Dystrophin Cardiomyopathies: Clinical Management, Molecular Pathogenesis and Evolution towards Precision Medicine


 , ,
, ,  ,
,
Abstract
:1. Introduction
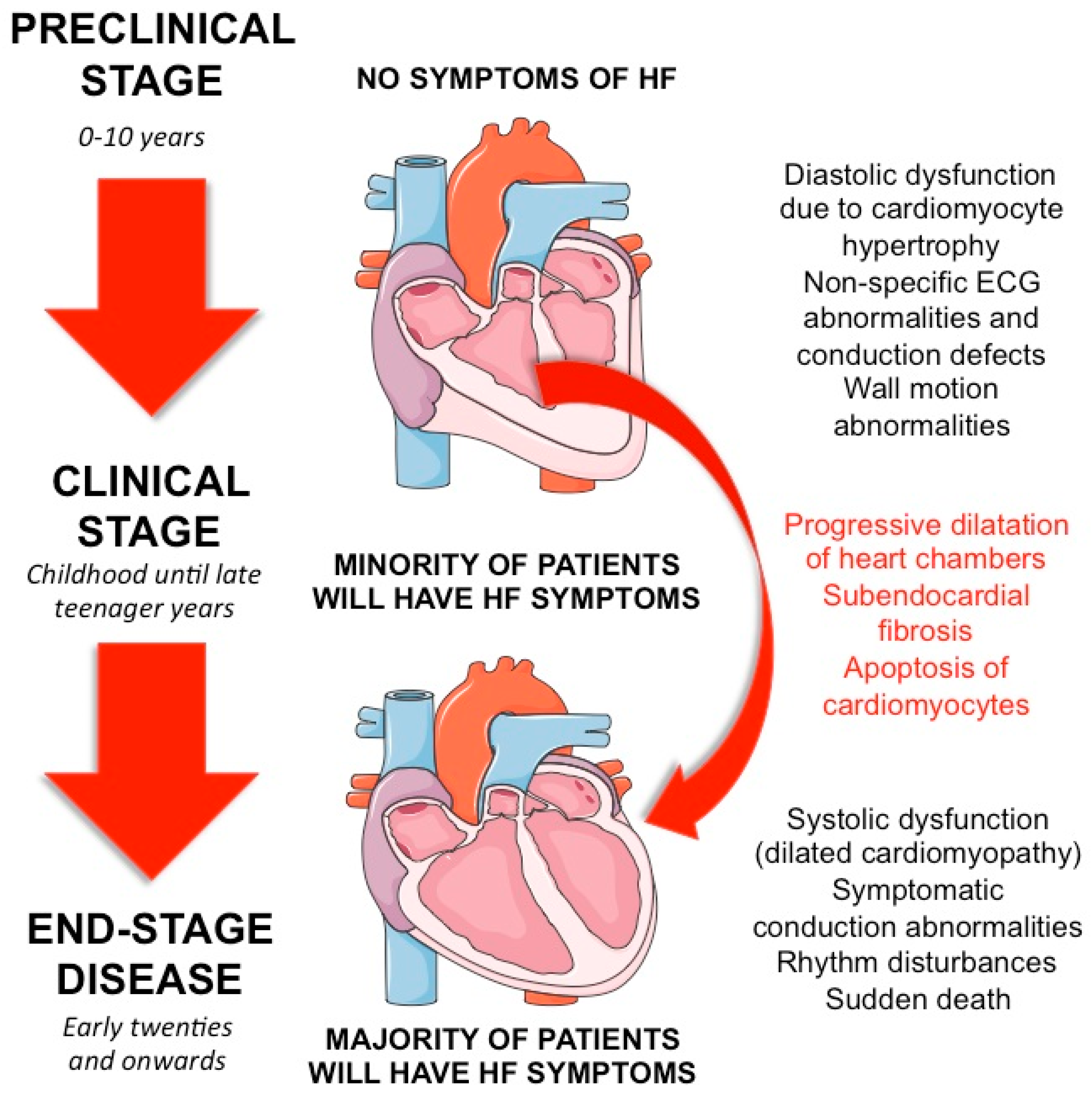
2. Clinical Aspects
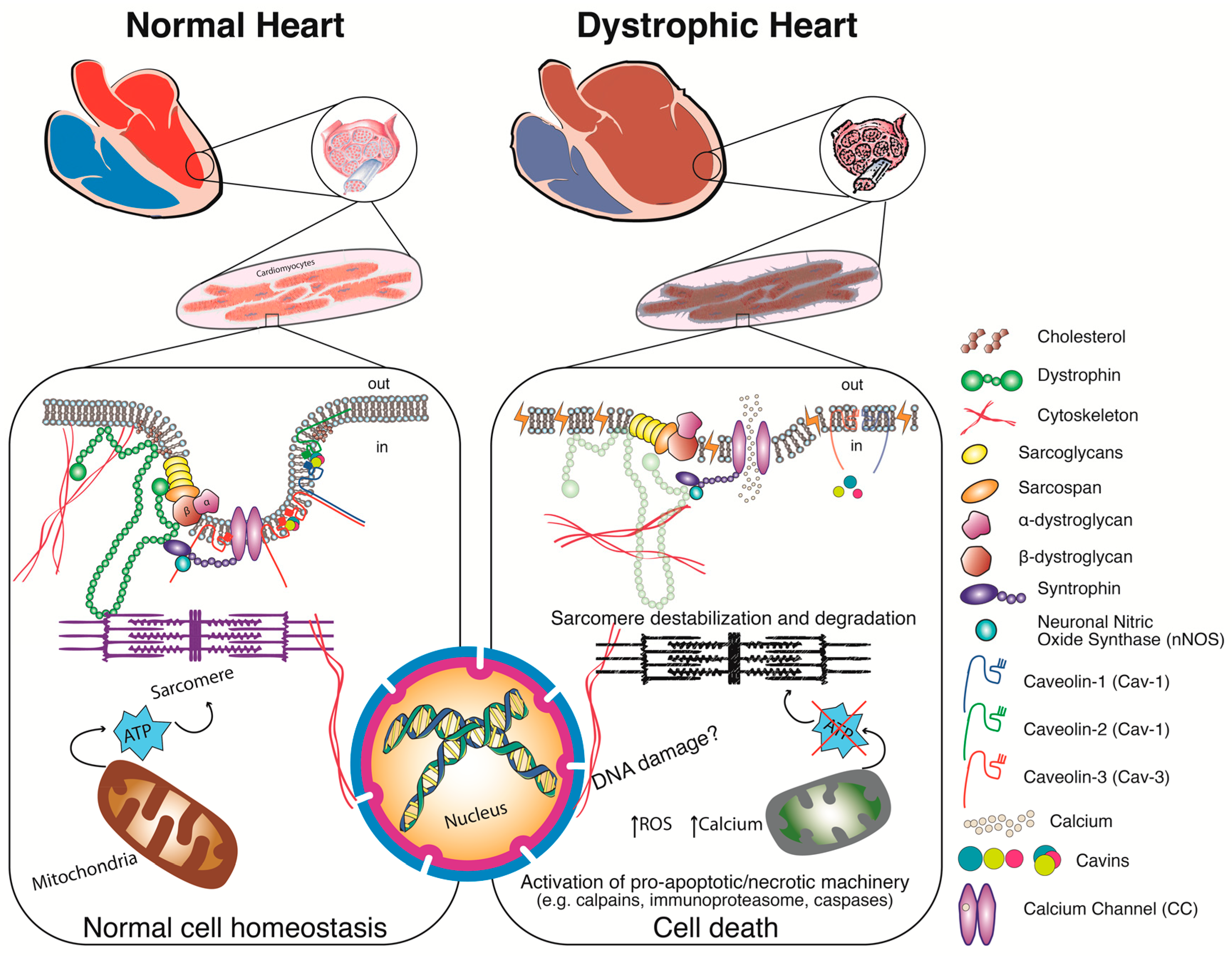
3. Dystrophin—From Gene to Protein
3.1. The Dystrophin Gene and Its Transcript
3.2. The Dystrophin Protein
3.3. DMD Mutations
4. Pathophysiology—Alternatives to the Structural Hypothesis
5. Current Pharmacotherapy
5.1. Corticosteroids
5.2. ACE Inhibitors
5.3. Beta-Blockers
5.4. Angiotensin-II-Receptor Blockers
5.5. Mineralocorticoid-Receptor Antagonist
6. Benefits of Future Therapies
6.1. Utrophin Upregulation
6.2. Stop Codon Read-Through Therapy
6.3. Viral Gene Therapy
6.4. Cell-Based Therapy
6.5. Antisense Oligonucleotides (AONs)
6.6. Possible Other Treatments and Drug Repositioning
7. Radical or Conservative Disease Management
8. Disease Modelling
8.1. Cell Source
8.2. Environment Matters
9. Future Perspectives
Author Contributions
Funding
Conflicts of Interest
References
- Finsterer, J.; Cripe, L. Treatment of dystrophin cardiomyopathies. Nat. Rev. Cardiol. 2014, 11, 168–179. [Google Scholar] [CrossRef] [PubMed]
- Shirokova, N.; Niggli, E. Cardiac phenotype of Duchenne muscular dystrophy: Insights from cellular studies. J. Mol. Cell. Cardiol. 2013, 58, 217–224. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Flanigan, K.M. The muscular dystrophies. Semin. Neurol. 2012, 32, 255–263. [Google Scholar] [CrossRef] [PubMed]
- Muntoni, F.; Torelli, S.; Ferlini, A. Dystrophin and mutations: One gene, several proteins, multiple phenotypes. Lancet Neurol. 2003, 2, 731–740. [Google Scholar] [CrossRef]
- D’Amario, D.; Amodeo, A.; Adorisio, R.; Tiziano, F.D.; Leone, A.M.; Perri, G.; Bruno, P.; Massetti, M.; Ferlini, A.; Pane, M.; et al. A current approach to heart failure in Duchenne muscular dystrophy. BMJ Heart 2017, 103, 1770–1779. [Google Scholar] [CrossRef] [PubMed]
- Verhaert, D.; Richards, K.; Rafael-Fortney, J.A.; Raman, S.V. Cardiac involvement in patients with muscular dystrophies: Magnetic resonance imaging phenotype and genotypic considerations. Circ. Cardiovasc. Imaging 2011, 4, 67–76. [Google Scholar] [CrossRef] [PubMed]
- Melacini, P.; Fanin, M.; Danieli, G.A.; Villanova, C.; Martinello, F.; Miorin, M.; Freda, M.P.; Miorelli, M.; Mostacciuolo, M.L.; Fasoli, G.; et al. Myocardial involvement is very frequent among patients affected with subclinical Becker’s muscular dystrophy. Circulation 1996, 94, 3168–3175. [Google Scholar] [CrossRef] [PubMed]
- Adachi, K.; Hashiguchi, S.; Saito, M.; Kashiwagi, S.; Miyazaki, T.; Kawai, H.; Yamada, H.; Iwase, T.; Akaike, M.; Takao, S.; et al. Detection and management of cardiomyopathy in female dystrophinopathy carriers. J. Neurol. Sci. 2018, 386, 74–80. [Google Scholar] [CrossRef] [PubMed]
- McNally, E.M.; Kaltman, J.R.; Benson, D.W.; Canter, C.E.; Cripe, L.H.; Duan, D.; Finder, J.D.; Groh, W.J.; Hoffman, E.P.; Judge, D.P.; et al. Contemporary cardiac issues in Duchenne muscular dystrophy. Circulation 2015, 131, 1590–1598. [Google Scholar] [CrossRef] [PubMed]
- Kamakura, K. Cardiac involvement of female carrier of Duchenne muscular dystrophy. Intern. Med. 2000, 39, 2–3. [Google Scholar] [CrossRef] [PubMed]
- Connuck, D.M.; Sleeper, L.A.; Colan, S.D.; Cox, G.F.; Towbin, J.A.; Lowe, A.M.; Wilkinson, J.D.; Orav, E.J.; Cuniberti, L.; Salbert, B.A.; et al. Characteristics and outcomes of cardiomyopathy in children with Duchenne or Becker muscular dystrophy: A comparative study from the pediatric cardiomyopathy registry. Am. Heart J. 2008, 155, 998–1005. [Google Scholar] [CrossRef] [PubMed]
- Romfh, A.; McNally, E.M. Cardiac assessment in duchenne and becker muscular dystrophies. Curr. Heart Fail. Rep. 2010, 7, 212–218. [Google Scholar] [CrossRef] [PubMed]
- Spurney, C.; Shimizu, R.; Morgenroth, L.P.; Kolski, H.; Gordish-Dressman, H.; Clemens, P.R.; CINRG Investigators. Cooperative International Neuromuscular Research Group Duchenne Natural History Study demonstrates insufficient diagnosis and treatment of cardiomyopathy in Duchenne muscular dystrophy. Muscle Nerv. 2014, 50, 250–256. [Google Scholar] [CrossRef] [PubMed]
- Kunkel, L.M.; Monaco, A.P.; Bertelson, C.J.; Colletti, C.A. Molecular genetics of Duchenne muscular dystrophy. Cold Spring Harb. Symp. Quant. Biol. 1986, 51, 349–351. [Google Scholar] [CrossRef] [PubMed]
- Monaco, A.P.; Neve, R.L.; Colletti-Feener, C.; Bertelson, C.J.; Kurnit, D.M.; Kunkel, L.M. Isolation of candidate cDNAs for portions of the Duchenne muscular dystrophy gene. Nature 1986, 323, 646–650. [Google Scholar] [CrossRef] [PubMed]
- Kunkel, L.M.; Hoffman, E.P. Duchenne/Becker muscular dystrophy: A short overview of the gene, the protein, and current diagnostics. Br. Med. Bull. 1989, 45, 63043. [Google Scholar] [CrossRef]
- Mandel, J.L. The gene and its product. Nature 1989, 339, 584–586. [Google Scholar] [CrossRef] [PubMed]
- Manole, E. The dystrophin gene and its product—A view. Rom. J. Neurol. Psychiatry 1995, 33, 109–119. [Google Scholar] [PubMed]
- Nudel, U.; Zuk, D.; Einat, P.; Zeelon, E.; Levy, Z.; Neuman, S.; Yaffe, D. Duchenne muscular dystrophy gene product is not identical in muscle and brain. Nature 1989, 337, 76–78. [Google Scholar] [CrossRef] [PubMed]
- Górecki, D.C.; Monaco, A.P.; Derry, J.M.; Walker, A.P.; Barnard, E.A.; Barnard, P.J. Expression of four alternative dystrophin transcripts in brain regions regulated by different promoters. Hum. Mol. Genet. 1992, 1, 505–510. [Google Scholar] [CrossRef] [PubMed]
- Bies, R.D.; Friedman, D.; Roberts, R.; Perryman, M.B.; Caskey, C.T. Expression and localization of dystrophin in human cardiac Purkinje fibres. Circulation 1992, 86, 147–153. [Google Scholar] [CrossRef] [PubMed]
- Bies, R.D.; Phelps, S.F.; Cortez, M.D.; Roberts, R.; Caskey, C.T.; Chamberlain, J.S. Human and murine dystrophin mRNA transcripts are differentially expressed during skeletal muscle, heart, and brain development. Nucleic Acids Res. 1992, 20, 1725–1731. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muntoni, F.; Melis, M.A.; Ganau, A.; Dubowitz, V. Transcription of the dystrophin gene in normal tissues and in skeletal muscle of a family with X-linked dilated cardiomyopathy. Am. J. Hum. Genet. 1995, 56, 151–157. [Google Scholar] [PubMed]
- Yaffe, D.; Makover, A.; Lederfein, D.; Rapaport, D.; Bar, S.; Barnea, E.; Nudel, U. Multiple products of the Duchenne muscular dystrophy gene. Symp. Soc. Exp. Biol. 1992, 46, 179–188. [Google Scholar] [PubMed]
- Ferlini, A.; Neri, M.; Gualandi, F. The medical genetics of dystrophinopathies: Molecular genetic diagnosis and its impact on clinical practice. Neuromuscul. Disord. 2013, 23, 4–14. [Google Scholar] [CrossRef] [PubMed]
- Blake, D.J.; Weir, A.; Newey, S.E.; Davies, K.E. Function and genetics of dystrophin and dystrophin-related proteins in muscle. Physiol. Rev. 2002, 82, 291–329. [Google Scholar] [CrossRef] [PubMed]
- Gao, Q.; McNally, E.M. The dystrophin complex: Structure, function and implications for therapy. Compr. Physiol. 2015, 5, 1223–1239. [Google Scholar] [CrossRef] [PubMed]
- Henderson, D.M.; Lin, A.Y.; Thomas, D.D.; Ervasti, J.M. The carboxy-terminal third of dystrophin enhances actin binding activity. J. Mol. Biol. 2012, 416, 414–424. [Google Scholar] [CrossRef] [PubMed]
- Koenig, M.; Monaco, A.P.; Kunkel, L.M. The complete sequence of dystrophin predicts a rod-shaped cytoskeletal protein. Cell 1988, 53, 219–228. [Google Scholar] [CrossRef]
- Sadoulet-Puccio, H.M.; Rajalaa, M.; Kunkel, L.M. Dystrobrevin and dystrophin: An interaction through coiled-coil motif. Proc. Natl. Acad. Sci. USA 1997, 94, 12413–12418. [Google Scholar] [CrossRef] [PubMed]
- Koenig, M.; Kunkel, L.M. Detailed analysis of the repeat domain of dystrophin reveals four potential hinge segments that may confer flexibility. J. Biol. Chem. 1990, 265, 4560–4566. [Google Scholar] [PubMed]
- Amann, K.J.; Renley, B.A.; Ervasti, J.M. A cluster of basic repeats in the dystrophin rod domain binds F-actin through an electrostatic interaction. J. Biol. Chem. 1998, 273, 28419–28423. [Google Scholar] [CrossRef] [PubMed]
- Legardinier, S.; Raguénès-Nicol, C.; Tascon, C.; Rocher, C.; Hardy, S.; Hubert, J.F.; Le Rumeur, E. Mapping of the lipid-binding and stability properties of the central rod domain of human dystrophin. J. Mol. Biol. 2009, 389, 546–558. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adams, M.E.; Mueller, H.A.; Froehner, S.C. In vivo requirement of the alpha-syntrophin PDZ domain for the sarcolemmal localization of nNOS and aquaporin-4. J. Cell Biol. 2001, 155, 113–122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brenman, J.E.; Chao, D.S.; Gee, S.H.; McGee, A.W.; Craven, S.E.; Santillano, D.R.; Wu, Z.; Huang, F.; Xia, H.; Peters, M.F.; et al. Interaction of nitric oxide synthase with the postsynaptic density protein PSD-95 and alpha1-syntrophin mediated by PDZ domains. Cell 1996, 84, 757–767. [Google Scholar] [CrossRef]
- Bork, P.; Sudol, M. The WW domain: A signalling site in dystrophin? Trends Biochem. Sci. 1994, 19, 531–533. [Google Scholar] [CrossRef]
- Ponting, C.P.; Blake, D.J.; Davies, K.E.; Kendrick-Jones, J.; Winder, S.J. ZZ and TAZ: New putative zinc fingers in dystrophin and other proteins. Trends Biochem. Sci. 1996, 21, 11–13. [Google Scholar] [CrossRef]
- Jung, D.; Yang, B.; Meyer, J.; Chamberlain, J.S.; Campbell, K.P. Identification and characterization of the dystrophin anchoring site on beta-dystroglycan. J. Biol. Chem. 1995, 270, 27305–27310. [Google Scholar] [CrossRef] [PubMed]
- Blake, D.J.; Tinsley, J.M.; Davies, K.E.; Knight, A.E.; Winder, S.J.; Kendrick-Jones, J. Coiled-coil regions in the carboxy-terminal domains of dystrophin and related proteins: Potentials for protein-protein interactions. Trends Biochem. Sci. 1995, 20, 133–135. [Google Scholar] [CrossRef]
- Ervasti, J.M.; Campbell, K.P. Membrane organization of the dystrophin-glycoprotein complex. Cell 1991, 66, 1121–1131. [Google Scholar] [CrossRef]
- Yoshida, M.; Suzuki, A.; Yamamoto, H.; Noguchi, S.; Mizuno, Y.; Ozawa, E. Dissociation of the complex of dystrophin and its associated proteins into several unique groups by n-octyl beta-D-glucoside. Eur. J. Biochem. 1994, 222, 1055–1061. [Google Scholar] [CrossRef] [PubMed]
