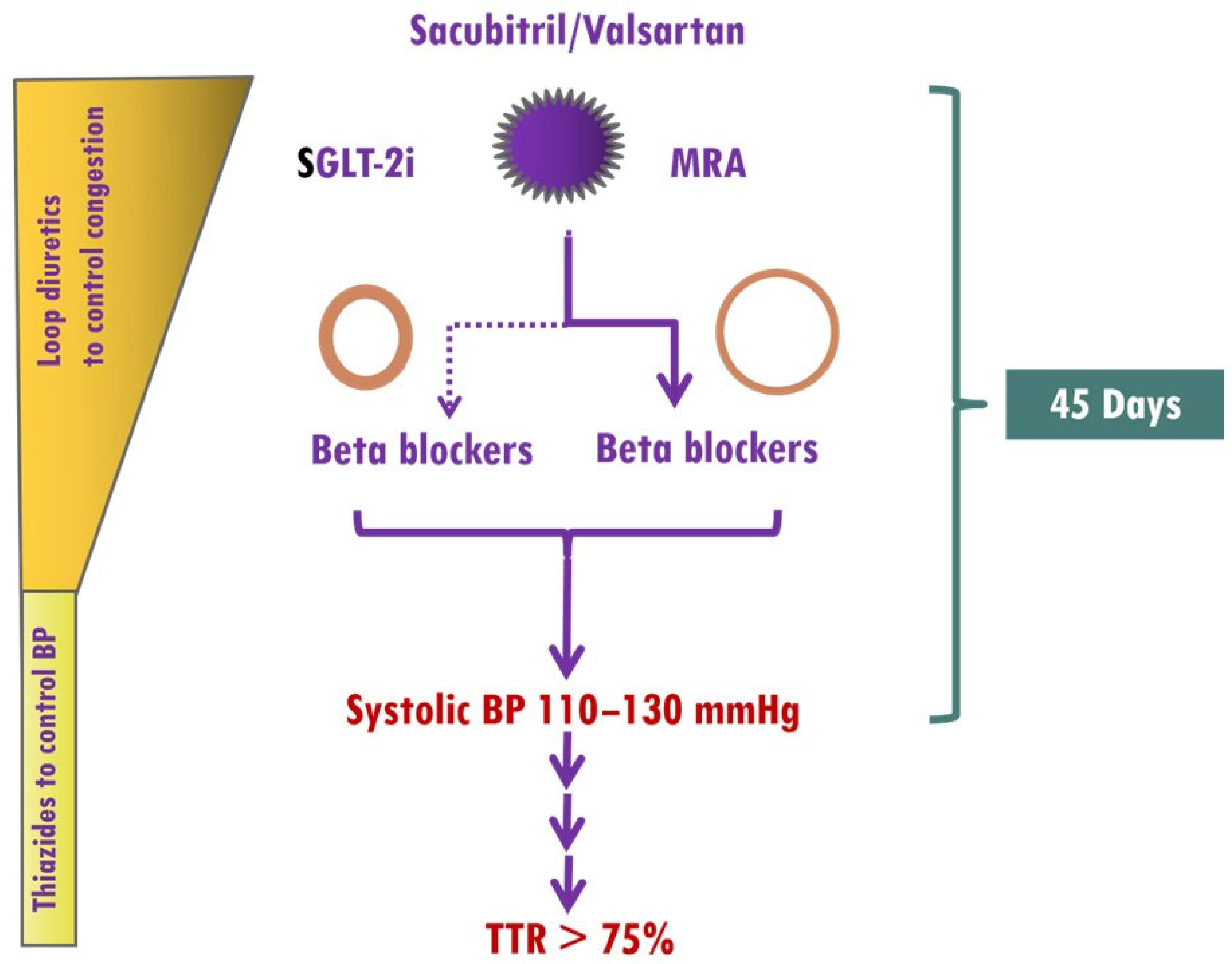
5.2.5. Implementation of Medical Treatment
5.3. Exercise
5.4. Emerging Treatments
6. Perspective
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Kearney, P.M.; Whelton, M.; Reynolds, K.; Muntner, P.; Whelton, P.K.; He, J. Global burden of hypertension: Analysis of worldwide data. Lancet 2005, 365, 217–223. [Google Scholar] [CrossRef] [PubMed]
- Murray, C.J.; Lopez, A.D. Measuring the global burden of disease. N. Engl. J. Med. 2013, 369, 448–457. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mills, K.T.; Stefanescu, A.; He, J. The global epidemiology of hypertension. Nat. Rev. Nephrol. 2020, 16, 223–237. [Google Scholar] [CrossRef] [PubMed]
- Conrad, N.; Judge, A.; Tran, J.; Mohseni, H.; Hedgecott, D.; Crespillo, A.P.; Allison, M.; Hemingway, H.; Cleland, J.G.; McMurray, J.J.V.; et al. Temporal trends and patterns in heart failure incidence: A population-based study of 4 million individuals. Lancet 2018, 391, 572–580. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rapsomaniki, E.; Timmis, A.; George, J.; Pujades-Rodriguez, M.; Shah, A.D.; Denaxas, S.; White, I.R.; Caulfield, M.J.; Deanfield, J.E.; Smeeth, L.; et al. Blood pressure and incidence of twelve cardiovascular diseases: Lifetime risks, healthy life-years lost, and age-specific associations in 1.25 million people. Lancet 2014, 383, 1899–1911. [Google Scholar] [CrossRef] [Green Version]
- Tromp, J.; Paniagua, S.M.A.; Lau, E.S.; Allen, N.B.; Blaha, M.J.; Gansevoort, R.T.; Hillege, H.L.; Lee, D.E.; Levy, D.; Vasan, R.S.; et al. Age dependent associations of risk factors with heart failure: Pooled population based cohort study. BMJ 2021, 372, n461. [Google Scholar] [CrossRef]
- Williams, B.; Mancia, G.; Spiering, W.; Agabiti Rosei, E.; Azizi, M.; Burnier, M.; Clement, D.L.; Coca, A.; de Simone, G.; Dominiczak, A.; et al. 2018 ESC/ESH Guidelines for the management of arterial hypertension. Eur. Heart J. 2018, 39, 3021–3104. [Google Scholar] [CrossRef] [Green Version]
- Yusuf, S.; Hawken, S.; Ounpuu, S.; Dans, T.; Avezum, A.; Lanas, F.; McQueen, M.; Budaj, A.; Pais, P.; Varigos, J.; et al. Effect of potentially modifiable risk factors associated with myocardial infarction in 52 countries (the INTERHEART study): Case-control study. Lancet 2004, 364, 937–952. [Google Scholar] [CrossRef]
- Nagueh, S.F. Heart failure with preserved ejection fraction: Insights into diagnosis and pathophysiology. Cardiovasc. Res. 2021, 117, 999–1014. [Google Scholar] [CrossRef]
- Steinberg, B.A.; Zhao, X.; Heidenreich, P.A.; Peterson, E.D.; Bhatt, D.L.; Cannon, C.P.; Hernandez, A.F.; Fonarow, G.C.; Get with the Guidelines Scientific Advisory Committee and Investigators. Trends in patients hospitalized with heart failure and preserved left ventricular ejection fraction: Prevalence, therapies, and outcomes. Circulation 2012, 126, 65–75. [Google Scholar] [CrossRef] [Green Version]
- Pitt, B.; Pfeffer, M.A.; Assmann, S.F.; Boineau, R.; Anand, I.S.; Claggett, B.; Clausell, N.; Desai, A.S.; Diaz, R.; Fleg, J.L.; et al. Spironolactone for heart failure with preserved ejection fraction. N. Engl. J. Med. 2014, 370, 1383–1392. [Google Scholar] [CrossRef] [Green Version]
- Solomon, S.D.; McMurray, J.J.V.; Anand, I.S.; Ge, J.; Lam, C.S.P.; Maggioni, A.P.; Martinez, F.; Packer, M.; Pfeffer, M.A.; Pieske, B.; et al. Angiotensin-Neprilysin Inhibition in Heart Failure with Preserved Ejection Fraction. N. Engl. J. Med. 2019, 381, 1609–1620. [Google Scholar] [CrossRef] [Green Version]
- Anker, S.D.; Butler, J.; Packer, M. Empagliflozin in Heart Failure with a Preserved Ejection Fraction. Reply. N. Engl. J. Med. 2022, 386, e57. [Google Scholar] [CrossRef]
- Solomon, S.D.; McMurray, J.J.V.; Claggett, B.; de Boer, R.A.; DeMets, D.; Hernandez, A.F.; Inzucchi, S.E.; Kosiborod, M.N.; Lam, C.S.P.; Martinez, F.; et al. Dapagliflozin in Heart Failure with Mildly Reduced or Preserved Ejection Fraction. N. Engl. J. Med. 2022, 387, 1089–1098. [Google Scholar] [CrossRef]
- Dandamudi, S.; Slusser, J.; Mahoney, D.W.; Redfield, M.M.; Rodeheffer, R.J.; Chen, H.H. The prevalence of diabetic cardiomyopathy: A population-based study in Olmsted County, Minnesota. J. Card. Fail. 2014, 20, 304–309. [Google Scholar] [CrossRef] [Green Version]
- Ren, J.; Wu, N.N.; Wang, S.; Sowers, J.R.; Zhang, Y. Obesity cardiomyopathy: Evidence, mechanisms, and therapeutic implications. Physiol. Rev. 2021, 101, 1745–1807. [Google Scholar] [CrossRef]
- Salvatore, T.; Pafundi, P.C.; Galiero, R.; Albanese, G.; Di Martino, A.; Caturano, A.; Vetrano, E.; Rinaldi, L.; Sasso, F.C. The Diabetic Cardiomyopathy: The Contributing Pathophysiological Mechanisms. Front. Med. 2021, 8, 695792. [Google Scholar] [CrossRef] [PubMed]