- Pillers, D.A.; Bulman, D.E.; Weleber, R.G.; Sigesmund, D.A.; Musarella, M.A.; Powell, B.R.; Murphey, W.H.; Westall, C.; Panton, C.; Becker, L.E.; et al. Dystrophin expression in the human retina is required for normal function as defined by electroretinography. Nat. Genet. 1993, 4, 82–86. [Google Scholar] [CrossRef] [PubMed]
- Ricotti, V.; Jagle, H.; Theodorou, M.; Moore, A.T.; Muntoni, F.; Thompson, D.A. Ocular and neurodevelopmental features of Duchenne muscular dystrophy: A signature of dystrophin function in the central nervous system. Eur. J. Hum. Genet. EJHG 2016, 24, 562–568. [Google Scholar] [CrossRef] [PubMed]
- D’Souza, V.N.; Nguyen, T.M.; Morris, G.E.; Karges, W.; Pillers, D.A.; Ray, P.N. A novel dystrophin isoform is required for normal retinal electrophysiology. Hum. Mol. Genet. 1995, 4, 837–842. [Google Scholar] [CrossRef] [PubMed]
- Lidov, H.G.; Selig, S.; Kunkel, L.M. Dp140: A novel 140 kDa CNS transcript from the dystrophin locus. Hum. Mol. Genet. 1995, 4, 329–335. [Google Scholar] [CrossRef] [PubMed]
- Doorenweerd, N.; Straathof, C.S.; Dumas, E.M.; Spitali, P.; Ginjaar, I.B.; Wokke, B.H.; Schrans, D.G.; van den Bergen, J.C.; van Zwet, E.W.; Webb, A.; et al. Reduced cerebral gray matter and altered white matter in boys with Duchenne muscular dystrophy. Ann. Neurol. 2014, 76, 403–411. [Google Scholar] [CrossRef] [PubMed]
- Doorenweerd, N.; Mahfouz, A.; van Putten, M.; Kaliyaperumal, R.; PAC, T.H.; Hendriksen, J.G.M.; Aartsma-Rus, A.M.; Verschuuren, J.; Niks, E.H.; Reinders, M.J.T.; et al. Timing and localization of human dystrophin isoform expression provide insights into the cognitive phenotype of Duchenne muscular dystrophy. Sci. Rep. 2017, 7, 12575. [Google Scholar] [CrossRef] [PubMed]
- Matsuo, M.; Awano, H.; Matsumoto, M.; Nagai, M.; Kawaguchi, T.; Zhang, Z.; Nishio, H. Dystrophin Dp116: A yet to be investigated product of the Duchenne muscular dystrophy gene. Genes 2017, 8, 251. [Google Scholar] [CrossRef] [PubMed]
- Tadayoni, R.; Rendon, A.; Soria-Jasso, L.E.; Cisneros, B. Dystrophin Dp71: The smallest but multifunctional product of the Duchenne muscular dystrophy gene. Mol. Neurobiol. 2012, 45, 43–60. [Google Scholar] [CrossRef] [PubMed]
- Aartsma-Rus, A.; Ginjaar, I.B.; Bushby, K. The importance of genetic diagnosis for Duchenne muscular dystrophy. J. Med. Genet. 2016, 53, 145–151. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Monaco, A.P.; Bertelson, C.J.; Liechti-Gallati, S.; Moser, H.; Kunkel, L.M. An explanation for the phenotypic differences between patients bearing partial deletions of the DMD locus. Genomics 1988, 2, 90–95. [Google Scholar] [CrossRef]
- Constantin, B. Dystrophin complex functions as a scaffold for signalling proteins. Biochim. Et Biophys. Acta 2014, 1838, 635–642. [Google Scholar] [CrossRef] [PubMed]
- van Westering, T.L.; Betts, C.A.; Wood, M.J. Current understanding of molecular pathology and treatment of cardiomyopathy in duchenne muscular dystrophy. Molecules 2015, 20, 8823–8855. [Google Scholar] [CrossRef] [PubMed]
- Gawlik, K.I. At the crossroads of clinical and preclinical research for muscular dystrophy-are we closer to effective treatment for patients? Int. J. Mol. Sci. 2018, 19, 1490. [Google Scholar] [CrossRef] [PubMed]
- Sahenk, Z.; Mendell, J.R. The muscular dystrophies: Distinct pathogenic mechanisms invite novel therapeutic approaches. Curr. Rheumatol. Rep. 2011, 13, 199–207. [Google Scholar] [CrossRef] [PubMed]
- Davies, K.E.; Nowak, K.J. Molecular mechanisms of muscular dystrophies: Old and new players. Nat. Rev. Mol. Cell. Biol. 2006, 7, 762–773. [Google Scholar] [CrossRef] [PubMed]
- Tsuda, T.; Fitzgerald, K.K. Dystrophic Cardiomyopathy: Complex pathobiological processes to generate clinical phenotype. J. Cardiovasc. Dev. Dis. 2017, 4, 14. [Google Scholar] [CrossRef] [PubMed]
- Gowran, A.; Spaltro, G.; Casalnuovo, F.; Vigorelli, V.; Spinelli, P.; Castiglioni, E.; Rovina, D.; Paganini, S.; Di Segni, M.; Gervasini, C.; et al. Generation of induced pluripotent stem cells from a Becker muscular dystrophy patient carrying a deletion of exons 45–55 of the dystrophin gene (CCMi002BMD-A-9 45-55). Stem Cell Res. 2018, 28, 21–24. [Google Scholar] [CrossRef] [PubMed]
- Eisen, B.; Ben Jehuda, R.; Cuttitta, A.J.; Mekies, L.N.; Reiter, I.; Ramchandren, S.; Arad, M.; Michele, D.E.; Binah, O. Generation of Duchenne muscular dystrophy patient-specific induced pluripotent stem cell line lacking exons 45-50 of the dystrophin gene (IITi001-A). Stem Cell Res. 2018, 29, 111–114. [Google Scholar] [CrossRef] [PubMed]
- Aminzadeh, M.A.; Rogers, R.G.; Fournier, M.; Tobin, R.E.; Guan, X.; Childers, M.K.; Andres, A.M.; Taylor, D.J.; Ibrahim, A.; Ding, X.; et al. Exosome-mediated benefits of cell therapy in mouse and human models of Duchenne muscular dystrophy. Stem Cell Rep. 2018, 10, 942–955. [Google Scholar] [CrossRef] [PubMed]
- Long, C.; Li, H.; Tiburcy, M.; Rodriguez-Caycedo, C.; Kyrychenko, V.; Zhou, H.; Zhang, Y.; Min, Y.L.; Shelton, J.M.; Mammen, P.P.A.; et al. Correction of diverse muscular dystrophy mutations in human engineered heart muscle by single-site genome editing. Sci. Adv. 2018, 4, eaap9004. [Google Scholar] [CrossRef] [PubMed]
- Kyrychenko, V.; Kyrychenko, S.; Tiburcy, M.; Shelton, J.M.; Long, C.; Schneider, J.W.; Zimmermann, W.H.; Bassel-Duby, R.; Olson, E.N. Functional correction of dystrophin actin binding domain mutations by genome editing. JCI Insight 2017, 2, e95918. [Google Scholar] [CrossRef] [PubMed]
- Spaltro, G.; Vigorelli, V.; Casalnuovo, F.; Spinelli, P.; Castiglioni, E.; Rovina, D.; Paganini, S.; Di Segni, M.; Nigro, P.; Gervasini, C.; et al. Derivation of the Duchenne muscular dystrophy patient-derived induced pluripotent stem cell line lacking DMD exons 49 and 50 (CCMi001DMD-A-3, ∆49, ∆50). Stem Cell Res. 2017, 25, 128–131. [Google Scholar] [CrossRef] [PubMed]
- Nanni, S.; Re, A.; Ripoli, C.; Gowran, A.; Nigro, P.; D’Amario, D.; Amodeo, A.; Crea, F.; Grassi, C.; Pontecorvi, A.; et al. The nuclear pore protein Nup153 associates with chromatin and regulates cardiac gene expression in dystrophic mdx hearts. Cardiovasc. Res. 2016, 112, 555–567. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hashimoto, A.; Naito, A.T.; Lee, J.K.; Kitazume-Taneike, R.; Ito, M.; Yamaguchi, T.; Nakata, R.; Sumida, T.; Okada, K.; Nakagawa, A.; et al. Generation of induced pluripotent stem cells from patients with Duchenne muscular dystrophy and their induction to cardiomyocytes. Int. Heart J. 2016, 57, 112–117. [Google Scholar] [CrossRef] [PubMed]
- Lin, B.; Li, Y.; Han, L.; Kaplan, A.D.; Ao, Y.; Kalra, S.; Bett, G.C.; Rasmusson, R.L.; Denning, C.; Yang, L. Modeling and study of the mechanism of dilated cardiomyopathy using induced pluripotent stem cells derived from individuals with Duchenne muscular dystrophy. Dis. Model. Mech. 2015, 8, 457–466. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guan, X.; Mack, D.L.; Moreno, C.M.; Strande, J.L.; Mathieu, J.; Shi, Y.; Markert, C.D.; Wang, Z.; Liu, G.; Lawlor, M.W.; et al. Dystrophin-deficient cardiomyocytes derived from human urine: New biologic reagents for drug discovery. Stem Cell Res. 2014, 12, 467–480. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zatti, S.; Martewicz, S.; Serena, E.; Uno, N.; Giobbe, G.; Kazuki, Y.; Oshimura, M.; Elvassore, N. Complete restoration of multiple dystrophin isoforms in genetically corrected Duchenne muscular dystrophy patient-derived cardiomyocytes. Mol. Ther. Method. Clin. Dev. 2014, 1, 1. [Google Scholar] [CrossRef] [PubMed]
- Dick, E.; Kalra, S.; Anderson, D.; George, V.; Ritso, M.; Laval, S.H.; Barresi, R.; Aartsma-Rus, A.; Lochmüller, H.; Denning, C. Exon skipping and gene transfer restore dystrophin expression in human induced pluripotent stem cells-cardiomyocytes harboring DMD mutations. Stem Cells Dev. 2013, 22, 2714–2724. [Google Scholar] [CrossRef] [PubMed]
- Jefferies, J.L.; Eidem, B.W.; Belmont, J.W.; Craigen, W.J.; Ware, S.M.; Fernbach, S.D.; Neish, S.R.; Smith, E.O.; Towbin, J.A. Genetic predictors and remodeling of dilated cardiomyopathy in muscular dystrophy. Circulation 2005, 112, 2799–2804. [Google Scholar] [CrossRef] [PubMed]
- Biggar, W.D.; Politano, L.; Harris, V.A.; Passamano, L.; Vajsar, J.; Alman, B.; Palladino, A.; Comi, L.I.; Nigro, G. Deflazacort in Duchenne muscular dystrophy: A comparison of two different protocols. Neuromuscul. Disord. 2004, 14, 476–482. [Google Scholar] [CrossRef] [PubMed]
- Griggs, R.C.; Moxley, R.T., 3rd; Mendell, J.R.; Fenichel, G.M.; Brooke, M.H.; Pestronk, A.; Miller, J.P. Prednisone in Duchenne dystrophy: A randomized, controlled trial defining the time course and dose response. Arch Neurol. 1991, 48, 383–388. [Google Scholar] [CrossRef] [PubMed]
- Alman, B.A.; Raza, S.N.; Biggar, W.D. Steroid treatment and the development of scoliosis in males with duchenne muscular dystrophy. J. Bone Joint Surg. Am. 2004, 86, 519–524. [Google Scholar] [CrossRef] [PubMed]
- Koeks, Z.; Bladen, C.L.; Salgado, D.; van Zwet, E.; Pogoryelova, O.; McMacken, G.; Monges, S.; Foncuberta, M.E.; Kekou, K.; Kosma, K.; et al. Clinical outcomes in Duchenne muscular dystrophy: A study of 5345 patients from the TREAT-NMD DMD Global Database. J. Neuromuscul. Dis. 2017, 4, 293–306. [Google Scholar] [CrossRef] [PubMed]
- Biggar, W.D.; Harris, V.A.; Eliasoph, L.; Alman, B. Long-term benefits of deflazacort treatment for boys with Duchenne muscular dystrophy in their second decade. Neuromuscul. Disord. 2006, 16, 249–255. [Google Scholar] [CrossRef] [PubMed]
- Barber, B.J.; Andrews, J.G.; Lu, Z.; West, N.A.; Meaney, F.J.; Price, E.T.; Gray, A.; Sheehan, D.W.; Pandya, S.; Yang, M.; et al. Oral corticosteroids and onset of cardiomyopathy in Duchenne muscular dystrophy. J. Pediatr. 2013, 163, 1080–1084. [Google Scholar] [CrossRef] [PubMed]
- Markham, L.W.; Spicer, R.L.; Khoury, P.R.; Wong, B.L.; Mathews, K.D.; Cripe, L.H. Steroid therapy and cardiac function in Duchenne muscular dystrophy. Pediatr. Cardiol. 2005, 26, 768–771. [Google Scholar] [CrossRef] [PubMed]
- Markham, L.W.; Kinnett, K.; Wong, B.L.; Woodrow Benson, D.; Cripe, L.H. Corticosteroid treatment retards development of ventricular dysfunction in Duchenne muscular dystrophy. Neuromuscul. Disord. 2008, 18, 365–370. [Google Scholar] [CrossRef] [PubMed]
- Buyse, G.M.; Goemans, N.; van den Hauwe, M.; Meier, T. Effects of glucocorticoids and idebenone on respiratory function in patients with duchenne muscular dystrophy. Pediatr. Pulmonol. 2013, 48, 912–920. [Google Scholar] [CrossRef] [PubMed]
- Gloss, D.; Moxley, R.T., 3rd; Ashwal, S.; Oskoui, M. Practice guideline update summary: Corticosteroid treatment of Duchenne muscular dystrophy: Report of the Guideline Development Subcommittee of the American Academy of Neurology. Neurology 2016, 86, 465–472. [Google Scholar] [CrossRef] [PubMed]
- Rutter, M.M.; Collins, J.; Rose, S.R.; Woo, J.G.; Sucharew, H.; Sawnani, H.; Hor, K.N.; Cripe, L.H.; Wong, B.L. Growth hormone treatment in boys with Duchenne muscular dystrophy and glucocorticoid-induced growth failure. Neuromuscul. Disord. 2012, 22, 1046–1056. [Google Scholar] [CrossRef] [PubMed]
- Duboc, D.; Meune, C.; Lerebours, G.; Devaux, J.Y.; Vaksmann, G.; Becane, H.M. Effect of perindopril on the onset and progression of left ventricular dysfunction in Duchenne muscular dystrophy. J. Am. Coll. Cardiol. 2005, 45, 855–857. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rafael-Fortney, J.A.; Chimanji, N.S.; Schill, K.E.; Martin, C.D.; Murray, J.D.; Ganguly, R.; Stangland, J.E.; Tran, T.; Xu, Y.; Canan, B.D.; et al. Early treatment with lisinopril and spironolactone preserves cardiac and skeletal muscle in Duchenne muscular dystrophy mice. Circulation 2011, 124, 582–588. [Google Scholar] [CrossRef] [PubMed]
- Duboc, D.; Meune, C.; Pierre, B.; Wahbi, K.; Eymard, B.; Toutain, A.; Berard, C.; Vaksmann, G.; Weber, S.; Bécane, H.M. Perindopril preventive treatment on mortality in Duchenne muscular dystrophy: 10 years’ follow-up. Am. Heart J. 2007, 154, 596–602. [Google Scholar] [CrossRef] [PubMed]
- Judge, D.P.; Kass, D.A.; Thompson, W.R.; Wagner, K.R. Pathophysiology and therapy of cardiac dysfunction in Duchenne muscular dystrophy. Am. J. Cardiovasc. Drugs 2011, 11, 287–294. [Google Scholar] [CrossRef] [PubMed]
- Barison, A.; Aquaro, G.D.; Passino, C.; Falorni, M.; Balbarini, A.; Lombardi, M.; Pasquali, L.; Emdin, M.; Siciliano, G. Cardiac magnetic resonance imaging and management of dilated cardiomyopathy in a Duchenne muscular dystrophy manifesting carrier. J. Neurol. 2009, 256, 283–284. [Google Scholar] [CrossRef] [PubMed]
- Deconinck, A.E.; Rafael, J.A.; Skinner, J.A.; Brown, S.C.; Potter, A.C.; Metzinger, L.; Watt, D.J.; Dickson, J.G.; Tinsley, J.M.; Davies, K.E. Utrophin-dystrophin-deficient mice as a model for Duchenne muscular dystrophy. Cell 1997, 90, 717–727. [Google Scholar] [CrossRef]
- Tinsley, J.M.; Fairclough, R.J.; Storer, R.; Wilkes, F.J.; Potter, A.C.; Squire, S.E.; Powell, D.S.; Cozzoli, A.; Capogrosso, R.F.; Lambert, A.; et al. Daily treatment with SMTC1100, a novel small molecule utrophin upregulator, dramatically reduces the dystrophic symptoms in the mdx mouse. PLoS ONE 2011, 6, e19189. [Google Scholar] [CrossRef] [PubMed]
- Ricotti, V.; Spinty, S.; Roper, H.; Hughes, I.; Tejura, B.; Robinson, N.; Layton, G.; Davies, K.; Muntoni, F.; Tinsley, J. Safety, tolerability, and pharmacokinetics of SMT C1100, a 2-Arylbenzoxazole utrophin modulator, following Single- and Multiple-Dose Administration to pediatric patients with Duchenne muscular dystrophy. PLoS ONE 2016, 11, e0152840. [Google Scholar] [CrossRef] [PubMed]
- Kaufman, R.J. Correction of genetic disease by making sense from nonsense. J. Clin. Invest. 1999, 104, 367–368. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malik, V.; Rodino-Klapac, L.R.; Viollet, L.; Wall, C.; King, W.; Al-Dahhak, R.; Lewis, S.; Shilling, C.J.; Kota, J.; Serrano-Munuera, C.; et al. Gentamicin-induced readthrough of stop codons in Duchenne muscular dystrophy. Ann. Neurol. 2010, 67, 771–780. [Google Scholar] [CrossRef] [PubMed]
- Welch, E.M.; Barton, E.R.; Zhuo, J.; Tomizawa, Y.; Friesen, W.J.; Trifillis, P.; Paushkin, S.; Patel, M.; Trotta, C.R.; Hwang, S.; et al. PTC124 targets genetic disorders caused by nonsense mutations. Nature 2007, 447, 87–91. [Google Scholar] [CrossRef] [PubMed]
- McDonald, C.M.; Campbell, C.; Torricelli, R.E.; Finkel, R.S.; Flanigan, K.M.; Goemans, N.; Heydemann, P.; Kaminska, A.; Kirschner, J.; Muntoni, F.; et al. Ataluren in patients with nonsense mutation Duchenne muscular dystrophy (ACT DMD): A multicenter, randomised, double-blind, placebo-controlled, phase 3 trial. Lancet 2017, 390, 1489–1498. [Google Scholar] [CrossRef]
- Kimura, E.; Li, S.; Gregorevic, P.; Fall, B.M.; Chamberlain, J.S. Dystrophin delivery to muscles of mdx mice using lentiviral vectors leads to myogenic progenitor targeting and stable gene expression. Mol. Ther. 2010, 18, 206–213. [Google Scholar] [CrossRef] [PubMed]
- Dello-Russo, C.; Scott, J.M.; Hartigan-O’Connor, D.; Salvatori, G.; Barjot, C.; Robinson, A.S.; Crawford, R.W.; Brooks, S.V.; Chamberlain, J.S. Functional correction of adult mdx mouse muscle using gutted adenoviral vectors expressing full-length dystrophin. Proc. Natl. Acad. Sci. USA 2002, 99, 12979–12984. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scott, J.M.; Li, S.; Harper, S.Q.; Welikson, R.; Bourque, D.; Dello-Russo, C.; Hauschka, S.D.; Chamberlain, J.S. Viral vectors for gene transfer of micro-, mini-, or full-length dystrophin. Neuromuscul. Disord. 2002, 12, S23–S29. [Google Scholar] [CrossRef]
- Duan, D. Systemic AAV micro-dystrophin gene therapy for Duchenne muscular dystrophy. Mol. Ther. 2018, 26, 10. [Google Scholar] [CrossRef] [PubMed]
- Xiao, X.; Li, J.; Samulski, R.J. Efficient long-term gene transfer into muscle tissue of immunocompetent mice by adeno-associated virus vector. J. Virol. 1996, 70, 8098–8108. [Google Scholar] [PubMed]
- Bish, L.T.; Sleeper, M.M.; Brainard, B.; Cole, S.; Russell, N.; Withnall, E.; Arndt, J.; Reynolds, C.; Davison, E.; Sanmiguel, J.; et al. Percutaneous transendocardial delivery of self-complementary adeno-associated virus 6 achieves global cardiac gene transfer in canines. Mol. Ther. J. Am. Soc. Gene Ther. 2008, 16, 1953–1959. [Google Scholar] [CrossRef] [PubMed]
- Daya, S.; Berns, K.I. Gene therapy using adeno-associated virus vectors. Clin. Microbiol. Rev. 2008, 21, 583–593. [Google Scholar] [CrossRef] [PubMed]
- Fisher, M.L.; Balke, C.W.; Freudenberger, R. Therapeutic options in advanced heart failure. Hosp. Pract. 1997, 32, 97–106. [Google Scholar] [CrossRef]
- Schultz, B.R.; Chamberlain, J.S. Recombinant adeno-associated virus transduction and integration. Mol. Ther. J. Am. Soc. Gene Ther. 2008, 16, 1189–1199. [Google Scholar] [CrossRef] [PubMed]
- Gregorevic, P.; Allen, J.M.; Minami, E.; Blankinship, M.J.; Haraguchi, M.; Meuse, L.; Finn, E.; Adams, M.E.; Froehner, S.C.; Murry, C.E.; et al. rAAV6-microdystrophin preserves muscle function and extends lifespan in severely dystrophic mice. Nat. Med. 2006, 12, 787–789. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gregorevic, P.; Blankinship, M.J.; Allen, J.M.; Chamberlain, J.S. Systemic microdystrophin gene delivery improves skeletal muscle structure and function in old dystrophic mdx mice. Mol. Ther. J. Am. Soc. Gene Ther. 2008, 16, 657–664. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Zhu, T.; Qiao, C.; Zhou, L.; Wang, B.; Zhang, J.; Chen, C.; Li, J.; Xiao, X. Adeno-associated virus serotype 8 efficiently delivers genes to muscle and heart. Nat. Biotechnol. 2005, 23, 321–328. [Google Scholar] [CrossRef] [PubMed]
- Inagaki, K.; Fuess, S.; Storm, T.A.; Gibson, G.A.; McTiernan, C.F.; Kay, M.A.; Nakai, H. Robust systemic transduction with AAV9 vectors in mice: Efficient global cardiac gene transfer superior to that of AAV8. Mol. Ther. J. Am. Soc. Gene Ther. 2006, 14, 45–53. [Google Scholar] [CrossRef] [PubMed]
- Schinkel, S.; Bauer, R.; Bekeredjian, R.; Stucka, R.; Rutschow, D.; Lochmuller, H.; Kleinschmidt, J.A.; Katus, H.A.; Muller, O.J. Long-term preservation of cardiac structure and function after adeno-associated virus serotype 9-mediated microdystrophin gene transfer in mdx mice. Hum. Gene Ther. 2012, 23, 566–575. [Google Scholar] [CrossRef] [PubMed]
- Bostick, B.; Shin, J.H.; Yue, Y.; Wasala, N.B.; Lai, Y.; Duan, D. AAV micro-dystrophin gene therapy alleviates stress-induced cardiac death but not myocardial fibrosis in >21-m-old mdx mice, an end-stage model of Duchenne muscular dystrophy cardiomyopathy. J. Mol. Cell. Cardiol. 2012, 53, 217–222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bostick, B.; Shin, J.H.; Yue, Y.; Duan, D. AAV-microdystrophin therapy improves cardiac performance in aged female mdx mice. Mol. Ther. J. Am. Soc. Gene Ther. 2011, 19, 1826–1832. [Google Scholar] [CrossRef] [PubMed]
- Kornegay, J.N.; Li, J.; Bogan, J.R.; Bogan, D.J.; Chen, C.; Zheng, H.; Wang, B.; Qiao, C.; Howard, J.F., Jr.; Xiao, X. Widespread muscle expression of an AAV9 human mini-dystrophin vector after intravenous injection in neonatal dystrophin-deficient dogs. Mol. Ther. J. Am. Soc. Gene Ther. 2010, 18, 1501–1508. [Google Scholar] [CrossRef] [PubMed]
- Yue, Y.; Pan, X.; Hakim, C.H.; Kodippili, K.; Zhang, K.; Shin, J.H.; Yang, H.T.; McDonald, T.; Duan, D. Safe and bodywide muscle transduction in young adult Duchenne muscular dystrophy dogs with adeno-associated virus. Hum. Mol. Genet. 2015, 24, 5880–5890. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Foster, H.; Sharp, P.S.; Athanasopoulos, T.; Trollet, C.; Graham, I.R.; Foster, K.; Wells, D.J.; Dickson, G. Codon and mRNA sequence optimization of microdystrophin transgenes improves expression and physiological outcome in dystrophic mdx mice following AAV2/8 gene transfer. Mol. Ther. J. Am. Soc. Gene Ther. 2008, 16, 1825–1832. [Google Scholar] [CrossRef] [PubMed]
- Le Guiner, C.; Servais, L.; Montus, M.; Larcher, T.; Fraysse, B.; Moullec, S.; Allais, M.; François, V.; Dutilleul, M.; Malerba, A.; et al. Long-term microdystrophin gene therapy is effective in a canine model of Duchenne muscular dystrophy. Nat. Commun. 2017, 8, 16105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Du, L.; Kido, M.; Lee, D.V.; Rabinowitz, J.E.; Samulski, R.J.; Jamieson, S.W.; Weitzman, M.D.; Thistlethwaite, P.A. Differential myocardial gene delivery by recombinant serotype-specific adeno-associated viral vectors. Mol. Ther. J. Am. Soc. Gene Ther. 2004, 10, 604–608. [Google Scholar] [CrossRef] [PubMed]
- Mendell, J.R.; Campbell, K.; Rodino-Klapac, L.; Sahenk, Z.; Shilling, C.; Lewis, S.; Bowles, D.; Gray, S.; Li, C.; Galloway, G.; et al. Dystrophin immunity in Duchenne’s muscular dystrophy. New Engl. J. Med. 2010, 363, 1429–1437. [Google Scholar] [CrossRef] [PubMed]