- Triposkiadis, F.; Xanthopoulos, A.; Bargiota, A.; Kitai, T.; Katsiki, N.; Farmakis, D.; Skoularigis, J.; Starling, R.C.; Iliodromitis, E. Diabetes Mellitus and Heart Failure. J. Clin. Med. 2021, 10, 3682. [Google Scholar] [CrossRef]
- Giamouzis, G.; Xanthopoulos, A.; Papamichalis, M.; Chroub-Papavaiou, A.N.; Pantziou, A.; Simou, A.; Dimos, A.; Bourazana, A.; Skoularigis, J.; Triposkiadis, F. Relative contribution of risk factors/co-morbidities to heart failure pathogenesis: Interaction with ejection fraction. ESC Heart Fail. 2020, 7, 4399–4403. [Google Scholar] [CrossRef]
- Triposkiadis, F.; Starling, R.C.; Boudoulas, H.; Giamouzis, G.; Butler, J. The cardiorenal syndrome in heart failure: Cardiac? renal? syndrome? Heart Fail. Rev. 2012, 17, 355–366. [Google Scholar] [CrossRef]
- Triposkiadis, F.; Giamouzis, G.; Parissis, J.; Starling, R.C.; Boudoulas, H.; Skoularigis, J.; Butler, J.; Filippatos, G. Reframing the association and significance of co-morbidities in heart failure. Eur. J. Heart Fail. 2016, 18, 744–758. [Google Scholar] [CrossRef]
- Kuan, V.; Denaxas, S.; Patalay, P.; Nitsch, D.; Mathur, R.; Gonzalez-Izquierdo, A.; Sofat, R.; Partridge, L.; Roberts, A.; Wong, I.C.K.; et al. Identifying and visualising multimorbidity and comorbidity patterns in patients in the English National Health Service: A population-based study. Lancet Digit. Health 2023, 5, e16–e27. [Google Scholar] [CrossRef]
- Whelton, P.K.; Carey, R.M.; Mancia, G.; Kreutz, R.; Bundy, J.D.; Williams, B. Harmonization of the American College of Cardiology/American Heart Association and European Society of Cardiology/European Society of Hypertension Blood Pressure/Hypertension Guidelines: Comparisons, Reflections, and Recommendations. Circulation 2022, 146, 868–877. [Google Scholar] [CrossRef]
- Triposkiadis, F.; Xanthopoulos, A.; Lampropoulos, K.; Briasoulis, A.; Sarafidis, P.; Skoularigis, J.; Boudoulas, H. Aortic Stiffness: A Major Risk Factor for Multimorbidity in the Elderly. J. Clin. Med. 2023, 12, 2321. [Google Scholar] [CrossRef]
- Frohlich, E.D.; Dustan, H.P.; Bumpus, F.M. Irvine H. Page: 1901–1991. The celebration of a leader. Hypertension 1991, 18, 443–445. [Google Scholar] [CrossRef] [Green Version]
- Harrison, D.G. The mosaic theory revisited: Common molecular mechanisms coordinating diverse organ and cellular events in hypertension. J. Am. Soc. Hypertens. 2013, 7, 68–74. [Google Scholar] [CrossRef] [Green Version]
- Harrison, D.G.; Coffman, T.M.; Wilcox, C.S. Pathophysiology of Hypertension: The Mosaic Theory and Beyond. Circ. Res. 2021, 128, 847–863. [Google Scholar] [CrossRef]
- Cryan, J.F.; O’Riordan, K.J.; Cowan, C.S.M.; Sandhu, K.V.; Bastiaanssen, T.F.S.; Boehme, M.; Codagnone, M.G.; Cussotto, S.; Fulling, C.; Golubeva, A.V.; et al. The Microbiota-Gut-Brain Axis. Physiol. Rev. 2019, 99, 1877–2013. [Google Scholar] [CrossRef]
- Tokarek, J.; Budny, E.; Saar, M.; Kucmierz, J.; Mlynarska, E.; Rysz, J.; Franczyk, B. Does the Composition of Gut Microbiota Affect Hypertension? Molecular Mechanisms Involved in Increasing Blood Pressure. Int. J. Mol. Sci. 2023, 24, 1377. [Google Scholar] [CrossRef]
- O’Donnell, J.A.; Zheng, T.; Meric, G.; Marques, F.Z. The gut microbiome and hypertension. Nat. Rev. Nephrol. 2023, 19, 153–167. [Google Scholar] [CrossRef]
- Richards, E.M.; Li, J.; Stevens, B.R.; Pepine, C.J.; Raizada, M.K. Gut Microbiome and Neuroinflammation in Hypertension. Circ. Res. 2022, 130, 401–417. [Google Scholar] [CrossRef]
- Madhur, M.S.; Elijovich, F.; Alexander, M.R.; Pitzer, A.; Ishimwe, J.; Van Beusecum, J.P.; Patrick, D.M.; Smart, C.D.; Kleyman, T.R.; Kingery, J.; et al. Hypertension: Do Inflammation and Immunity Hold the Key to Solving this Epidemic? Circ. Res. 2021, 128, 908–933. [Google Scholar] [CrossRef]
- Bostick, J.W.; Schonhoff, A.M.; Mazmanian, S.K. Gut microbiome-mediated regulation of neuroinflammation. Curr. Opin. Immunol. 2022, 76, 102177. [Google Scholar] [CrossRef]
- Jaworska, K.; Koper, M.; Ufnal, M. Gut microbiota and renin-angiotensin system: A complex interplay at local and systemic levels. Am. J. Physiol. Gastrointest. Liver Physiol. 2021, 321, G355–G366. [Google Scholar] [CrossRef]
- Paulus, W.J.; Tschope, C. A novel paradigm for heart failure with preserved ejection fraction: Comorbidities drive myocardial dysfunction and remodeling through coronary microvascular endothelial inflammation. J. Am. Coll. Cardiol. 2013, 62, 263–271. [Google Scholar] [CrossRef] [Green Version]
- Xanthopoulos, A.; Triposkiadis, F.; Starling, R.C. Heart failure with preserved ejection fraction: Classification based upon phenotype is essential for diagnosis and treatment. Trends Cardiovasc. Med. 2018, 28, 392–400. [Google Scholar] [CrossRef]
- Adua, E. Decoding the mechanism of hypertension through multiomics profiling. J. Hum. Hypertens. 2023, 37, 253–264. [Google Scholar] [CrossRef]