- Bowles, D.E.; McPhee, S.W.; Li, C.; Gray, S.J.; Samulski, J.J.; Camp, A.S.; Li, J.; Wang, B.; Monahan, P.E.; Rabinowitz, J.E.; et al. Phase 1 gene therapy for Duchenne muscular dystrophy using a translational optimized AAV vector. Mol. Ther. J. Am. Soc. Gene Ther. 2012, 20, 443–455. [Google Scholar] [CrossRef] [PubMed]
- Montarras, D.; Morgan, J.; Collins, C.; Relaix, F.; Zaffran, S.; Cumano, A.; Partridge, T.; Buckingham, M. Direct isolation of satellite cells for skeletal muscle regeneration. Science 2005, 309, 2064–2067. [Google Scholar] [CrossRef] [PubMed]
- Cerletti, M.; Jurga, S.; Witczak, C.A.; Hirshman, M.F.; Shadrach, J.L.; Goodyear, L.J.; Wagers, A.J. Highly efficient, functional engraftment of skeletal muscle stem cells in dystrophic muscles. Cell 2008, 134, 37–47. [Google Scholar] [CrossRef] [PubMed]
- Qu-Petersen, Z.; Deasy, B.; Jankowski, R.; Ikezawa, M.; Cummins, J.; Pruchnic, R.; Mytinger, J.; Cao, B.; Gates, C.; Wernig, A.; et al. Identification of a novel population of muscle stem cells in mice: Potential for muscle regeneration. J. Cell Biol. 2002, 157, 851–864. [Google Scholar] [CrossRef] [PubMed]
- Torrente, Y.; Tremblay, J.P.; Pisati, F.; Belicchi, M.; Rossi, B.; Sironi, M.; Fortunato, F.; El Fahime, M.; D’Angelo, M.G.; Caron, N.J.; et al. Intraarterial injection of muscle-derived CD34(+) Sca-1(+) stem cells restores dystrophin in mdx mice. J. Cell Biol. 2001, 152, 335–348. [Google Scholar] [CrossRef] [PubMed]
- Gussoni, E.; Soneoka, Y.; Strickland, C.D.; Buzney, E.A.; Khan, M.K.; Flint, A.F.; Kunkel, L.M.; Mulligan, R.C. Dystrophin expression in the mdx mouse restored by stem cell transplantation. Nature 1999, 401, 390–394. [Google Scholar] [CrossRef] [PubMed]
- Asakura, A.; Seale, P.; Girgis-Gabardo, A.; Rudnicki, M.A. Myogenic specification of side population cells in skeletal muscle. J. Cell Biol. 2002, 159, 123–134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McKinney-Freeman, S.L.; Jackson, K.A.; Camargo, F.D.; Ferrari, G.; Mavilio, F.; Goodell, M.A. Muscle-derived hematopoietic stem cells are hematopoietic in origin. Proc. Natl. Acad. Sci. USA 2002, 99, 1341–1346. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- LaBarge, M.A.; Blau, H.M. Biological progression from adult bone marrow to mononucleate muscle stem cell to multinucleate muscle fiber in response to injury. Cell 2002, 111, 589–601. [Google Scholar] [CrossRef]
- Ferrari, G.; Cusella-De Angelis, G.; Coletta, M.; Paolucci, E.; Stornaiuolo, A.; Cossu, G.; Mavilio, F. Muscle regeneration by bone marrow-derived myogenic progenitors. Science 1998, 279, 1528–1530. [Google Scholar] [CrossRef] [PubMed]
- Sampaolesi, M.; Blot, S.; D’Antona, G.; Granger, N.; Tonlorenzi, R.; Innocenzi, A.; Mognol, P.; Thibaud, J.L.; Galvez, B.G.; Barthelemy, I.; et al. Mesoangioblast stem cells ameliorate muscle function in dystrophic dogs. Nature 2006, 444, 574–579. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tedesco, F.S.; Hoshiya, H.; D’Antona, G.; Gerli, M.F.; Messina, G.; Antonini, S.; Tonlorenzi, R.; Benedetti, S.; Berghella, L.; Torrente, Y.; et al. Stem cell-mediated transfer of a human artificial chromosome ameliorates muscular dystrophy. Sci. Transl. Med. 2011, 3, 96ra78. [Google Scholar] [CrossRef] [PubMed]
- Dellavalle, A.; Sampaolesi, M.; Tonlorenzi, R.; Tagliafico, E.; Sacchetti, B.; Perani, L.; Innocenzi, A.; Galvez, B.G.; Messina, G.; Morosetti, R.; et al. Pericytes of human skeletal muscle are myogenic precursors distinct from satellite cells. Nat. Cell Biol. 2007, 9, 255–267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benchaouir, R.; Meregalli, M.; Farini, A.; D’Antona, G.; Belicchi, M.; Goyenvalle, A.; Battistelli, M.; Bresolin, N.; Bottinelli, R.; Garcia, L.; et al. Restoration of human dystrophin following transplantation of exon-skipping-engineered DMD patient stem cells into dystrophic mice. Cell Stem Cell 2007, 1, 646–657. [Google Scholar] [CrossRef] [PubMed]
- Torrente, Y.; Belicchi, M.; Marchesi, C.; D’Antona, G.; Cogiamanian, F.; Pisati, F.; Gavina, M.; Giordano, R.; Tonlorenzi, R.; Fagiolari, G.; et al. Autologous transplantation of muscle-derived CD133+ stem cells in Duchenne muscle patients. Cell Transplant. 2007, 16, 563–577. [Google Scholar] [CrossRef] [PubMed]
- Darabi, R.; Arpke, R.W.; Irion, S.; Dimos, J.T.; Grskovic, M.; Kyba, M.; Perlingeiro, R.C. Human ES- and iPS-derived myogenic progenitors restore DYSTROPHIN and improve contractility upon transplantation in dystrophic mice. Cell Stem Cell 2012, 10, 610–619. [Google Scholar] [CrossRef] [PubMed]
- Alter, J.; Lou, F.; Rabinowitz, A.; Yin, H.; Rosenfeld, J.; Wilton, S.D.; Partridge, T.A.; Lu, Q.L. Systemic delivery of morpholino oligonucleotide restores dystrophin expression bodywide and improves dystrophic pathology. Nat. Med. 2006, 12, 175–177. [Google Scholar] [CrossRef] [PubMed]
- Aartsma-Rus, A.; Krieg, A.M. FDA Approves Eteplirsen for Duchenne Muscular Dystrophy: The Next Chapter in the Eteplirsen Saga. Nucleic Acids Ther. 2017, 27, 1–3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shimizu-Motohashi, Y.; Murakami, T.; Kimura, E.; Komaki, H.; Watanabe, N. Exon skipping for Duchenne muscular dystrophy: A systematic review and meta-analysis. Orphanet. J. Rare Dis. 2018, 13, 93. [Google Scholar] [CrossRef] [PubMed]
- Kesselheim, A.S.; Avorn, J. Approving a problematic muscular dystrophy drug: Implications for FDA policy. JAMA 2016, 316, 2357–2358. [Google Scholar] [CrossRef] [PubMed]
- Goemans, N.M.; Tulinius, M.; van den Akker, J.T.; Burm, B.E.; Ekhart, P.F.; Heuvelmans, N.; Holling, T.; Janson, A.A.; Platenburg, G.J.; Sipkens, J.A.; et al. Systemic administration of PRO051 in Duchenne’s muscular dystrophy. New. Engl. J. Med. 2011, 364, 1513–1522. [Google Scholar] [CrossRef] [PubMed]
- Lu, Q.L.; Rabinowitz, A.; Chen, Y.C.; Yokota, T.; Yin, H.; Alter, J.; Jadoon, A.; Bou-Gharios, G.; Partridge, T. Systemic delivery of antisense oligoribonucleotide restores dystrophin expression in body-wide skeletal muscles. Proc. Natl. Acad. Sci. USA 2005, 102, 198–203. [Google Scholar] [CrossRef] [PubMed]
- Goyenvalle, A.; Griffith, G.; Babbs, A.; El Andaloussi, S.; Ezzat, K.; Avril, A.; Dugovic, B.; Chaussenot, R.; Ferry, A.; Voit, T.; et al. Functional correction in mouse models of muscular dystrophy using exon-skipping tricyclo-DNA oligomers. Nat. Med. 2015, 21, 270–275. [Google Scholar] [CrossRef] [PubMed]
- Betts, C.A.; Saleh, A.F.; Carr, C.A.; Hammond, S.M.; Coenen-Stass, A.M.; Godfrey, C.; McClorey, G.; Varela, M.A.; Roberts, T.C.; Clarke, K.; et al. Prevention of exercised induced cardiomyopathy following Pip-PMO treatment in dystrophic mdx mice. Sci. Rep. 2015, 5, 8986. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yin, H.; Saleh, A.F.; Betts, C.; Camelliti, P.; Seow, Y.; Ashraf, S.; Arzumanov, A.; Hammond, S.; Merritt, T.; Gait, M.J.; et al. Pip5 transduction peptides direct high efficiency oligonucleotide-mediated dystrophin exon skipping in heart and phenotypic correction in mdx mice. Mol. Ther. J. Am. Soc. Gene Ther. 2011, 19, 1295–1303. [Google Scholar] [CrossRef] [PubMed]
- Jirka, S.M.; Heemskerk, H.; Tanganyika-de Winter, C.L.; Muilwijk, D.; Pang, K.H.; de Visser, P.C.; Janson, A.; Karnaoukh, T.G.; Vermue, R.; ’t Hoen, P.A.; et al. Peptide conjugation of 2′-O-methyl phosphorothioate antisense oligonucleotides enhances cardiac uptake and exon skipping in mdx mice. Nucleic Acids Ther. 2014, 24, 25–36. [Google Scholar] [CrossRef] [PubMed]
- Yasuda, S.; Townsend, D.; Michele, D.E.; Favre, E.G.; Day, S.M.; Metzger, J.M. Dystrophic heart failure blocked by membrane sealant poloxamer. Nature 2005, 436, 1025–1029. [Google Scholar] [CrossRef] [PubMed]
- Weisleder, N.; Takizawa, N.; Lin, P.; Wang, X.; Cao, C.; Zhang, Y.; Tan, T.; Ferrante, C.; Zhu, H.; Chen, P.J.; et al. Recombinant MG53 protein modulates therapeutic cell membrane repair in treatment of muscular dystrophy. Sci. Transl. Med. 2012, 4, 139ra85. [Google Scholar] [CrossRef] [PubMed]
- Khairallah, M.; Khairallah, R.J.; Young, M.E.; Allen, B.G.; Gillis, M.A.; Danialou, G.; Deschepper, C.F.; Petrof, B.J.; Des Rosiers, C. Sildenafil and cardiomyocyte-specific cGMP signaling prevent cardiomyopathic changes associated with dystrophin deficiency. Proc. Natl. Acad. Sci. USA 2008, 105, 7028–7033. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Percival, J.M.; Whitehead, N.P.; Adams, M.E.; Adamo, C.M.; Beavo, J.A.; Froehner, S.C. Sildenafil reduces respiratory muscle weakness and fibrosis in the mdx mouse model of Duchenne muscular dystrophy. J. Pathol. 2012, 228, 77–87. [Google Scholar] [CrossRef] [PubMed]
- Leung, D.G.; Herzka, D.A.; Thompson, W.R.; He, B.; Bibat, G.; Tennekoon, G.; Russell, S.D.; Schuleri, K.H.; Lardo, A.C.; Kass, D.A.; et al. Sildenafil does not improve cardiomyopathy in Duchenne/Becker muscular dystrophy. Ann. Neurol. 2014, 76, 541–549. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spurney, C.F.; Sali, A.; Guerron, A.D.; Iantorno, M.; Yu, Q.; Gordish-Dressman, H.; Rayavarapu, S.; van der Meulen, J.; Hoffman, E.P.; Nagaraju, K. Losartan decreases cardiac muscle fibrosis and improves cardiac function in dystrophin-deficient mdx mice. J. Cardiovasc. Pharmacol. Ther. 2011, 16, 87–95. [Google Scholar] [CrossRef] [PubMed]
- van Erp, C.; Irwin, N.G.; Hoey, A.J. Long-term administration of pirfenidone improves cardiac function in mdx mice. Muscle Nerv. 2006, 34, 327–334. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amthor, H.; Hoogaars, W.M. Interference with myostatin/ActRIIB signaling as a therapeutic strategy for Duchenne muscular dystrophy. Curr. Gene Ther. 2012, 12, 245–259. [Google Scholar] [CrossRef] [PubMed]
- Wagner, K.R.; Fleckenstein, J.L.; Amato, A.A.; Barohn, R.J.; Bushby, K.; Escolar, D.M.; Flanigan, K.M.; Pestronk, A.; Tawil, R.; Wolfe, G.I.; et al. A phase I/IItrial of MYO-029 in adult subjects with muscular dystrophy. Ann. Neurol. 2008, 63, 561–571. [Google Scholar] [CrossRef] [PubMed]
- Reutenauer-Patte, J.; Boittin, F.X.; Patthey-Vuadens, O.; Ruegg, U.T.; Dorchies, O.M. Urocortins improve dystrophic skeletal muscle structure and function through both PKA- and Epac-dependent pathways. Am. J. Pathol. 2012, 180, 749–762. [Google Scholar] [CrossRef] [PubMed]
- Bostick, B.; Yue, Y.; Lai, Y.; Long, C.; Li, D.; Duan, D. Adeno-associated virus serotype-9 microdystrophin gene therapy ameliorates electrocardiographic abnormalities in mdx mice. Hum. Gene Ther. 2008, 19, 851–856. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Allen, J.M.; Riddell, S.R.; Gregorevic, P.; Storb, R.; Tapscott, S.J.; Chamberlain, J.S.; Kuhr, C.S. Immunity to adeno-associated virus-mediated gene transfer in a random-bred canine model of Duchenne muscular dystrophy. Hum. Gene Ther. 2007, 18, 18–26. [Google Scholar] [CrossRef] [PubMed]
- Mendell, J.R.; Rodino-Klapac, L.; Sahenk, Z.; Malik, V.; Kaspar, B.K.; Walker, C.M.; Clark, K.R. Gene therapy for muscular dystrophy: Lessons learned and path forward. Neurosci. Lett. 2012, 527, 90–99. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramos, J.; Chamberlain, J.S. Gene therapy for Duchenne muscular dystrophy. Expert Opin. Orphan. Drugs 2015, 3, 1255–1266. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bittner, R.E.; Shorny, S.; Streubel, B.; Hübner, C.; Voit, T.; Kress, W. Serum antibodies to the deleted dystrophin sequence after cardiac transplantation in a patient with Becker’s muscular dystrophy. New Engl. J. Med. 1995, 333, 732–733. [Google Scholar] [CrossRef] [PubMed]
- Lorain, S.; Gross, D.A.; Goyenvalle, A.; Danos, O.; Davoust, J.; Garcia, L. Transient immunomodulation allows repeated injections of AAV1 and correction of muscular dystrophy in multiple muscles. Mol. Ther. J. Am. Soc. Gene Ther. 2008, 16, 541–547. [Google Scholar] [CrossRef] [PubMed]
- Foster, H.; Popplewell, L.; Dickson, G. Genetic therapeutic approaches for Duchenne muscular dystrophy. Hum. Gene Ther. 2012, 23, 676–687. [Google Scholar] [CrossRef] [PubMed]
- Seale, P.; Sabourin, L.A.; Girgis-Gabardo, A.; Mansouri, A.; Gruss, P.; Rudnicki, M.A. Pax7 is required for the specification of myogenic satellite cells. Cell 2000, 102, 777–786. [Google Scholar] [CrossRef]
- Ferrer, A.; Wells, K.E.; Wells, D.J. Immune responses to dystropin: Implications for gene therapy of Duchenne muscular dystrophy. Gene Ther. 2000, 7, 1439–1446. [Google Scholar] [CrossRef] [PubMed]
- Peault, B.; Rudnicki, M.; Torrente, Y.; Cossu, G.; Tremblay, J.P.; Partridge, T.; Gussoni, E.; Kunkel, L.M.; Huard, J. Stem and progenitor cells in skeletal muscle development, maintenance, and therapy. Mol. Ther. J. Am. Soc. Gene Ther. 2007, 15, 867–877. [Google Scholar] [CrossRef] [PubMed]
- Tamaki, T.; Okada, Y.; Uchiyama, Y.; Tono, K.; Masuda, M.; Wada, M.; Hoshi, A.; Ishikawa, T.; Akatsuka, A. Clonal multipotency of skeletal muscle-derived stem cells between mesodermal and ectodermal lineage. Stem Cells 2007, 25, 2283–2290. [Google Scholar] [CrossRef] [PubMed]
- Deasy, B.M.; Jankowski, R.J.; Huard, J. Muscle-derived stem cells: Characterization and potential for cell-mediated therapy. Blood Cell. Mol. Dis. 2001, 27, 924–933. [Google Scholar] [CrossRef] [PubMed]
- Li, H.L.; Fujimoto, N.; Sasakawa, N.; Shirai, S.; Ohkame, T.; Sakuma, T.; Tanaka, M.; Amano, N.; Watanabe, A.; Sakurai, H.; et al. Precise correction of the dystrophin gene in duchenne muscular dystrophy patient induced pluripotent stem cells by TALEN and CRISPR-Cas9. Stem Cell Rep. 2015, 4, 143–154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Konieczny, P.; Swiderski, K.; Chamberlain, J.S. Gene and cell-mediated therapies for muscular dystrophy. Muscle Nerv. 2013, 47, 649–663. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Douglas, A.G.; Wood, M.J. Splicing therapy for neuromuscular disease. Mol. Cell Neurosci. 2013, 56, 169–185. [Google Scholar] [CrossRef] [PubMed]
- Wilton, S.D.; Lloyd, F.; Carville, K.; Fletcher, S.; Honeyman, K.; Agrawal, S.; Kole, R. Specific removal of the nonsense mutation from the mdx dystrophin mRNA using antisense oligonucleotides. Neuromuscul. Disord. 1999, 9, 330–338. [Google Scholar] [CrossRef]
- Niks, E.H.; Aartsma-Rus, A. Exon skipping: A first in class strategy for Duchenne muscular dystrophy. Expert Opin. Biol. Ther. 2017, 17, 225–236. [Google Scholar] [CrossRef] [PubMed]
- Aartsma-Rus, A.; Fokkema, I.; Verschuuren, J.; Ginjaar, I.; van Deutekom, J.; van Ommen, G.J.; den Dunnen, J.T. Theoretic applicability of antisense-mediated exon skipping for Duchenne muscular dystrophy mutations. Hum. Mutat. 2009, 30, 293–299. [Google Scholar] [CrossRef] [PubMed]
- Aartsma-Rus, A.; Straub, V.; Hemmings, R.; Haas, M.; Schlosser-Weber, G.; Stoyanova-Beninska, V.; Mercuri, E.; Muntoni, F.; Sepodes, B.; Vroom, E.; et al. Development of exon skipping therapies for Duchenne muscular dystrophy: A critical review and a perspective on the outstanding issues. Nucleic Acids Ther. 2017, 27, 251–259. [Google Scholar] [CrossRef] [PubMed]
- Mitrpant, C.; Fletcher, S.; Iversen, P.L.; Wilton, S.D. By-passing the nonsense mutation in the 4 CV mouse model of muscular dystrophy by induced exon skipping. J. Gene. Med. 2009, 11, 46–56. [Google Scholar] [CrossRef] [PubMed]
- Aartsma-Rus, A.; van Deutekom, J.C.; Fokkema, I.F.; Van Ommen, G.J.; Den Dunnen, J.T. Entries in the Leiden Duchenne muscular dystrophy mutation database: An overview of mutation types and paradoxical cases that confirm the reading-frame rule. Muscle Nerv. 2006, 34, 135–144. [Google Scholar] [CrossRef] [PubMed]
- Aartsma-Rus, A. Dystrophin analysis in clinical trials. J. Neuromuscul. Dis. 2014, 1, 41–53. [Google Scholar] [PubMed]
- Malerba, A.; Sharp, P.S.; Graham, I.R.; Arechavala-Gomeza, V.; Foster, K.; Muntoni, F.; Wells, D.J.; Dickson, G. Chronic systemic therapy with low-dose morpholino oligomers ameliorates the pathology and normalizes locomotor behavior in mdx mice. Mol. Ther. J. Am. Soc. Gene Ther. 2011, 19, 345–354. [Google Scholar] [CrossRef] [PubMed]
- Moulton, H.M.; Moulton, J.D. Morpholinos and their peptide conjugates: Therapeutic promise and challenge for Duchenne muscular dystrophy. Biochim. Et Biophys. Acta 2010, 1798, 2296–2303. [Google Scholar] [CrossRef] [PubMed]
- Lehto, T.; Castillo Alvarez, A.; Gauck, S.; Gait, M.J.; Coursindel, T.; Wood, M.J.; Lebleu, B.; Boisguerin, P. Cellular trafficking determines the exon skipping activity of Pip6a-PMO in mdx skeletal and cardiac muscle cells. Nucl. Acids Res. 2014, 42, 3207–3217. [Google Scholar] [CrossRef] [PubMed]
- Samoylova, T.I.; Smith, B.F. Elucidation of muscle-binding peptides by phage display screening. Muscle Nerv. 1999, 22, 460–466. [Google Scholar] [CrossRef]
- Yin, H.; Moulton, H.M.; Betts, C.; Seow, Y.; Boutilier, J.; Iverson, P.L.; Wood, M.J. A fusion peptide directs enhanced systemic dystrophin exon skipping and functional restoration in dystrophin-deficient mdx mice. Hum. Mol. Genet. 2009, 18, 4405–4414. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McPherron, A.C.; Lawler, A.M.; Lee, S.J. Regulation of skeletal muscle mass in mice by a new TGF-beta superfamily member. Nature 1997, 387, 83–90. [Google Scholar] [CrossRef] [PubMed]
- Ríos, R.; Carneiro, I.; Arce, V.M.; Devesa, J. Myostatin is an inhibitor of myogenic differentiation. Am. J. Physiol. Cell Physiol. 2002, 282, C993–C999. [Google Scholar] [CrossRef] [PubMed]
- Trendelenburg, A.U.; Meyer, A.; Rohner, D.; Boyle, J.; Hatakeyama, S.; Glass, D.J. Myostatin reduces Akt/TORC1/p70S6K signaling, inhibiting myoblast differentiation and myotube size. Am. J. Physiol. Cell Physiol. 2009, 296, C1258–C1270. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McFarlane, C.; Hui, G.Z.; Amanda, W.Z.; Lau, H.Y.; Lokireddy, S.; Xiaojia, G.; Mouly, V.; Butler-Browne, G.; Gluckman, P.D.; Sharma, M.; et al. Human myostatin negatively regulates human myoblast growth and differentiation. Am. J. Physiol. Cell Physiol. 2011, 301, C195–C203. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, S.J.; Reed, L.A.; Davies, M.V.; Girgenrath, S.; Goad, M.E.; Tomkinson, K.N.; Wright, J.F.; Barker, C.; Ehrmantraut, G.; Holmstrom, J.; et al. Regulation of muscle growth by multiple ligands signaling through activin type II receptors. Proc. Natl. Acad. Sci. USA 2005, 102, 18117–18122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Attie, K.M.; Borgstein, N.G.; Yang, Y.; Condon, C.H.; Wilson, D.M.; Pearsall, A.E.; Kumar, R.; Willins, D.A.; Seehra, J.S.; Sherman, M.L. A single ascending-dose study of muscle regulator ACE-031 in healthy volunteers. Muscle Nerv. 2013, 47, 416–423. [Google Scholar] [CrossRef] [PubMed]
- Matthews, E.; Brassington, R.; Kuntzer, T.; Jichi, F.; Manzur, A.Y. Corticosteroids for the treatment of Duchenne muscular dystrophy. Cochrane. Database Syst. Rev. 2016, 5, CD003725. [Google Scholar] [CrossRef] [PubMed]
- Bushby, K.; Finkel, R.; Birnkrant, D.J.; Case, L.E.; Clemens, P.R.; Cripe, L.; Kaul, A.; Kinnett, K.; McDonald, C.; Pandya, S.; et al. Diagnosis and management of Duchenne muscular dystrophy, part 2: Implementation of multidisciplinary care. Lancet Neurol. 2010, 9, 177–189. [Google Scholar] [CrossRef]
- Ramaciotti, C.; Heistein, L.C.; Coursey, M.; Lemler, M.S.; Eapen, R.S.; Iannaccone, S.T.; Scott, W.A. Left ventricular function and response to enalapril in patients with duchenne muscular dystrophy during the second decade of life. Am. J. Cardiol. 2006, 98, 825–827. [Google Scholar] [CrossRef] [PubMed]
- Shaddy, R.E.; Boucek, M.M.; Hsu, D.T.; Boucek, R.J.; Canter, C.E.; Mahony, L.; Ross, R.D.; Pahl, E.; Blume, E.D.; Dodd, D.A.; et al. Carvedilol for children and adolescents with heart failure: A randomized controlled trial. JAMA 2007, 298, 1171–1179. [Google Scholar] [CrossRef] [PubMed]
- Ogata, H.; Ishikawa, Y.; Ishikawa, Y.; Minami, R. Beneficial effects of beta-blockers and angiotensin-converting enzyme inhibitors in Duchenne muscular dystrophy. J. Cardiol. 2009, 53, 72–78. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Viollet, L.; Thrush, P.T.; Flanigan, K.M.; Mendell, J.R.; Allen, H.D. Effects of angiotensin-converting enzyme inhibitors and/or beta blockers on the cardiomyopathy in Duchenne muscular dystrophy. Am. J. Cardiol. 2012, 110, 98–102. [Google Scholar] [CrossRef] [PubMed]
- Uaesoontrachoon, K.; Quinn, J.L.; Tatem, K.S.; Van Der Meulen, J.H.; Yu, Q.; Phadke, A.; Miller, B.K.; Gordish-Dressman, H.; Ongini, E.; Miglietta, D.; et al. Long-term treatment with naproxcinod significantly improves skeletal and cardiac disease phenotype in the mdx mouse model of dystrophy. Hum. Mol. Genet. 2014, 23, 3239–3249. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cripe, L.; Kinnett, K.; Uzark, K.; Eghtesady, P.; Wong, B.; Spicer, R. P1.14 Cardiac transplantation in Duchenne muscular dystrophy: A case report. Neuromuscul. Disorder. 2011, 21, 645. [Google Scholar] [CrossRef]