- Seidel, E.; Scholl, U.I. Genetic mechanisms of human hypertension and their implications for blood pressure physiology. Physiol. Genom. 2017, 49, 630–652. [Google Scholar] [CrossRef] [Green Version]
- Masenga, S.K.; Kirabo, A. Hypertensive heart disease: Risk factors, complications and mechanisms. Front. Cardiovasc. Med. 2023, 10, 1205475. [Google Scholar] [CrossRef]
- Nadruz, W. Myocardial remodeling in hypertension. J. Hum. Hypertens. 2015, 29, 1–6. [Google Scholar] [CrossRef]
- Osler, W. The Principles and Practice of Medicine; Appleton: New York, NY, USA, 1892. [Google Scholar]
- Messerli, F.H.; Rimoldi, S.F.; Bangalore, S. The Transition From Hypertension to Heart Failure: Contemporary Update. JACC Heart Fail. 2017, 5, 543–551. [Google Scholar] [CrossRef]
- Cuspidi, C.; Sala, C.; Negri, F.; Mancia, G.; Morganti, A.; Italian Society of Hypertension. Prevalence of left-ventricular hypertrophy in hypertension: An updated review of echocardiographic studies. J. Hum. Hypertens. 2012, 26, 343–349. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Drazner, M.H. The progression of hypertensive heart disease. Circulation 2011, 123, 327–334. [Google Scholar] [CrossRef]
- Velagaleti, R.S.; Gona, P.; Pencina, M.J.; Aragam, J.; Wang, T.J.; Levy, D.; D’Agostino, R.B.; Lee, D.S.; Kannel, W.B.; Benjamin, E.J.; et al. Left ventricular hypertrophy patterns and incidence of heart failure with preserved versus reduced ejection fraction. Am. J. Cardiol. 2014, 113, 117–122. [Google Scholar] [CrossRef] [Green Version]
- Vedin, O.; Lam, C.S.P.; Koh, A.S.; Benson, L.; Teng, T.H.K.; Tay, W.T.; Braun, O.O.; Savarese, G.; Dahlstrom, U.; Lund, L.H. Significance of Ischemic Heart Disease in Patients with Heart Failure and Preserved, Midrange, and Reduced Ejection Fraction: A Nationwide Cohort Study. Circ. Heart Fail. 2017, 10, e003875. [Google Scholar] [CrossRef]
- Yamanaka, S.; Sakata, Y.; Nochioka, K.; Miura, M.; Kasahara, S.; Sato, M.; Aoyanagi, H.; Fujihashi, T.; Hayashi, H.; Shiroto, T.; et al. Prognostic impacts of dynamic cardiac structural changes in heart failure patients with preserved left ventricular ejection fraction. Eur. J. Heart Fail. 2020, 22, 2258–2268. [Google Scholar] [CrossRef]
- Milani, R.V.; Lavie, C.J.; Mehra, M.R.; Ventura, H.O.; Kurtz, J.D.; Messerli, F.H. Left ventricular geometry and survival in patients with normal left ventricular ejection fraction. Am. J. Cardiol. 2006, 97, 959–963. [Google Scholar] [CrossRef]
- Lamirault, G.; Artifoni, M.; Daniel, M.; Barber-Chamoux, N.; Nantes University Hospital Working Group On, H. Resistant Hypertension: Novel Insights. Curr. Hypertens. Rev. 2020, 16, 61–72. [Google Scholar] [CrossRef]
- De Keulenaer, G.W.; Brutsaert, D.L. Systolic and diastolic heart failure are overlapping phenotypes within the heart failure spectrum. Circulation 2011, 123, 1996–2004; discussion 2005. [Google Scholar] [CrossRef] [Green Version]
- Triposkiadis, F.; Butler, J.; Abboud, F.M.; Armstrong, P.W.; Adamopoulos, S.; Atherton, J.J.; Backs, J.; Bauersachs, J.; Burkhoff, D.; Bonow, R.O.; et al. The continuous heart failure spectrum: Moving beyond an ejection fraction classification. Eur. Heart J. 2019, 40, 2155–2163. [Google Scholar] [CrossRef]
- Triposkiadis, F.; Xanthopoulos, A.; Parissis, J.; Butler, J.; Farmakis, D. Pathogenesis of chronic heart failure: Cardiovascular aging, risk factors, comorbidities, and disease modifiers. Heart Fail. Rev. 2022, 27, 337–344. [Google Scholar] [CrossRef]
- Triposkiadis, F.; Xanthopoulos, A.; Butler, J. Cardiovascular Aging and Heart Failure: JACC Review Topic of the Week. J. Am. Coll. Cardiol. 2019, 74, 804–813. [Google Scholar] [CrossRef]
- Holwerda, S.W.; Luehrs, R.E.; DuBose, L.; Collins, M.T.; Wooldridge, N.A.; Stroud, A.K.; Fadel, P.J.; Abboud, F.M.; Pierce, G.L. Elevated Muscle Sympathetic Nerve Activity Contributes to Central Artery Stiffness in Young and Middle-Age/Older Adults. Hypertension 2019, 73, 1025–1035. [Google Scholar] [CrossRef]
- Nardone, M.; Floras, J.S.; Millar, P.J. Sympathetic neural modulation of arterial stiffness in humans. Am. J. Physiol. Heart Circ. Physiol. 2020, 319, H1338–H1346. [Google Scholar] [CrossRef]
- Zern, E.K.; Ho, J.E.; Panah, L.G.; Lau, E.S.; Liu, E.; Farrell, R.; Sbarbaro, J.A.; Schoenike, M.W.; Pappagianopoulos, P.P.; Namasivayam, M.; et al. Exercise Intolerance in Heart Failure with Preserved Ejection Fraction: Arterial Stiffness and Aabnormal Left Ventricular Hemodynamic Responses During Exercise. J. Card. Fail. 2021, 27, 625–634. [Google Scholar] [CrossRef]
- Floras, J.S.; Ponikowski, P. The sympathetic/parasympathetic imbalance in heart failure with reduced ejection fraction. Eur. Heart J. 2015, 36, 1974–1982. [Google Scholar] [CrossRef] [Green Version]
- Gronda, E.; Dusi, V.; D’Elia, E.; Iacoviello, M.; Benvenuto, E.; Vanoli, E. Sympathetic activation in heart failure. Eur. Heart J. Suppl. 2022, 24, E4–E11. [Google Scholar] [CrossRef]
- Bencivenga, L.; Palaia, M.E.; Sepe, I.; Gambino, G.; Komici, K.; Cannavo, A.; Femminella, G.D.; Rengo, G. Why Do We Not Assess Sympathetic Nervous System Activity in Heart Failure Management: Might GRK2 Serve as a New Biomarker? Cells 2021, 10, 457. [Google Scholar] [CrossRef]
- Boussi, L.; Frishman, W.H. beta-Arrestin as a Therapeutic Target in Heart Failure. Cardiol. Rev. 2021, 29, 223–229. [Google Scholar] [CrossRef]
- Floras, J.S. Alterations in the sympathetic and parasympathetic nnervous systems in heart failure. In Heart Failure: A Companion to Braunwald’s Heart Disease; Felker, M.G., Mann, D.L., Eds.; Elsevier: Toronto, ON, Canada, 2019; pp. 181–200. [Google Scholar]
- Grassi, G.; Seravalle, G.; Quarti-Trevano, F.; Dell’Oro, R.; Arenare, F.; Spaziani, D.; Mancia, G. Sympathetic and baroreflex cardiovascular control in hypertension-related left ventricular dysfunction. Hypertension 2009, 53, 205–209. [Google Scholar] [CrossRef] [Green Version]
- Verloop, W.L.; Beeftink, M.M.; Santema, B.T.; Bots, M.L.; Blankestijn, P.J.; Cramer, M.J.; Doevendans, P.A.; Voskuil, M. A systematic review concerning the relation between the sympathetic nervous system and heart failure with preserved left ventricular ejection fraction. PLoS ONE 2015, 10, e0117332. [Google Scholar] [CrossRef]
- Kaye, D.M.; Nanayakkara, S.; Wang, B.; Shihata, W.; Marques, F.Z.; Esler, M.; Lambert, G.; Mariani, J. Characterization of Cardiac Sympathetic Nervous System and Inflammatory Activation in HFpEF Patients. JACC Basic. Transl. Sci. 2022, 7, 116–127. [Google Scholar] [CrossRef]
- Seo, M.; Yamada, T.; Tamaki, S.; Watanabe, T.; Morita, T.; Furukawa, Y.; Kawasaki, M.; Kikuchi, A.; Kawai, T.; Nakamura, J.; et al. Prognostic Significance of Cardiac (123)I-MIBG SPECT Imaging in Heart Failure Patients with Preserved Ejection Fraction. JACC Cardiovasc. Imaging 2022, 15, 655–668. [Google Scholar] [CrossRef]
- Olshansky, B.; Sabbah, H.N.; Hauptman, P.J.; Colucci, W.S. Parasympathetic nervous system and heart failure: Pathophysiology and potential implications for therapy. Circulation 2008, 118, 863–871. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramos-Martinez, I.E.; Rodriguez, M.C.; Cerbon, M.; Ramos-Martinez, J.C.; Ramos-Martinez, E.G. Role of the Cholinergic Anti-Inflammatory Reflex in Central Nervous System Diseases. Int. J. Mol. Sci. 2021, 22, 13427. [Google Scholar] [CrossRef] [PubMed]
- Kittipibul, V.; Fudim, M. Tackling Inflammation in Heart Failure with Preserved Ejection Fraction: Resurrection of Vagus Nerve Stimulation? J. Am. Heart Assoc. 2022, 11, e024481. [Google Scholar] [CrossRef]
- Adamo, L.; Rocha-Resende, C.; Prabhu, S.D.; Mann, D.L. Reappraising the role of inflammation in heart failure. Nat. Rev. Cardiol. 2020, 17, 269–285. [Google Scholar] [CrossRef]
- Pinho-Gomes, A.C.; Rahimi, K. Management of blood pressure in heart failure. Heart 2019, 105, 589–595. [Google Scholar] [CrossRef]
- Singhania, N.; Bansal, S.; Mohandas, S.; Nimmatoori, D.P.; Ejaz, A.A.; Singhania, G. Role of renin-angiotensin-aldosterone system inhibitors in heart failure and chronic kidney disease. Drugs Context 2020, 9, 2020-7-3. [Google Scholar] [CrossRef]
- Sano, M. A new class of drugs for heart failure: SGLT2 inhibitors reduce sympathetic overactivity. J. Cardiol. 2018, 71, 471–476. [Google Scholar] [CrossRef] [Green Version]
- Moser, M.; Hebert, P.R. Prevention of disease progression, left ventricular hypertrophy and congestive heart failure in hypertension treatment trials. J. Am. Coll. Cardiol. 1996, 27, 1214–1218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mancia Chairperson, G.; Kreutz Co-Chair, R.; Brunstrom, M.; Burnier, M.; Grassi, G.; Januszewicz, A.; Muiesan, M.L.; Tsioufis, K.; Agabiti-Rosei, E.; Algharably, E.A.E.; et al. 2023 ESH Guidelines for the management of arterial hypertension The Task Force for the management of arterial hypertension of the European Society of Hypertension Endorsed by the European Renal Association (ERA) and the International Society of Hypertension (ISH). J. Hypertens. 2023. [Google Scholar] [CrossRef]
- Moser, M.; Gifford, R.W. Hypertension: Steps forward and steps backward. Arch. Intern. Med. 1993, 153, 1843–1846. [Google Scholar] [CrossRef]
- Maeda, D.; Dotare, T.; Matsue, Y.; Teramoto, K.; Sunayama, T.; Tromp, J.; Minamino, T. Blood pressure in heart failure management and prevention. Hypertens. Res. 2023, 46, 817–833. [Google Scholar] [CrossRef] [PubMed]
- Group, S.R.; Wright, J.T., Jr.; Williamson, J.D.; Whelton, P.K.; Snyder, J.K.; Sink, K.M.; Rocco, M.V.; Reboussin, D.M.; Rahman, M.; Oparil, S.; et al. A Randomized Trial of Intensive versus Standard Blood-Pressure Control. N. Engl. J. Med. 2015, 373, 2103–2116. [Google Scholar] [CrossRef]