- Burkhoff, D.; Sayer, G.; Doshi, D.; Uriel, N. Hemodynamics of Mechanical Circulatory Support. J. Am. Coll. Cardiol. 2015, 66, 2663–2674. [Google Scholar] [CrossRef] [PubMed]
- Silva, E.J. Mechanical Circulatory support: Current status and future directions. Prog. Cardiovasc. Dis. 2016, 58, 444–454. [Google Scholar] [CrossRef] [PubMed]
- Kirklin, J.K.; Naftel, D.C.; Kormos, R.L.; Stevenson, L.W.; Pagani, F.D.; Miller, M.A.; Baldwin, J.T.; Young, J.B. Fifth INTERMACS annual report: Risk factor analysis from more than 6,000 mechanical circulatory support patients. J. Heart Lung Transplant. 2013, 32, 141–156. [Google Scholar] [CrossRef] [PubMed]
- Miller, L.W.; Guglin, M. Patient selection for ventricular assist devices: A moving target. J. Am. Coll. Cardiol. 2013, 61, 1209–1221. [Google Scholar] [CrossRef] [PubMed]
- Ryan, T.D.; Jefferies, J.L.; Sawnani, H.; Wong, B.L.; Gardner, A.; Del Corral, M.; Lorts, A.; Morales, D.L. Implantation of the HeartMate II and HeartWare left ventricular assist devices in patients with duchenne muscular dystrophy: Lessons learned from the first applications. ASAIO J. 2014, 60, 246–248. [Google Scholar] [CrossRef] [PubMed]
- Brancaccio, G.; Filippelli, S.; Michielon, G.; Iacobelli, R.; Alfieri, S.; Gandolfo, F.; Pongiglione, G.; Albanese, S.; Perri, G.; Parisi, F.; et al. Ventricular assist devices as a bridge to heart transplantation or as destination therapy in pediatric patients. Transplant. Proc. 2012, 44, 2007–2012. [Google Scholar] [CrossRef] [PubMed]
- Amodeo, A.; Adorisio, R. Left ventricular assist device in duchenne cardiomyopathy: Can we change the natural history of cardiac disease? Int. J. Cardiol. 2012, 161, e43. [Google Scholar] [CrossRef] [PubMed]
- Iodice, F.; Testa, G.; Averardi, M.; Brancaccio, G.; Amodeo, A.; Cogo, P. Implantation of a left ventricular assist device as a destination therapy in Duchenne muscular dystrophy patients with end stage cardiac failure: Management and lessons learned. Neuromuscul. Disord. 2015, 25, 19–23. [Google Scholar] [CrossRef] [PubMed]
- Perri, G.; Filippelli, S.; Adorisio, R.; Iacobelli, R.; Iodice, F.; Testa, G.; Paglietti, M.G.; D’Amario, D.; Massetti, M.; Amodeo, A. Left ventricular assist device as destination therapy in cardiac end-stage dystrophinopathies: Midterm results. J. Thorac. Cardiovasc. Surg. 2017, 153, 669–674. [Google Scholar] [CrossRef] [PubMed]
- Gowran, A.; Rasponi, M.; Visone, R.; Nigro, P.; Perrucci, G.L.; Righetti, S.; Zanobini, M.; Pompilio, G. Young at heart: Pioneering approaches to model nonischaemic cardiomyopathy with induced pluripotent stem cells. Stem Cells Int. 2016, 2016, 4287158. [Google Scholar] [CrossRef] [PubMed]
- Kalra, S.; Montanaro, F.; Denning, C. Can human pluripotent stem cell-derived cardiomyocytes advance understanding of muscular dystrophies? J. Neuromuscul. Dis. 2016, 3, 309–332. [Google Scholar] [CrossRef] [PubMed]
- Musunuru, K.; Sheikh, F.; Gupta, R.M.; Houser, S.R.; Maher, K.O.; Milan, D.J.; Terzic, A.; Wu, J.C.; American Heart Association Council on Functional Genomics and Translational Biology; Council on Cardiovascular Disease in the Young; et al. Induced pluripotent stem cells for cardiovascular disease modeling and precision medicine: A scientific statement from the American Heart Association. Circ. Genom. Precis. Med. 2018, 11, e000043. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.H. Maintenance of undifferentiated state of human induced pluripotent stem cells through cytoskeleton-driven force acting to secreted fibronectin on a dendrimer-immobilized surface. J. Biosci. Bioeng. 2014, 118, 716–722. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, I.G.; Gil, C.H.; Seo, J.; Park, S.J.; Subbiah, R.; Jung, T.H.; Kim, J.S.; Jeong, Y.H.; Chung, H.M.; Lee, J.H.; et al. Echanotransduction of human pluripotent stem cells cultivated on tunable cell-derived extracellular matrix. Biomaterials 2018, 150, 100–111. [Google Scholar] [CrossRef] [PubMed]
- Musah, S. Substratum-induced differentiation of human pluripotent stem cells reveals the coactivator YAP is a potent regulator of neuronal specification. Proc. Natl. Acad. Sci. USA 2014, 111, 13805–13810. [Google Scholar] [CrossRef] [PubMed]
- Rao, L.; Qian, Y.; Khodabukus, A.; Ribar, T.; Bursac, N. Engineering human pluripotent stem cells into a functional skeletal muscle tissue. Nature Commun. 2018, 9, 126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cimetta, E. Production of arrays of cardiac and skeletal muscle myofibres by micropatterning techniques on a soft substrate. Biomed. Microdevices 2009, 11, 389–400. [Google Scholar] [CrossRef] [PubMed]
- Batalov, I.; Feinberg, A.W. Differentiation of cardiomyocytes from human pluripotent stem cells using monolayer culture. Biomark. Insights 2015, 10, 71–76. [Google Scholar] [PubMed]
- Macrí-Pellizzeri, L.; Pelacho, B.; Sancho, A.; Iglesias-García, O.; Simón-Yarza, A.M.; Soriano-Navarro, M.; González-Granero, S.; García-Verdugo, J.M.; De-Juan-Pardo, E.M.; Prosper, F. Substrate stiffness and composition specifically direct differentiation of induced pluripotent stem cells. Tissue Eng. Part A 2015, 21, 1633–1641. [Google Scholar] [CrossRef] [PubMed]
- Kshitiz; Park, J.; Kim, P.; Helen, W.; Engler, A.J.; Levchenko, A.; Kim, D.H. Control of stem cell fate and function by engineering physical microenvironments. Integr. Biol. 2012, 4, 1008–1018. [Google Scholar] [CrossRef]
- Serena, E.; Zatti, S.; Reghelin, E.; Pasut, A.; Cimetta, E.; Elvassore, N. Soft substrates drive optimal differentiation of human healthy and dystrophic myotubes. Integr. Biol. 2010, 2, 193–201. [Google Scholar] [CrossRef] [PubMed]
- Lian, X.; Zhang, J.; Azarin, S.M.; Zhu, K.; Hazeltine, L.B.; Bao, X.; Hsiao, C.; Kamp, T.J.; Palecek, S.P. Directed cardiomyocyte differentiation from human pluripotent stem cells by modulating Wnt/beta-catenin signaling under fully defined conditions. Nat. Protoc. 2013, 8, 162–175. [Google Scholar] [CrossRef] [PubMed]
- Burridge, P.W.; Zambidis, E.T. Highly efficient directed differentiation of human induced pluripotent stem cells into cardiomyocytes. Methods Mol. Biol. 2013, 997, 149–161. [Google Scholar] [CrossRef] [PubMed]
- Carmen, L.; Maria, V.; Morales-Medina, J.C.; Vallelunga, A.; Palmieri, B.; Iannitti, T. Role of proteoglycans and glycosaminoglycans in Duchenne muscular dystrophy. Glycobiology 2018. [Google Scholar] [CrossRef] [PubMed]
- Arshi, A.; Nakashima, Y.; Nakano, H.; Eaimkhong, S.; Evseenko, D.; Reed, J.; Stieg, A.Z.; Gimzewski, J.K.; Nakano, A. Rigid microenvironments promote cardiac differentiation of mouse and human embryonic stem cells. Sci. Technol. Adv. Mater. 2013, 14, 025003. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santoro, R.; Scaini, D.; Severino, L.U.; Amadeo, F.; Ferrari, S.; Bernava, G.; Garoffolo, G.; Agrifoglio, M.; Casalis, L.; Pesce, M. Activation of human aortic valve interstitial cells by local stiffness involves YAP-dependent transcriptional signaling. Biomaterials 2018, 181, 268–279. [Google Scholar] [CrossRef] [PubMed]
- Acevedo-Acevedo, S.; Crone, W.C. Substrate stiffness effect and chromosome missegregation in hIPS cells. J. Negat. Results Biomed. 2015, 14, 22. [Google Scholar] [CrossRef] [PubMed]
- Choi, K.H.; Park, J.K.; Son, D.; Hwang, J.Y.; Lee, D.K.; Ka, H.; Park, J.; Lee, C.K. Reactivation of Endogenous Genes and Epigenetic Remodeling Are Barriers for Generating Transgene-Free Induced Pluripotent Stem Cells in Pig. PLoS ONE 2016, 11, e0158046. [Google Scholar] [CrossRef] [PubMed]
- Pennarossa, G.; Santoro, R.; Manzoni, E.F.M.; Pesce, M.; Gandaolfi, F.; Brevini, T.A.L. Epigenetic erasing and pancreatic differentiation of dermal fibroblasts into insulin-producing cells are boosted by the use of low-stiffness substrate. Stem Cell Rev. 2018, 14, 398–411. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Serpooshan, V.; Tong, X.; Venkatraman, S.; Lee, M.; Lee, J.; Chirikian, O.; Wu, J.C.; Wu, S.M.; Yang, F. Contractile force generation by 3D hiPSC-derived cardiac tissues is enhanced by rapid establishment of cellular interconnection in matrix with muscle-mimicking stiffness. Biomaterials 2017, 131, 111–120. [Google Scholar] [CrossRef] [PubMed]
- Engler, A.J.; Carag-Krieger, C.; Johnson, C.P.; Raab, M.; Tang, H.Y.; Speicher, D.W.; Sanger, J.W.; Sanger, J.M.; Discher, D.E. Embryonic cardiomyocytes beat best on a matrix with heart-like elasticity: Scar-like rigidity inhibits beating. J. Cell sci. 2008, 121, 3794–3802. [Google Scholar] [CrossRef] [PubMed]
- Serena, E.; Cimetta, E.; Zatti, S.; Zaglia, T.; Zagallo, M.; Keller, G.; Elvassore, N. Micro-arrayed human embryonic stem cells-derived cardiomyocytes for in vitro functional assay. PLoS ONE 2012, 7, e48483. [Google Scholar] [CrossRef] [PubMed]
- Grespan, E. Effect of geometrical constraints on human pluripotent stem cell nuclei in pluripotency and differentiation. Integr. Biol. 2018, 10, 278–289. [Google Scholar] [CrossRef] [PubMed]
- Tijore, A.; Irvine, S.A.; Sarig, U.; Mhaisalkar, P.; Baisane, V.; Venkatraman, S. Contact guidance for cardiac tissue engineering using 3D bioprinted gelatin patterned hydrogel. Biofabrication 2018, 10, 025003. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Happe, C.L.; Engler, A.J. Mechanical forces reshape differentiation cues that guide cardiomyogenesis. Circ. Res. 2016, 118, 296–310. [Google Scholar] [CrossRef] [PubMed]
- Marsano, A. Beating heart on a chip: A novel microfluidic platform to generate functional 3D cardiac microtissues. Lab Chip 2016, 16, 599–610. [Google Scholar] [CrossRef] [PubMed]
- Occhetta, P.; Sadr, N.; Piraino, F.; Redaelli, A.; Moretti, M.; Rasponi, M. Fabrication of 3D cell-laden hydrogel microstructures through photo-mold patterning. Biofabrication 2013, 5, 035002. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giacomelli, E.; Bellin, M.; Sala, L.; van Meer, B.J.; Tertoolen, L.G.; Orlova, V.V.; Mummery, C.L. Three-dimensional cardiac microtissues composed of cardiomyocytes and endothelial cells co-differentiated from human pluripotent stem cells. Development 2017, 144, 1008–1017. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schaefer, J.A. A cardiac patch from aligned microvessel and cardiomyocyte patches. J. Tissue Eng. Regen. Med. 2018, 12, 546–556. [Google Scholar] [CrossRef] [PubMed]