- Buckley, L.F.; Dixon, D.L.; Wohlford, G.F.t.; Wijesinghe, D.S.; Baker, W.L.; Van Tassell, B.W. Intensive Versus Standard Blood Pressure Control in SPRINT-Eligible Participants of ACCORD-BP. Diabetes Care 2017, 40, 1733–1738. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wright, J.T., Jr.; Whelton, P.K.; Johnson, K.C.; Snyder, J.K.; Reboussin, D.M.; Cushman, W.C.; Williamson, J.D.; Pajewski, N.M.; Cheung, A.K.; Lewis, C.E.; et al. SPRINT Revisited: Updated Results and Implications. Hypertension 2021, 78, 1701–1710. [Google Scholar] [CrossRef]
- Blood Pressure Lowering Treatment Trialists, C. Pharmacological blood pressure lowering for primary and secondary prevention of cardiovascular disease across different levels of blood pressure: An individual participant-level data meta-analysis. Lancet 2021, 397, 1625–1636. [Google Scholar] [CrossRef]
- Razo, C.; Welgan, C.A.; Johnson, C.O.; McLaughlin, S.A.; Iannucci, V.; Rodgers, A.; Wang, N.; LeGrand, K.E.; Sorensen, R.J.D.; He, J.; et al. Effects of elevated systolic blood pressure on ischemic heart disease: A Burden of Proof study. Nat. Med. 2022, 28, 2056–2065. [Google Scholar] [CrossRef]
- Mancia, G.; Messerli, F.; Bakris, G.; Zhou, Q.; Champion, A.; Pepine, C.J. Blood pressure control and improved cardiovascular outcomes in the International Verapamil SR-Trandolapril Study. Hypertension 2007, 50, 299–305. [Google Scholar] [CrossRef] [Green Version]
- Doumas, M.; Tsioufis, C.; Fletcher, R.; Amdur, R.; Faselis, C.; Papademetriou, V. Time in Therapeutic Range, as a Determinant of All-Cause Mortality in Patients with Hypertension. J. Am. Heart Assoc. 2017, 6, e007131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, R.; Lin, Y.; Liu, M.; Xiong, Z.; Zhang, S.; Zhong, X.; Ye, X.; Huang, Y.; Zhuang, X.; Liao, X. Time in Target Range for Systolic Blood Pressure and Cardiovascular Outcomes in Patients with Heart Failure with Preserved Ejection Fraction. J. Am. Heart Assoc. 2022, 11, e022765. [Google Scholar] [CrossRef]
- Chen, K.; Li, C.; Cornelius, V.; Yu, D.; Wang, Q.; Shi, R.; Wu, Z.; Su, H.; Yan, J.; Chen, T.; et al. Prognostic Value of Time in Blood Pressure Target Range Among Patients with Heart Failure. JACC Heart Fail. 2022, 10, 369–379. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.J.; Kim, K.I. Blood Pressure Target in Type 2 Diabetes Mellitus. Diabetes Metab. J. 2022, 46, 667–674. [Google Scholar] [CrossRef]
- Burnier, M.; Damianaki, A. Hypertension as Cardiovascular Risk Factor in Chronic Kidney Disease. Circ. Res. 2023, 132, 1050–1063. [Google Scholar] [CrossRef]
- Te Riet, L.; van Esch, J.H.; Roks, A.J.; van den Meiracker, A.H.; Danser, A.H. Hypertension: Renin-angiotensin-aldosterone system alterations. Circ. Res. 2015, 116, 960–975. [Google Scholar] [CrossRef] [PubMed]
- Lund, L.H.; Claggett, B.; Liu, J.; Lam, C.S.; Jhund, P.S.; Rosano, G.M.; Swedberg, K.; Yusuf, S.; Granger, C.B.; Pfeffer, M.A.; et al. Heart failure with mid-range ejection fraction in CHARM: Characteristics, outcomes and effect of candesartan across the entire ejection fraction spectrum. Eur. J. Heart Fail. 2018, 20, 1230–1239. [Google Scholar] [CrossRef] [Green Version]
- Solomon, S.D.; Vaduganathan, M.; Claggett, B.L.; Packer, M.; Zile, M.; Swedberg, K.; Rouleau, J.; Pfeffer, M.A.; Desai, A.; Lund, L.H.; et al. Sacubitril/Valsartan Across the Spectrum of Ejection Fraction in Heart Failure. Circulation 2020, 141, 352–361. [Google Scholar] [CrossRef]
- Pfeffer, M.A.; Claggett, B.; Assmann, S.F.; Boineau, R.; Anand, I.S.; Clausell, N.; Desai, A.S.; Diaz, R.; Fleg, J.L.; Gordeev, I.; et al. Regional variation in patients and outcomes in the Treatment of Preserved Cardiac Function Heart Failure with an Aldosterone Antagonist (TOPCAT) trial. Circulation 2015, 131, 34–42. [Google Scholar] [CrossRef] [Green Version]
- Jackson, A.M.; Jhund, P.S.; Anand, I.S.; Dungen, H.D.; Lam, C.S.P.; Lefkowitz, M.P.; Linssen, G.; Lund, L.H.; Maggioni, A.P.; Pfeffer, M.A.; et al. Sacubitril-valsartan as a treatment for apparent resistant hypertension in patients with heart failure and preserved ejection fraction. Eur. Heart J. 2021, 42, 3741–3752. [Google Scholar] [CrossRef]
- Carey, R.M.; Calhoun, D.A.; Bakris, G.L.; Brook, R.D.; Daugherty, S.L.; Dennison-Himmelfarb, C.R.; Egan, B.M.; Flack, J.M.; Gidding, S.S.; Judd, E.; et al. Resistant Hypertension: Detection, Evaluation, and Management: A Scientific Statement From the American Heart Association. Hypertension 2018, 72, e53–e90. [Google Scholar] [CrossRef]
- Gregg, L.P.; Navaneethan, S.D. Steroidal or non-steroidal MRAs: Should we still enable RAASi use through K binders? Nephrol. Dial. Transplant. 2023, 38, 1355–1365. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, D.; Winocour, P.; Chowdhury, T.A.; De, P.; Wahba, M.; Montero, R.; Fogarty, D.; Frankel, A.H.; Karalliedde, J.; Mark, P.B.; et al. Management of hypertension and renin-angiotensin-aldosterone system blockade in adults with diabetic kidney disease: Association of British Clinical Diabetologists and the Renal Association UK guideline update 2021. BMC Nephrol. 2022, 23, 9. [Google Scholar] [CrossRef]