- Nunes, S.S.; Miklas, J.W.; Liu, J.; Aschar-Sobbi, R.; Xiao, Y.; Zhang, B.; Jiang, J.; Masse, S.; Gagliardi, M.; Hsieh, A.; et al. Biowire: A platform for maturation of human pluripotent stem cell-derived cardiomyocytes. Nat. Methods 2013, 10, 781–787. [Google Scholar] [CrossRef] [PubMed]
- Tandon, N.; Marsano, A.; Maidhof, R.; Numata, K.; Montouri-Sorrentino, C.; Cannizzaro, C.; Voldman, J.; Vunjak-Novakovic, G. Surface-patterned electrode bioreactor for electrical stimulation. Lab Chip 2010, 10, 692–700. [Google Scholar] [CrossRef] [PubMed]
- Pavesi, A.; Adriani, G.; Rasponi, M.; Zervantonakis, I.K.; Fiore, G.B.; Kamm, R.D. Controlled electromechanical cell stimulation on-a-chip. Sci. Rep. 2015, 5, 11800. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wheelwright, M.; Win, Z.; Mikkila, J.L.; Amen, K.Y.; Alford, P.W.; Metzger, J.M. Investigation of human iPSC-derived cardiac myocyte functional maturation by single cell traction force microscopy. PLoS ONE 2018, 13, e0194909. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, D.; Matsuura, K.; Seta, H.; Haraguchi, Y.; Okano, T.; Shimizu, T. Contractile force measurement of human induced pluripotent stem cell-derived cardiac cell sheet-tissue. PLoS ONE 2018, 13, e0198026. [Google Scholar] [CrossRef] [PubMed]
- Sala, L.; van Meer, B.J.; Tertoolen, L.G.J.; Bakkers, J.; Bellin, M.; Davis, R.P.; Denning, C.; Dieben, M.A.E.; Eschenhagen, T.; Giacomelli, E.; et al. Musclemotion: A versatile open software tool to quantify cardiomyocyte and cardiac muscle contraction in vitro and in vivo. Circ. Res. 2018, 122, e5–e16. [Google Scholar] [CrossRef] [PubMed]
- Macadangdang, J.; Guan, X.; Smith, A.S.; Lucero, R.; Czerniecki, S.; Childers, M.K.; Mack, D.L.; Kim, D.H. Nanopatterned human iPSC-based model of a dystrophin-null cardiomyopathic phenotype. Cell. Mol. Bioeng. 2015, 8, 320–332. [Google Scholar] [CrossRef] [PubMed]
- Hamazaki, T.; El Rouby, N.; Fredette, N.C.; Santostefano, K.E.; Terada, N. Concise review: Induced pluripotent stem cell research in the era of precision medicine. Stem Cells 2017, 35, 545–550. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Treatment | Research Stage | Methods and Results | References |
|---|---|---|---|
| Utrophin up-regulation | |||
| SMT C1100 | Preclinical—mdx mice. Clinical, Phase I—DMD children | Adult mdx mice were daily administered with SMT C1100 (ezutromid, 50 mg/kg), an utrophin modulator, for 4 weeks: increased mRNA and protein levels of utrophin; reduction of skeletal muscle inflammation and fibrosis; protection form exercise-induced injury. In a clinical trial (NCT02383511), 12 DMD patients were treated with SMT C1100 (50 and 100 mg/kg bid or 100 mg/kg tid): assess safety and tolerability. | [88,89] |
| Stop codon read-through therapy | |||
| Ataluren | Clinical, Phase III—DMD boys | In a clinical trial (NCT01826487), Ataluren (PTC124) was administered to DMD boys with a nonsense mutation for 48 weeks (40 mg/kg daily): assessed tolerability; positive effects, particularly on the subgroup of patients with baseline 6MWD between 300 m and 400 m (least square mean difference of 42.9 m; p = 0.007). Another trial (NCT03179631) started to examine long-term effects of Ataluren in 250 DMD 5-year and older patients. | [93] |
| Viral gene therapy | |||
| Lentivirus | Preclinical—mdx | Micro-dystrophin IM injection into TA muscle of neonatal mdx mice: stable expression (20–25% of CSA) of dystrophin up to 2 years; ameliorated pathophysiology but no protection from c.i. injury; transduced both myofibres and satellite cells that contributes to muscle regeneration. | [94] |
| ‘Gutted’ adenovirus | Preclinical—mdx | IM injection of full-length dystrophin cDNA into TA muscle of 1-year-old mdx mice: dystrophin expression 1 month after injection (25–30% of CSA) and ≈ 40% correction of susceptibility of muscles to c.i. injury. | [95] |
| rAAV6 | Preclinical—mdx/utr-/- and mdx mice | Single IV administration of micro-dystrophin in 1-month-old mdx/utr-/- and 20-month-old mdx mice. In young mice: body-wide dystrophin expression 1 year post-injection; efficient transduction of diaphragm with improved resistance to c.i. injury; heart transduction and normal heart mass but no alteration of myocardial performance index; increased size of TA muscle, peak force production and resistance to c.i. injury. Increased body mass and extended lifespan of treated mice. In old mice: widespread expression of dystrophin 4 months after injection in skeletal muscle, diaphragm and heart; increased resistance to c.i. injury of diaphragm; increased peak force production of TA muscle. | [103,104] |
| rAAV8 | Preclinical—normal mice | Single IV and IP administrations of the viral vector in neonatal and adult mice: efficient gene transfer in skeletal muscles and heart. | [105] |
| rAAV-rh74 | Clinical trial, Phase I/II—DMD infants and children | One clinical trial (Jerry R. Mendell, NCT03375164, drug name rAAV-rh74.MHCK7.Micro-dystrophin) started in USA to assess the safety of micro-dystrophin delivery with rAAV-rh74 in very young dystrophic patients (from 3 months to 7 years). Only ambulatory patients with frameshift or nonsense mutation within exon 18–58 are recruited. | [97] |
| rAAV9 | Preclinical—normal mice, mdx mice, GRMD dogs and DMD dogs. Clinical trial, Phase I—DMD children and boys | Single IV administration of the viral vector in six to eight-week-old mice resulted in an efficient heart transduction. Neonatal and 6-week-old mdx mice were treated with micro-dystrophin through single IV administration: heart transduction and improvement of cardiac pathology. Single IV administration of micro-dystrophin in 16 to 20-month-old female mdx mice with severe cardiomyopathy: efficient cardiac transduction after 2–8 months and improvement of cardiac pathology. Treatment of >21-month-old mdx mice with micro-dystrophin revealed strong dystrophin expression in the heart but only partial correction of ECG abnormalities and no improvement in cardiac fibrosis. Single IV administration of mini-dystrophin in neonatal GRMD dogs: efficient limb muscles, diaphragm and heart transduction after 16 weeks (from 15 to 100% dystrophin-positive myofibres); inflammatory myopathy, contractures and growth retardation were observed. Micro-dystrophin was administered to 2-month-old DMD dogs through single IV injection of tyrosine-engineered vector carrying micro-dystrophin; immunosuppression was performed: widespread transduction in skeletal muscles, diaphragm and heart after 16 weeks without adverse reactions. Two clinical trials started in USA to assess the safety of systemic micro-dystrophin delivery with AAV9 vector. The first (Solid Biosciences, NCT03368742, drug name SGT-001) recruits both ambulatory and non-ambulatory patients, the second (Pfizer, NCT03362502, drug name PF-06939926) recruits only ambulatory patients. In both cases, children and adolescents with any DMD mutation are recruited. | [97,106,107,108,109,110,111] |
| rAAV2/8 | Preclinical—mdx mice and GRMD dogs | IM and IV administrations of codon-optimized micro-dystrophin in neonatal and adult mdx mice: dystrophin expression in both heart and skeletal muscles after systemic administration (12 weeks post injection); improved muscle function and protection from c.i. injury; no immunological response was observed. Juvenile GRMD dogs were treated with IV and LR (i.e., forelimb) administrations of canine micro-dystrophin without immunosuppression: high protein expression (50% on average) in the treated limb and recover of function after LR perfusion; significant amelioration of the clinical status and gait quality (up to 2 years) following IV injection. No data regarding heart pathology. | [112,113] |
| rAAV2/1-5 | Preclinical—primary neonatal and adult mice CMs, primary human CMs and adult mice | Murine and human CMs were cultured in vitro and infected with five rAAV serotypes (1 to 5). In detail, murine neonatal CMs were cultured for 7 days after infection while primary human and murine CMs only for 72 h. IC injection of five serotypes of the viral vector was performed in 10-week-old mice. Serotype rAAV1 has shown a good cardiac transduction efficacy in vitro (12 and 10% positive CMs in murine and adult CMs respectively) and also in vivo (40% positive CMs 1 month after infection). | [114] |
| rAAV2.5 | Clinical trial, Phase I—DMD boys | Six DMD boys were treated through IM administration of mini-dystrophin in the bicep muscle (Jerry R. Mendell, NCT00428935). Only few dystrophin-positive myofibres were detected in two patients and T cell-mediated immune response against mini-dystrophin and the viral capsid was observed. | [115,116] |
| Cell-based therapy | |||
| Satellite cells (SCs) | Preclinical—mdx and mdx nu/nu mice | Isolation of SCs from the diaphragm of Pax3GFP/+ mice through FACS and injection in the irradiated TA muscle of 3-week-old mdx nu/nu mice: dystrophin expression three weeks after transplantation and contribution to the satellite cell pool. Reduced efficiency after culturing was observed. A subpopulation of stem cells, namely skeletal muscle precursors (SMPs), were purified through FACS from normal mice and engrafted into limb muscles of mdx mice: high efficiency (up to 94% of engrafted myofibres); restored dystrophin expression; improved muscle functionality and renewing of the satellite cell niche. No heart data available. | [117,118] |
| Muscle-derived stem cells (MD-SCs) | Preclinical—normal, scid/bg and mdx mice | Hindlimb IA injection of purified CD34+/Sca-1+ MD-SCs isolated from newborn mice into 3-month-old mdx mice: adhesion to the endothelium of muscle microcirculations; migration to limb muscles and dystrophin expression. A CD34+/-/Sca-1+/c-kit-/CD45- MD-SC population was purified from mice relying on their adhesion behaviour and transplanted into the limb muscles of 6–8-week-old mdx mice: dystrophin positive myofibres were detected 90 days after implantation; proliferation, self renewal capability and multipotency were assessed both in vitro and in vivo. Muscle side population (m-SP) cells were obtained from normal mice and injected IV into irradiated female mdx mice: up to 6% dystrophin positive myofibres after 30 days. FACS/Hoechst-purified m-SP cells were obtained from transgenic mice and injected into the regenerating TA of scid/bg mice: differentiation into myocytes and satellite cells and fiber regeneration in the injured site. Purified m-SP cells were isolated from donor mice and injected into damaged TA of irradiated mice: CD45+ m-SP integrated into regenerating myofibres. No heart data available. | [119,120,121,122,123] |