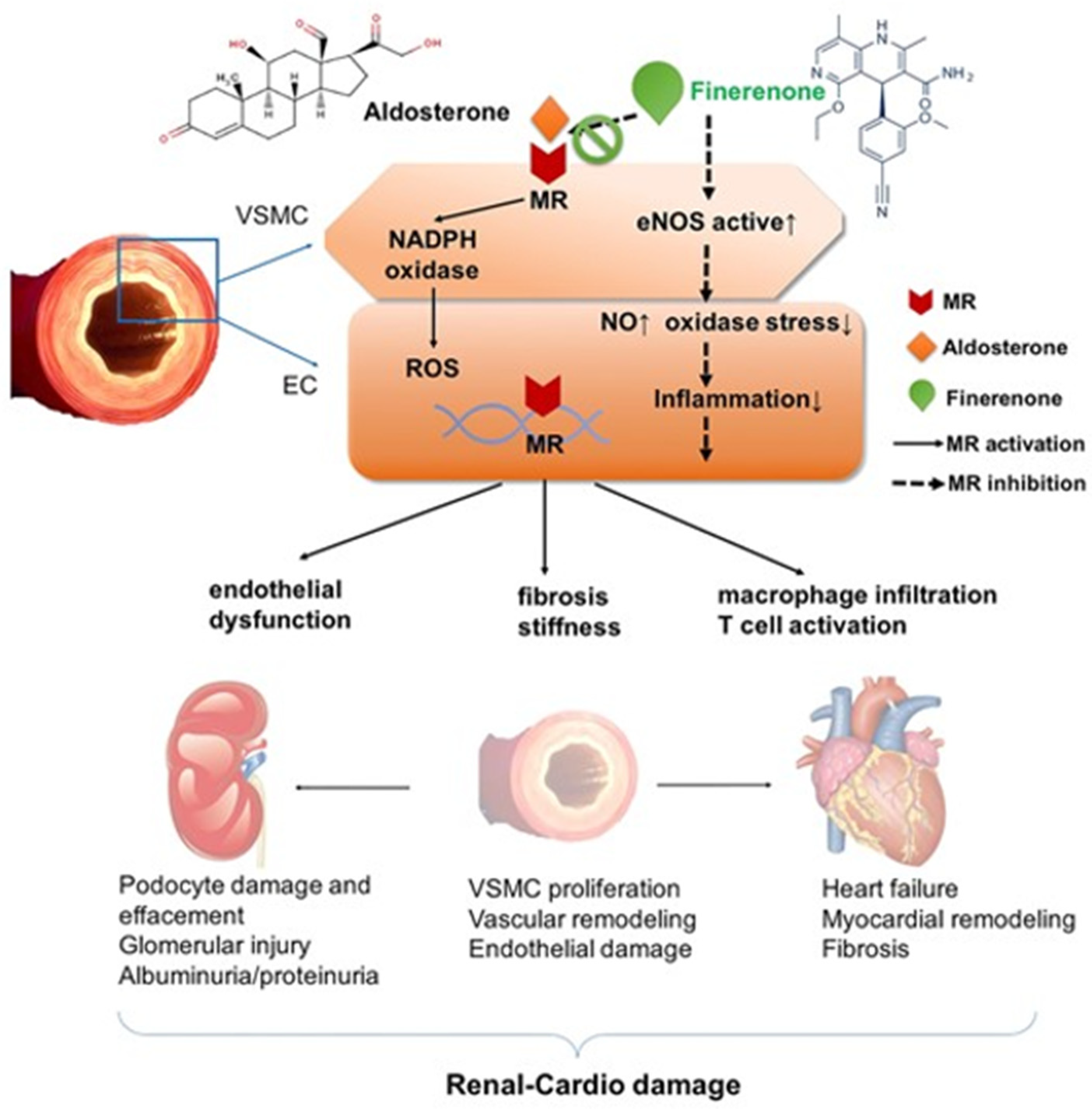
- Bakris, G.L.; Agarwal, R.; Anker, S.D.; Pitt, B.; Ruilope, L.M.; Rossing, P.; Kolkhof, P.; Nowack, C.; Schloemer, P.; Joseph, A.; et al. Effect of Finerenone on Chronic Kidney Disease Outcomes in Type 2 Diabetes. N. Engl. J. Med. 2020, 383, 2219–2229. [Google Scholar] [CrossRef] [PubMed]
- Pitt, B.; Filippatos, G.; Agarwal, R.; Anker, S.D.; Bakris, G.L.; Rossing, P.; Joseph, A.; Kolkhof, P.; Nowack, C.; Schloemer, P.; et al. Cardiovascular Events with Finerenone in Kidney Disease and Type 2 Diabetes. N. Engl. J. Med. 2021, 385, 2252–2263. [Google Scholar] [CrossRef]
- Ruolin, L.; Lili, X.; Lin, C.; Song, L.; Yangang, W.; Bingzi, D. Cardiovascular-renal protective effect and molecular mechanism of finerenone in type 2 diabetic mellitus. Front. Endocrinol. 2023, 14, 1125693. [Google Scholar] [CrossRef]
- Jia, G.; Sowers, J.R. Hypertension in Diabetes: An Update of Basic Mechanisms and Clinical Disease. Hypertension 2021, 78, 1197–1205. [Google Scholar] [CrossRef]
- Agarwal, R.; Ruilope, L.M.; Ruiz-Hurtado, G.; Haller, H.; Schmieder, R.E.; Anker, S.D.; Filippatos, G.; Pitt, B.; Rossing, P.; Lambelet, M.; et al. Effect of finerenone on ambulatory blood pressure in chronic kidney disease in type 2 diabetes. J. Hypertens. 2023, 41, 295–302. [Google Scholar] [CrossRef]
- Wojcik, C.; Warden, B.A. Mechanisms and Evidence for Heart Failure Benefits from SGLT2 Inhibitors. Curr. Cardiol. Rep. 2019, 21, 130. [Google Scholar] [CrossRef]
- Gronda, E.; Vanoli, E.; Iacoviello, M.; Caldarola, P.; Gabrielli, D.; Tavazzi, L. The Benefit of Sodium-Glucose Co-Transporter Inhibition in Heart Failure: The Role of the Kidney. Int. J. Mol. Sci. 2022, 23, 11987. [Google Scholar] [CrossRef]
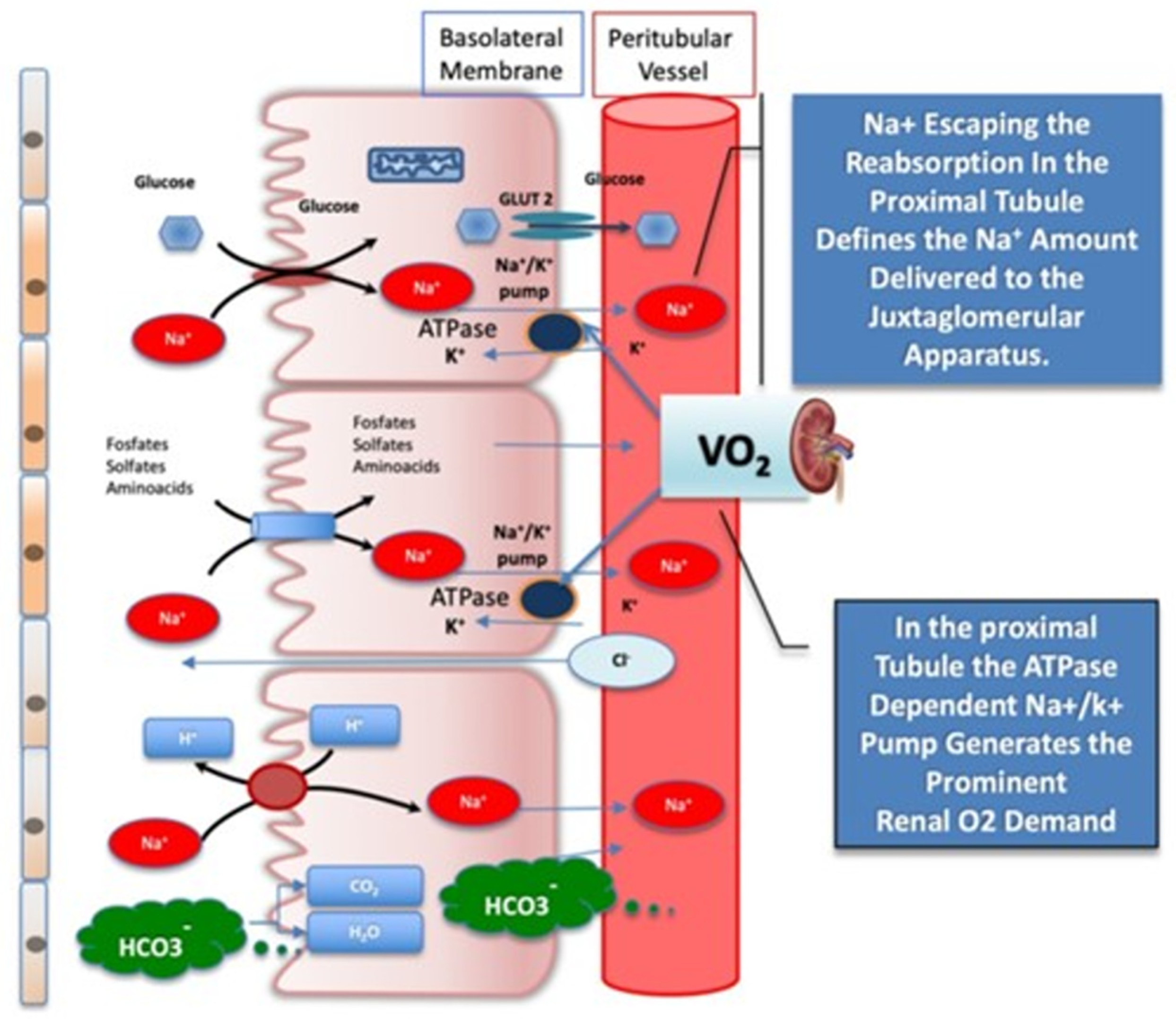
- Bjornstad, P.; Greasley, P.J.; Wheeler, D.C.; Chertow, G.M.; Langkilde, A.M.; Heerspink, H.J.L.; Van Raalte, D.H. The Potential Roles of Osmotic and Nonosmotic Sodium Handling in Mediating the Effects of Sodium-Glucose Cotransporter 2 Inhibitors on Heart Failure. J. Card. Fail. 2021, 27, 1447–1455. [Google Scholar] [CrossRef] [PubMed]
- Verma, S.; McMurray, J.J.V. SGLT2 inhibitors and mechanisms of cardiovascular benefit: A state-of-the-art review. Diabetologia 2018, 61, 2108–2117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zelniker, T.A.; Braunwald, E. Mechanisms of Cardiorenal Effects of Sodium-Glucose Cotransporter 2 Inhibitors: JACC State-of-the-Art Review. J. Am. Coll. Cardiol. 2020, 75, 422–434. [Google Scholar] [CrossRef] [PubMed]
- Varadhan, A.; Stephan, K.; Gupta, R.; Vyas, A.V.; Ranchal, P.; Aronow, W.S.; Hawwa, N.; Lanier, G.M. Growing role of SGLT2i in heart failure: Evidence from clinical trials. Expert. Rev. Clin. Pharmacol. 2022, 15, 147–159. [Google Scholar] [CrossRef]
- Gupta, R.; Maitz, T.; Egeler, D.; Mehta, A.; Nyaeme, M.; Hajra, A.; Goel, A.; Sreenivasan, J.; Patel, N.; Aronow, W.S. SGLT2 inhibitors in hypertension: Role beyond diabetes and heart failure. Trends Cardiovasc. Med. 2022, in press. [Google Scholar] [CrossRef]
- Kario, K.; Ferdinand, K.C.; O’Keefe, J.H. Control of 24-hour blood pressure with SGLT2 inhibitors to prevent cardiovascular disease. Prog. Cardiovasc. Dis. 2020, 63, 249–262. [Google Scholar] [CrossRef]
- Kario, K.; Ferdinand, K.C.; Vongpatanasin, W. Are SGLT2 Inhibitors New Hypertension Drugs? Circulation 2021, 143, 1750–1753. [Google Scholar] [CrossRef]
- Goldberg, L.R. The Pleiotropic Effects of SGLT2 Inhibitors: Remodeling the Treatment of Heart Failure. J. Am. Coll. Cardiol. 2021, 77, 256–258. [Google Scholar] [CrossRef]
- Zannad, F.; Ferreira, J.P.; Pocock, S.J.; Anker, S.D.; Butler, J.; Filippatos, G.; Brueckmann, M.; Ofstad, A.P.; Pfarr, E.; Jamal, W.; et al. SGLT2 inhibitors in patients with heart failure with reduced ejection fraction: A meta-analysis of the EMPEROR-Reduced and DAPA-HF trials. Lancet 2020, 396, 819–829. [Google Scholar] [CrossRef]
- Salazar, R.A.; Stroud, S.C.; DeFilippis, E.M. A Sweet Solution for Heart Failure with Preserved Ejection Fraction: The Role of Sodium-Glucose Cotransporter-2 Inhibitors. Circ. Heart Fail. 2023, 16, e010283. [Google Scholar] [CrossRef]
- Li, M.; Yi, T.; Fan, F.; Qiu, L.; Wang, Z.; Weng, H.; Ma, W.; Zhang, Y.; Huo, Y. Effect of sodium-glucose cotransporter-2 inhibitors on blood pressure in patients with heart failure: A systematic review and meta-analysis. Cardiovasc. Diabetol. 2022, 21, 139. [Google Scholar] [CrossRef] [PubMed]
- Packer, M. How can sodium-glucose cotransporter 2 inhibitors stimulate erythrocytosis in patients who are iron-deficient? Implications for understanding iron homeostasis in heart failure. Eur. J. Heart Fail. 2022, 24, 2287–2296. [Google Scholar] [CrossRef]