| Bone marrow-derived stem cells (BM-MSCs) | Preclinical—normal, scid/bg and mdx mice | Haematopoietic stem cells from normal mice were administered to irradiated female mdx mice through IV administration: up to 4% dystrophin positive myofibres detected 12 weeks after transplant in the TA muscle. BM-MSCs (GFP+) from donor mice were injected IV into irradiated mice: GFP+ cells were found into the TA muscle after 6 months, occupying the ablated satellite cell niche; regeneration of muscle fibres after exercise-induced damage. BM-MSCs cells from transgenic mice were injected IV into irradiated scid/bg mice with chemically induced TA muscle degeneration: migration of BM-MSCs to the injured site; myogenic differentiation and regeneration of the damaged fibres. No heart data available. | [121,124,125] |
| Mesoangioblasts | Preclinical—scid/mdx mice and GRMD dogs | Mesoangioblasts from mdx mice were genetically corrected with human artificial chromosome carrying the whole human dystrophin genetic locus; cells were lentivirally transduced with MyoD and nLacZ and transplanted into 4-month-old scid/mdx mice through IM and IA delivery: dystrophin positive myofibres; contribution to satellite cell pool and improved muscle function up to 8 months after administration. GRMD dogs received wild-type heterologous and genetically-corrected autologous mesoangioblasts after IA delivery: increased dystrophin expression; improved muscle function and mobility; low immune reaction. No heart data available. | [126,127] |
| Pericytes | Preclinical—scid/mdx mice | Pericyte-derived cells were isolated from muscular biopsies of human healthy and dystrophic subjects and transplanted into irradiated scid/mdx mice: colonization of host muscle and dystrophin expression. No heart data available. | [128] |
| CD133+ stem cells | Preclinical—scid/mdx mice. Clinical, Phase I—DMD boys | Blood- and muscle-derived CD133+ cells were isolated from human dystrophic subjects, genetically corrected to re-express dystrophin and injected into scid/mdx mice: restored dystrophin expression and recovery of muscle function. In a clinical trial, eight DMD boys were treated with autologous muscle-derived CD133+ cells through injection in the ADM muscle: increased number of capillaries per muscle fiber; switch from slow to fast myofibre type; assessed safety of the procedure; no alterations in the cardiac dimensions and function. | [129,130] |
| iPSCs | Preclinical—NSG/mdx mice | Pax7-derived myogenic progenitor cells were generated from human ESC and iPSC cells and injected in the TA of adult NSG/mdx mice: stable engraftment (up to 11 months); dystrophin expression; enrichment of satellite cell niche; improved muscle strength. No heart data available. | [131] |
| Antisense oligonucleotides (AONs) | |||
| PMO | Preclinical—mdx mice. Clinical, Phase IV | Weekly IV injections of PMO (skipping exon 23) into adult mdx mice: body-wide dystrophin expression in the skeletal muscles (>70% in quadriceps and gastrocnemius after seven injections), although low in the diaphragm and absent in the heart; transcript lacking exon 23. Another AON targeting the exon 51, namely eteplirsen (AVI-4658) was approved by FDA in 2016, although its effectiveness for the treatment of DMD remains controversial. | [132,133,134,135] |
| 2OMePS | Preclinical—mdx mice. Clinical, Phase III | Young to adult mdx mice were treated with IV injections with 2OMePs together with tryblock copolymer F127: exon 23 skipping confirmed; induced dystrophin expression in skeletal muscles, notably in the diaphragm, but not in the heart; more effectiveness in older mice; no toxic effects. In a Phase I/II clinical trial (NTR1241) 12 DMD patients were treated with PRO051 (drisapersen) (0.5, 2, 4 and 6 mg/kg, SC weekly injections) for 5 weeks to induce exon 51 skipping; extension of the treatment for 12 weeks (6 mg/kg per week): assessed safety; detectable exon 51 skipping; restored dystrophin expression; positive effects in 6MWD. A randomized, placebo-controlled Phase III trial (NCT01254019) in 186 ambulant boys aged ≥5 years again evaluated the long-term efficacy and safety of subcutaneous drisapersen (6 mg/kg/week, 48-week); a favourable response time in the 6MWD was recorded for drisapersen at 48 weeks with further analysis concluding suggesting drisapersen could specifically benefit a patient subpopulation with a milder disease impairment. Notably, drisapersen did not get FDA approval in 2016. | [134,136,137] |
| Tricyclo-DNA | Preclinical—mdx mice | The efficacy of tricyclo-DNA was compared with PMO and 2OMePS in mdx mice (weekly IV injections for 12 weeks, up to 200 mg/kg/week): greater efficacy in exon skipping compared with the other treatments; restored dystrophin expression, particularly in the heart (up to 53%); improved respiratory function and skeletal muscle function. | [138] |
| CPP-AOs-Pips | Preclinical—mdx mice | Adult mdx mice were treated with different peptide-PMO (i.e., Pip5-PMO) conjugates, IM or IV administered: Pip5e-PMO was the most efficient in terms of exon 23 skipping (in skeletal muscle and heart); high dystrophin expression in skeletal muscle and particularly in the heart (>90%) after single IV injection (25 mg/kg). Repeated IV administrations of Pip6f-PMOs were delivered to mdx mice (10 mg/kg each dose): restored dystrophin in the skeletal muscles and heart and prevented exercise-induced cardiomyopathy. | [139,140] |
| CPP-AOs-Phage Peptides | Preclinical—mdx mice | Adult mdx mice were treated with SC injection of 2OMePS conjugated to 7-mer peptide (50 mg/kg, four times a week for 6 weeks): conjugation promoted uptake and exon skipping in skeletal muscles and heart. | [141] |
| Possible other treatments and drug repositioning | |||
| P188 | Preclinical—mdx CMs and mice | Copolymer poloxamer P188 was administered both in isolated dystrophic CMs from mdx mice and to mdx mice: reduction of stretch-induced calcium overload and increased compliance in vitro; improved cardiac haemodynamic performance in vivo also after dobutamine stress induction. | [142] |
| MG53 | Preclinical—mdx mice | Recombinant human mitsugumin 53 (rhMG53) was injected IV and SC into adult mdx mice: reduced muscular pathology; prevention of exercise-induced damaged; no toxic effects. | [143] |
| Sildenafil | Preclinical—mdx mice. Clinical, Phase II—DBMD adults | Phosphodiesterase 5 inhibitor sildenafil was administered daily to mdx mice for 6 weeks: no alteration of cardiac haemodynamic parameters; decreased CM sarcolemmal injury (≈44%) after damage with isoproterenol. Sildenafil was daily administered for 14 weeks to mdx mice: increased diaphragm strength (≈15%) and anti-fibrotic effect; no impact on fatigue resistance; decreased muscular membrane permeability. In a clinical trial (NCT01168908), 20 adults MD patients were randomized to be treated with sildenafil (20 mg 3x daily) for 6 months, then additional 6 month treatment was administered to all patients: treated patients showed worsening of cardiac conditions (increased LVESV) and no statistically significan effect of the treatment was found; premature termination due to futility. | [144,145,146] |
| Losartan | Preclinical—mdx mice | Female mdx mice were treated with losartan for 6 months: decreased skeletal and cardiac muscle fibrosis; improved cardiac function (increased shortening fraction and reduced systolic blood pressure). | [147] |
| Pirfenidone | Preclinical—mdx mice | Aged mdx mice were treated with pirfenidone for 7 months: improved cardiac contractility and developed pressure and relaxation over time (±dP/dt); decreased level of TGF-β mRNA but no antifibrotic effect. | [148] |
| Myostatin | Clinical, Phase I/II—MD adults. Clinical, Phase II—DMD boys | Inhibition of myostatin/ActRIIB signaling pathway has been proven to improve dystrophic pathology in mdx mice and GRMD dogs preclinical models. In a clinical trial (NCT00104078), MYO-029, a neutralizing antibody for myostatin, was employed to treat 116 subjects with different types of MD; the compound was administered in different dosage (1, 3, 10 and 30 mg/kg) every two weeks for 6 months, followed by 3 months of follow up: assessed tolerability and safety; no improvement of muscle function. In another trial (NCT01239758) 11 DMD boys were treated with ACE-031 (0.5, 1.0 and 2.5 mg/kg) every 4 for 24 weeks: significant side effects were observed (e.g., nosebleeds, gum bleeding and dilated skin blood vessels); premature stop of the trial due to preliminary safety data. | [149,150] |
| Urocortin | Preclinical—mdx mice | Urocortins Ucn1 and Ucn2 were daily administered SC to young mdx mice for 2 weeks: improved muscle function; reduction of necrosis, particularly in the diaphragm; resistance to mechanical stress provoked by repetitive tetanization; contribution to calcium homeostasis. | [151] |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
D’Amario, D.; Gowran, A.; Canonico, F.; Castiglioni, E.; Rovina, D.; Santoro, R.; Spinelli, P.; Adorisio, R.; Amodeo, A.; Perrucci, G.L.; et al. Dystrophin Cardiomyopathies: Clinical Management, Molecular Pathogenesis and Evolution towards Precision Medicine. J. Clin. Med. 2018, 7, 291. https://0-doi-org.brum.beds.ac.uk/10.3390/jcm7090291
D’Amario D, Gowran A, Canonico F, Castiglioni E, Rovina D, Santoro R, Spinelli P, Adorisio R, Amodeo A, Perrucci GL, et al. Dystrophin Cardiomyopathies: Clinical Management, Molecular Pathogenesis and Evolution towards Precision Medicine. Journal of Clinical Medicine. 2018; 7(9):291. https://0-doi-org.brum.beds.ac.uk/10.3390/jcm7090291
Chicago/Turabian StyleD’Amario, Domenico, Aoife Gowran, Francesco Canonico, Elisa Castiglioni, Davide Rovina, Rosaria Santoro, Pietro Spinelli, Rachele Adorisio, Antonio Amodeo, Gianluca Lorenzo Perrucci, and et al. 2018. "Dystrophin Cardiomyopathies: Clinical Management, Molecular Pathogenesis and Evolution towards Precision Medicine" Journal of Clinical Medicine 7, no. 9: 291. https://0-doi-org.brum.beds.ac.uk/10.3390/jcm7090291