- Mancia, G.; Kjeldsen, S.E.; Kreutz, R.; Pathak, A.; Grassi, G.; Esler, M. Individualized Beta-Blocker Treatment for High Blood Pressure Dictated by Medical Comorbidities: Indications Beyond the 2018 European Society of Cardiology/European Society of Hypertension Guidelines. Hypertension 2022, 79, 1153–1166. [Google Scholar] [CrossRef] [PubMed]
- Masarone, D.; Martucci, M.L.; Errigo, V.; Pacileo, G. The Use of beta-Blockers in Heart Failure with Reduced Ejection Fraction. J. Cardiovasc. Dev. Dis. 2021, 8, 101. [Google Scholar] [CrossRef] [PubMed]
- Cleland, J.G.F.; Bunting, K.V.; Flather, M.D.; Altman, D.G.; Holmes, J.; Coats, A.J.S.; Manzano, L.; McMurray, J.J.V.; Ruschitzka, F.; van Veldhuisen, D.J.; et al. Beta-blockers for heart failure with reduced, mid-range, and preserved ejection fraction: An individual patient-level analysis of double-blind randomized trials. Eur. Heart J. 2018, 39, 26–35. [Google Scholar] [CrossRef] [Green Version]
- Hogg, K.; McMurray, J. Neurohumoral pathways in heart failure with preserved systolic function. Prog. Cardiovasc. Dis. 2005, 47, 357–366. [Google Scholar] [CrossRef]
- Meyer, M.; LeWinter, M.M. Heart Rate and Heart Failure with Preserved Ejection Fraction: Time to Slow beta-Blocker Use? Circ. Heart Fail. 2019, 12, e006213. [Google Scholar] [CrossRef]
- Triposkiadis, F.; Xanthopoulos, A.; Starling, R.C. Medical Treatment of Heart Failure: Ignore the Ejection Fraction and Treat All? J. Card. Fail. 2021, 27, 907–909. [Google Scholar] [CrossRef]
- Mullens, W.; Damman, K.; Harjola, V.P.; Mebazaa, A.; Brunner-La Rocca, H.P.; Martens, P.; Testani, J.M.; Tang, W.H.W.; Orso, F.; Rossignol, P.; et al. The use of diuretics in heart failure with congestion—A position statement from the Heart Failure Association of the European Society of Cardiology. Eur. J. Heart Fail. 2019, 21, 137–155. [Google Scholar] [CrossRef] [Green Version]
- Wilcox, C.S.; Testani, J.M.; Pitt, B. Pathophysiology of Diuretic Resistance and Its Implications for the Management of Chronic Heart Failure. Hypertension 2020, 76, 1045–1054. [Google Scholar] [CrossRef]
- Shah, S.U.; Anjum, S.; Littler, W.A. Use of diuretics in cardiovascular diseases: (1) heart failure. Postgrad. Med. J. 2004, 80, 201–205. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weir, M.R.; Zappe, D.; Orloski, L.A.; Sowers, J.R. How early should blood pressure control be achieved for optimal cardiovascular outcomes? J. Hum. Hypertens. 2011, 25, 211–217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Agarwal, R.; Joseph, A.; Anker, S.D.; Filippatos, G.; Rossing, P.; Ruilope, L.M.; Pitt, B.; Kolkhof, P.; Scott, C.; Lawatscheck, R.; et al. Hyperkalemia Risk with Finerenone: Results from the FIDELIO-DKD Trial. J. Am. Soc. Nephrol. 2022, 33, 225–237. [Google Scholar] [CrossRef]
- Paczkowska-Walendowska, M.; Sip, S.; Staszewski, R.; Cielecka-Piontek, J. Single-Pill Combination to Improve Hypertension Treatment: Pharmaceutical Industry Development. Int. J. Environ. Res. Public. Health 2022, 19, 4156. [Google Scholar] [CrossRef] [PubMed]
- Perrone, V.; Veronesi, C.; Gambera, M.; Nati, G.; Perone, F.; Tagliabue, P.F.; Degli Esposti, L.; Volpe, M. Treatment with Free Triple Combination Therapy of Atorvastatin, Perindopril, Amlodipine in Hypertensive Patients: A Real-World Population Study in Italy. High. Blood Press. Cardiovasc. Prev. 2019, 26, 399–404. [Google Scholar] [CrossRef]
- Gaciong, Z. Preference and Adherence to a Fixed-Dose Combination of Bisoprolol-Aspirin and Blood Pressure Control: Results of an Open-Label, Multicentre Study. J. Clin. Med. 2022, 12, 17. [Google Scholar] [CrossRef]
- Taylor, J.L.; Myers, J.; Bonikowske, A.R. Practical guidelines for exercise prescription in patients with chronic heart failure. Heart Fail. Rev. 2023. [Google Scholar] [CrossRef]
- Crisci, G.; De Luca, M.; D’Assante, R.; Ranieri, B.; D’Agostino, A.; Valente, V.; Giardino, F.; Capone, V.; Chianese, S.; Rega, S.; et al. Effects of Exercise on Heart Failure with Preserved Ejection Fraction: An Updated Review of Literature. J. Cardiovasc. Dev. Dis. 2022, 9, 241. [Google Scholar] [CrossRef]
- Olsen, L.N.; Fischer, M.; Evans, P.A.; Gliemann, L.; Hellsten, Y. Does Exercise Influence the Susceptibility to Arterial Thrombosis? An Integrative Perspective. Front. Physiol. 2021, 12, 636027. [Google Scholar] [CrossRef]
- Daniela, M.; Catalina, L.; Ilie, O.; Paula, M.; Daniel-Andrei, I.; Ioana, B. Effects of Exercise Training on the Autonomic Nervous System with a Focus on Anti-Inflammatory and Antioxidants Effects. Antioxidants 2022, 11, 350. [Google Scholar] [CrossRef]
- Boulmpou, A.; Theodorakopoulou, M.P.; Boutou, A.K.; Alexandrou, M.E.; Papadopoulos, C.E.; Bakaloudi, D.R.; Pella, E.; Sarafidis, P.; Vassilikos, V. Effects of different exercise programs on the cardiorespiratory reserve in HFpEF patients: A systematic review and meta-analysis. Hell. J. Cardiol. 2022, 64, 58–66. [Google Scholar] [CrossRef]
- Lopes, S.; Afreixo, V.; Teixeira, M.; Garcia, C.; Leitao, C.; Gouveia, M.; Figueiredo, D.; Alves, A.J.; Polonia, J.; Oliveira, J.; et al. Exercise training reduces arterial stiffness in adults with hypertension: A systematic review and meta-analysis. J. Hypertens. 2021, 39, 214–222. [Google Scholar] [CrossRef]
- Rahman, M.M.; Islam, F.; Or-Rashid, M.H.; Mamun, A.A.; Rahaman, M.S.; Islam, M.M.; Meem, A.F.K.; Sutradhar, P.R.; Mitra, S.; Mimi, A.A.; et al. The Gut Microbiota (Microbiome) in Cardiovascular Disease and Its Therapeutic Regulation. Front. Cell Infect. Microbiol. 2022, 12, 903570. [Google Scholar] [CrossRef] [PubMed]
- Yuan, L.; Li, Y.; Chen, M.; Xue, L.; Wang, J.; Ding, Y.; Gu, Q.; Zhang, J.; Yang, R.; Zhao, H.; et al. Effects of probiotics on hypertension. Appl. Microbiol. Biotechnol. 2023, 107, 1107–1117. [Google Scholar] [CrossRef] [PubMed]
- Zarezadeh, M.; Musazadeh, V.; Ghalichi, F.; Kavyani, Z.; Nasernia, R.; Parang, M.; Jamilian, P.; Jamilian, P.; Fakhr, L.; Ostadrahimi, A.; et al. Effects of probiotics supplementation on blood pressure: An umbrella meta-analysis of randomized controlled trials. Nutr. Metab. Cardiovasc. Dis. 2023, 33, 275–286. [Google Scholar] [CrossRef]
- Nagai, M.; Dote, K.; Forster, C.Y. Denervation or stimulation? Role of sympatho-vagal imbalance in HFpEF with hypertension. Hypertens. Res. 2023, 46, 1727–1737. [Google Scholar] [CrossRef] [PubMed]
- Barbato, E.; Azizi, M.; Schmieder, R.E.; Lauder, L.; Bohm, M.; Brouwers, S.; Bruno, R.M.; Dudek, D.; Kahan, T.; Kandzari, D.E.; et al. Renal denervation in the management of hypertension in adults. A clinical consensus statement of the ESC Council on Hypertension and the European Association of Percutaneous Cardiovascular Interventions (EAPCI). Eur. Heart J. 2023, 44, 1313–1330. [Google Scholar] [CrossRef]
- Pavlov, V.A.; Tracey, K.J. The vagus nerve and the inflammatory reflex—Linking immunity and metabolism. Nat. Rev. Endocrinol. 2012, 8, 743–754. [Google Scholar] [CrossRef]
- Stavrakis, S.; Elkholey, K.; Morris, L.; Niewiadomska, M.; Asad, Z.U.A.; Humphrey, M.B. Neuromodulation of Inflammation to Treat Heart Failure with Preserved Ejection Fraction: A Pilot Randomized Clinical Trial. J. Am. Heart Assoc. 2022, 11, e023582. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Systolic BP | Patient A | Patient B | Patient C |
|---|---|---|---|
| Measurement 1 | 135 | 125 | 115 |
| Measurement 2 | 125 | 125 | 120 |
| Measurement 3 | 135 | 120 | 125 |
| Measurement 4 | 140 | 120 | 120 |
| Measurement 5 | 100 | 140 | 125 |
| Measurement 6 | 100 | 100 | 135 |
| Measurement 7 | 125 | 125 | 115 |
| Measurement 8 | 135 | 115 | 125 |
| Measurement 9 | 135 | 140 | 135 |
| Measurement 10 | 105 | 100 | 120 |
| Average BP (mmHg) | 123.5 | 121 | 123. 5 |
| Time to target (%) | 30% (3/10) | 60% (6/10) | 80% (8/10) |
| Congestion | Blood Pressure Lowering and Cardioprotection | |
|---|---|---|
bumetanide) |
| |
| Blood Pressure Targets Systolic BP 110–130 mmHg Time in therapeutic range > 75 % | Comments: 1. Additional use of thiazide diuretics or carbonic anhydrase inhibitors in cases with diuretic resistance 2. Thiazide diuretics may be considered in decongested patients instead of loop diuretics for blood pressure control. | Comments: 1. Besides effectively lowering BP, all the aforementioned classes of antihypertensives are cardioprotective. 2. Vasodilating beta-blockers with a favorable metabolic profile (e.g, carvedilol, nebivolol) may be preferable. 3. Beta-blockers are first-line agents in eccentric hypertrophy. 4. B-blockers should be used in selected patients with concentric hypertrophy (e.g., atrial fibrillation, angina, resistant hypertension). 5. The additional use of dihydropyridine calcium channel blockers should be considered when BP cannot be otherwise controlled. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Triposkiadis, F.; Sarafidis, P.; Briasoulis, A.; Magouliotis, D.E.; Athanasiou, T.; Skoularigis, J.; Xanthopoulos, A. Hypertensive Heart Failure. J. Clin. Med. 2023, 12, 5090. https://0-doi-org.brum.beds.ac.uk/10.3390/jcm12155090
Triposkiadis F, Sarafidis P, Briasoulis A, Magouliotis DE, Athanasiou T, Skoularigis J, Xanthopoulos A. Hypertensive Heart Failure. Journal of Clinical Medicine. 2023; 12(15):5090. https://0-doi-org.brum.beds.ac.uk/10.3390/jcm12155090
Chicago/Turabian StyleTriposkiadis, Filippos, Pantelis Sarafidis, Alexandros Briasoulis, Dimitrios E. Magouliotis, Thanos Athanasiou, John Skoularigis, and Andrew Xanthopoulos. 2023. "Hypertensive Heart Failure" Journal of Clinical Medicine 12, no. 15: 5090. https://0-doi-org.brum.beds.ac.uk/10.3390/jcm12155090